
INTRODUCTION
Plasma cell leukemia (PCL) is a highly aggressive form of plasma cell (PC) dyscrasia defined by the presence of more than 20% of circulating PCs in peripheral blood and/or an absolute circulating PC count greater than 2 × 109/l [1]. PCL can be classified as either primary (pPCL), originating de novo at diagnosis, or secondary (sPCL), as a progression from a previous multiple myeloma (MM). PCLs occur rarely, being observed in approximately 2–4% of PC dyscrasias, with pPCL accounting for approximately 50–70% of all cases [2–4]. PCL prognosis is very poor, with a median survival of 7–13 months for pPCL, and even worse for sPCL (2–7 months) [4, 5]. The pathogenetic mechanisms involved in either the primary or secondary forms remain to be fully elucidated [1, 4, 5].
As demonstrated by us and others, PCL shows more complex and heterogeneous molecular patterns than MM, with a greater number of associated genomic aberrations and, in particular, a higher incidence of 13q and 17p deletions and translocations involving immunoglobulin heavy chain locus, whereas very few patients with hyperdiploidy have been described [3, 6–10]. Overall, these findings also suggest that pPCL may represent a valid model for genetic and molecular alterations associated with high-risk MM.
Recently, we provided an extensive biological and molecular characterization of a panel of 23 pPCLs included in a Phase II prospective trial, aiming to evaluate the efficacy of novel biological drugs in the treatment of this aggressive form [11]. In particular, our data based on FISH and SNP array techniques showed the presence of IgH translocations in 87% of pPCL cases, with prevalence of t(11;14) and t(14;16), the occurrence of chr 13 deletion in 74% and of TP53 deletion/mutations in approximately 40% of cases, and a low frequency of BRAF and RAS gene mutations [10]. Furthermore, microarray expression profiling revealed the presence of specific transcriptional signatures compared to MM, identifying genes and miRNAs potentially associated with clinical outcome in pPCL [12, 13].
Recently, next-generation sequencing (NGS) studies of MM enlarged the knowledge about the genetic landscape of this disease. Indeed, they collectively provided remarkable information concerning gene mutation signatures as well as the clonal and subclonal heterogeneous structure at diagnosis and its evolution during disease progression [14–17]. However, to the best of our knowledge, no NGS data are currently available in representative series of PCL. Here, we took advantage of our prospective series to decipher, by means of whole-exome sequencing (WES), the pattern of coding somatic mutations in pPCL. Sequencing results were combined with copy number and gene expression data to achieve a comprehensive and integrated view of the molecular landscape of pPCL.
RESULTS
Identification of coding somatic variants in pPCL
WES of 12 highly purified PC samples, together with five matched normal controls, was performed by Illumina technology. A mean of 95 M raw reads was generated per sample, of which 98% uniquely mapped to the GRCh37 reference genome. Referring to captured regions (62 Mb), we achieved a target coverage of 97% and a mean depth of 42.3x for both tumor and control cases (Suppl Table S1).
Since matched controls were not available in all pPCL cases, traditional somatic callers such as Mutect could not be used in our study. Therefore, to analyze all the 12 samples using the same method, we implemented a custom pipeline based on GATK caller using at first public databases and then the pool of control samples to filter out germline variants. Following this approach, we initially identified more than 52, 000 raw variants per tumor sample, including both single nucleotide variants (SNVs) and insertions/deletions (indels) (Suppl Table S2). Since our primary interest was in coding regions and splice sites, we applied a series of stringent filters to select coding non-silent variants, including non-synonymous (non-syn) SNVs and indels. First of all, after evaluation of target coverage in relation to sequencing depth, we chose to set at 10x the threshold to reduce the false positive rate. At this depth, all samples (both tumor and control cases) had at least 85% of covered target (Suppl Figure S1). Then, we removed all positions mapping out of coding regions and all variants already annotated in public databases, unless the same variant was also included in COSMIC or NCBI ClinVar catalogues (see Materials and Methods). Thus, we obtained a mean of 1, 525 coding non-syn SNVs and 130 coding indels per tumor sample (Suppl Table S2). The CD138-negative bone marrow populations, available for five patients, were used at this step to filter out germline background, thus obtaining a mean of 149 and 9 coding somatic non-syn SNVs and indels per sample, respectively (Table 1). Lastly, we addressed specific analyses to the identification of somatic sub-clonal variants in the five tumor/normal pairs, and we found 93 sub-clonal non-syn SNVs and 1 sub-clonal insertion in 94 genes (Table 1). All but one (in TCHH gene) were private variants occurring in individual cases (details in Suppl File S1). Full description of bioinformatics pipeline is provided in Supplementary Methods.
Table 1: Coding somatic non-silent variants identified in pPCL panel
Sample |
Somatic |
Somatic sub-clonal |
Total No. Non-syn SNVs |
Total No. Indels |
Total No. Somatic Variants |
||
Non-syn SNVs |
Indels |
Non-syn SNVs |
Indels |
||||
PCL-016 |
148 |
4 |
17 |
0 |
165 |
4 |
169 |
PCL-017 |
160 |
11 |
0 |
0 |
160 |
11 |
171 |
PCL-018 |
25 |
2 |
9 |
1 |
34 |
3 |
37 |
PCL-019 |
133 |
5 |
17 |
0 |
150 |
5 |
155 |
PCL-020 |
195 |
15 |
0 |
0 |
195 |
15 |
210 |
PCL-026 |
232 |
6 |
34 |
0 |
266 |
6 |
272 |
PCL-027 |
156 |
16 |
0 |
0 |
156 |
16 |
172 |
PCL-030 |
179 |
11 |
0 |
0 |
179 |
11 |
190 |
PCL-032 |
176 |
8 |
0 |
0 |
176 |
8 |
184 |
PCL-035 |
203 |
14 |
0 |
0 |
203 |
14 |
217 |
PCL-036 |
146 |
16 |
0 |
0 |
146 |
16 |
162 |
PCL-038 |
40 |
2 |
17 |
0 |
57 |
2 |
59 |
Globally, we identified a total of 1, 928 coding somatic non-silent variants (mean 166 per sample), including 1, 831 non-syn SNVs (missense and nonsense), 90 indels and 7 splice site variants, distributed on 1, 643 protein-coding genes (Figure 1A). All variants are provided with full annotation in Supplementary File S1 and summarized by gene in Supplementary File S2. Eighty-one variants were validated by independent sequencing experiments, with a confirmation rate of 95%, in line with other cancer genome sequencing studies (Suppl File S3).
Concerning nucleotide changes, we observed an excess of C > T transitions, in agreement with what recently reported in MM [16] (Figure 1B). Furthermore, when looking at nucleotide context for coding somatic mutations in the whole dataset, we observed the occurrence of two main mutational signatures (Figure 1C). The first signature (corresponding to Signature 1A, according to the current classification [18]) was dominated by C > T transitions at NpCpG sites resulting from the spontaneous deamination of 5-methyl cytosine associated with aging. The second signature (Signature 2) was characterized by the prevalence of C > T and C > G in a TpCpN context, resulting from the over-activity of APOBEC cytidine deaminase enzymes [18]. Both signatures were recently described in MM [16] as well as in other cancer types [18]. Evaluating the contribution of each nucleotide context in individual pPCL samples, few cases appeared driven by one single mutational signature, while the majority showed an admixture of the two processes (Suppl Figure S2). In line with recently published data, this seems to be a common feature in cancer genomes, regardless of tumor type [18]. Interestingly, when considering the translocation groups of our dataset, three out of the four samples carrying the t(14;16) translocation involving MAF gene showed a distinct APOBEC mutational signature, while among the other five samples carrying t(11;14) or t(4;14), one case presented a distinct age-related signature and three other samples showed an admixture of both Signatures 1A and 2, with the predominant contribution of age-related process. Thus, in agreement with what recently presented for MM by Walker et al. [19], we can confirm also for pPCL (although in a relatively small dataset) that t(14;16) cases predominantly had an APOBEC signature.
Integration with copy number alterations
Somatic copy number alterations (CNAs) in the 12 pPCLs were identified from WES data using EXCAVATOR software. Globally, we found a mean of 20 CNA traits per sample, with more frequent deletions than amplifications (Figure 2; Suppl File S4). The CNAs here identified were concordant to those previously characterized in the same samples by using Affymetrix SNP Array [10] and in agreement with the known PCL genomic signature, including deletions of chrs 1p, 8p, 13q and 17p, and gain of chr 1q. To evaluate the global amount of somatic alterations carried by each of the 12 pPCLs, coding somatic variants (non-syn SNVs and indels) and CNA results were summed up by sample (Figure 3). PCL-026 and PCL-020 resulted the most impacted samples, with the highest number of variants and CNAs, respectively.
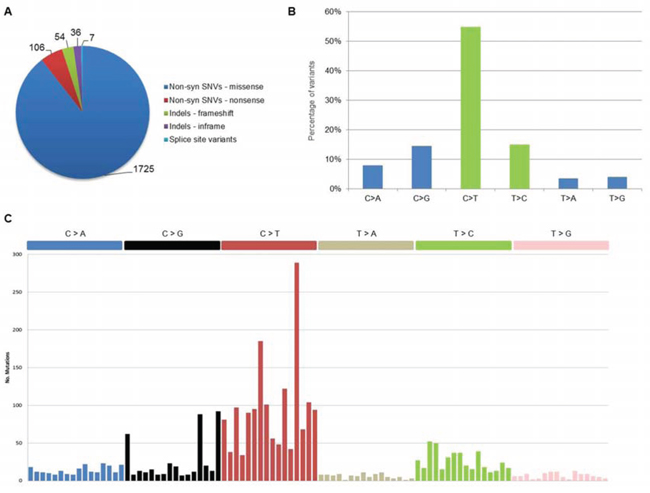
Figure 1: Coding somatic variants identified in the 12 pPCL samples. A. Distribution of coding somatic non-silent variants according to amino acid changes or reading frameshifts. B. Classification of coding somatic SNVs (both syn and non-syn) by nucleotide change. Transitions are in green (C > T, T > C) and transversions are in blue (C > A, C > G, T > A, T > G). C. Mutational signatures in pPCL. Coding somatic SNVs identified in the 12 samples were classified according to nucleotide change and sequence context into the 96 possible mutated trinucleotides (listed horizontally according to the conventional order). Substitutions are displayed in different colors along the horizontal axis and number of mutations attributed to each type is shown on the vertical axis.
Lastly, in order to assess CN state of the 1, 643 mutated genes, WES and CNA results were integrated for all the 12 pPCLs (Suppl File S5). We identified three homozygous deletions involving three genes, namely BIRC2 on chr 11 and TRPM6 on chr 9 (both in PCL-019) and TRAF3 on chr 14 (in PCL-017). Furthermore, we found 77 genes concomitantly affected by mutation and 1-copy loss, suggesting a potential tumor suppressor gene (TSG) function (Suppl File S5). Among them, TP53 resulted the most recurrently disrupted gene (three missense variants and one frameshift indel in four cases, all associated with deletion of the other allele (see below for further details)), while BIRC2 and TRAF3, besides the homozygous deletions above described, were found biallelically affected in two additional cases, presenting respectively a truncating mutation and a frameshift deletion, both associated with loss of the remaining allele.
Candidate driver genes in pPCL
Using DOTS-Finder tool and taking into account gene mutation rates calculated in our samples with respect to COSMIC and TCGA databases, we identified 14 statistically significant recurrently affected genes with potential driver role in pPCL. Among them, we found genes involved in cell-matrix adhesion and membrane organization (SPTB, CELA1), cell cycle and apoptosis (CIDEC), genome stability (KIF2B), RNA binding and degradation (DIS3, RPL17), and protein folding (CMYA5) (Table 2). In particular, DIS3, coding for an RNA exonuclease catalytic subunit of the exosome complex and described as one of the most recurrently mutated genes in MM [14, 17, 20], resulted affected in three PCLs (25%). In two samples the mutation involved the same aminoacid position but introducing two different changes (R780K and R780T), both already reported in literature for MM [17, 20]; in the third case, the mutation was in homozygous state, associated with deletion of the other allele. In addition, the gene showed hemizygous deletion in other seven pPCLs. Although deserving investigations in larger collections to assess their actual recurrence, these genes might represent novel potential markers for translational clinical applications.
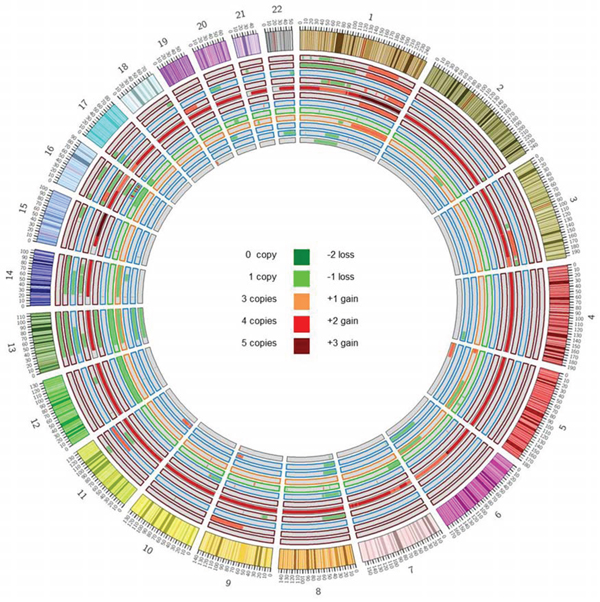
Figure 2: Copy number analysis. Copy number alterations were identified starting from WES data by using EXCAVATOR software. The Circos plot represents somatic amplifications (red) and deletions (green) found in each case, distributed across all chromosomes. Samples are displayed according to increasing case number from outer to inner track. Colors of track borders indicate translocation groups: t(14;16) in red, t(11;14) in blue, t(14;20) in green, t(4;14) in orange. PCL-020 resulted as outlier with the greatest number of CNAs and a basal tetraploidy with regions till to five copies.
Pathways affected by somatic mutations in pPCL
After removal of potentially spurious genes frequently mutated in cancer but not necessarily relevant to tumor biology [21], functional enrichment analysis of the mutated genes using ToppGene evidenced five significantly enriched pathways, including cadherin and Wnt signaling, extracellular matrix (ECM)-receptor interaction, ECM organization, and G2/M cell cycle checkpoint (Table 3).
In cadherin/Wnt signaling pathways, we globally recognized 51 genes, 30 of which (59%) showed a cadherin domain. Among cadherin genes, the most recurrent ones were PCDH15 (four cases), DCHS1 and FAT3 (each in three samples). Ten pPCLs showed mutations in two or more cadherin genes. Furthermore, cadherin genes were principally identified in the protocadherin (PCDH) family (19/30, 63%). Most PCDH mutated genes (15/19, 79%) belonged to the three tandem 5q31 region gene clusters [22].
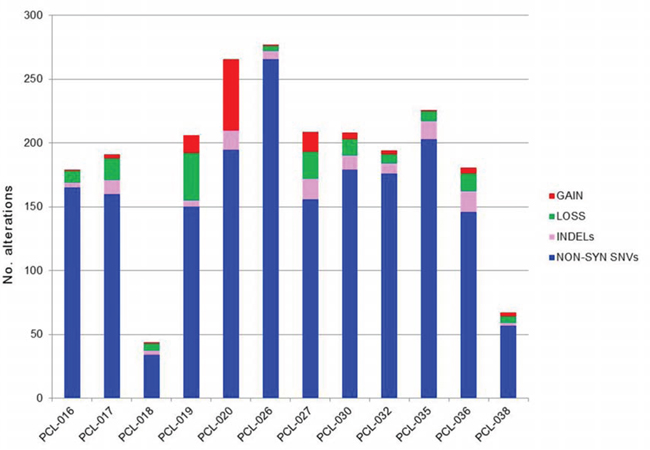
Figure 3: Global amount of somatic alterations per sample. The numbers of coding somatic non-silent variants (non-synonymous SNVs and indels) and CNA regions (gains and losses) were summed up by sample, to have an overview of the global burden of somatic alterations carried by each case.
Notably, 49 genes were involved in ECM-receptor interaction and organization pathways, 34 of which (69%) annotated as proteinaceous ECM cellular components, including collagen and laminin genes, ADAM metallopeptidases and members of the aggrecan/versican proteoglycan family. Ten PCLs resulted affected in these ECM cellular components, and six cases presented mutations in five genes or more.
Finally, 10 genes were involved in G2/M cell cycle checkpoint pathway in nine PCLs. Among them, TP53 and ATM (four cases) were the most recurrent ones, followed by ATR and CDKN1A (three cases) and BRCA1 (two cases). Notably, six PCL cases showed the involvement of the kinase ATM (PCL-017, PCL-026, PCL-030) or ATR (PCL-019, PCL-035) in a mutually exclusive fashion, or in a combined manner (PCL-020). Additionally, TP53 gene was frequently found altered in association with at least one another gene of this pathway: ATM (PCL-017), CDKN1A and RPS6KA1 (PCL-027), or both ATM and CDKN1A, together with CDC25A, CHEK2 and PRKDC genes, in the most affected case (PCL-030). No concurrent mutations in ATR and TP53 were evidenced.
Furthermore, we identified other eight affected pathways that, although did not reach statistical significance after p-value correction, are noteworthy since notoriously involved in MM and lymphoid malignancies [23, 24] and targeted by current molecular therapies [25]. They included the proteasome and ubiquitin-mediated proteolysis machineries and the TNF, NF-kB, PI3k-Akt, MAPK and Hippo signaling pathways (Suppl Table S3).
Interestingly, if looking at impact prediction of variants affecting all these pathways, we observed a statistically significant enrichment for damaging variants (P < 0.01, Fisher exact test) (Suppl Figure S3A). The probability of having the same fraction of damaging variants randomly sampling the same number of mutations in our dataset was estimated to be 10−5 and 10−4 for the five enriched and the eight selected pathways, respectively (Suppl Figure S3B–S3C). Moreover, when tested individually, Wnt signaling and G2/M cell cycle checkpoint (q-value < 0.05) and MAPK pathway (q-value < 0.01) maintained a significant enrichment for damaging variants with respect to background.
Comparison with multiple myeloma recurrent genes
We evaluated in our series the occurrence of somatic variants in 11 genes recurrently mutated in MM [17]. Globally, seven genes were affected in our samples (TP53, DIS3, BRAF, KRAS, NRAS, FAM46C, TRAF3), while ACTG1, CYLD, PRDM1 and RB1 carried no variant (Suppl Table S4). TP53 was the most frequent, completely inactivated in four pPCLs (33%) as above mentioned. Three pPCLs (25%) carried variants in DIS3, in addition to other seven samples showing hemizygous deletion, and this gene resulted a statistically significant driver gene in our dataset. The remaining five genes showed individual missense and frameshift variants. BRAF carried two missense mutations in the same sample, both already described in MM. FAM46C presented a novel variant, never previously reported for other tumors, besides being deleted in other five PCLs. KRAS and NRAS resulted affected in two distinct samples and both mutations were already described for MM; moreover, NRAS resulted deleted in five PCLs, while amplified only in the sample harboring the mutation. Finally, TRAF3 was completely inactivated in two samples, as aforementioned. All variants were confirmed by Sanger sequencing (Suppl File S3). Overall, we observed that KRAS and NRAS were three-fold less frequent, while DIS3 and TP53 were till to two-fold more recurrent in pPCL than MM considered as a whole [16, 17, 20] (Figure 4).
Table 2: Statistically significant recurrently affected genes with potential driver role in pPCL
Gene |
Gene Name |
No. Cases (%) |
No. Variants |
No. Non-syn SNVs |
No. Indels |
q-value |
Function |
HLA-DQA1 |
major histocompatibility complex, class II, DQ alpha 1 |
3 (25%) |
3 |
- |
3* |
3.89E-09 |
immune response |
CIDEC |
cell death-inducing DFFA-like effector c |
3 (25%) |
1 |
1* |
- |
3.07E-05 |
apoptosis |
CELA1 |
chymotrypsin-like elastase family, member 1 |
3 (25%) |
1 |
- |
1 |
7.42E-05 |
proteolysis, cell membrane and matrix organization |
SRRM5 |
serine/arginine repetitive matrix 5 |
3 (25%) |
2 |
2* |
- |
7.89E-05 |
unknown |
CCDC144NL |
coiled-coil domain containing 144 family, N-terminal like |
3 (25%) |
2 |
- |
2 |
8.59E-05 |
unknown |
SPTB |
spectrin, beta, erythrocytic |
3 (25%) |
5 |
5 |
- |
0.00280 |
cell membrane organization and stability |
DIS3 |
exosome catalytic subunit DIS3 |
3 (25%) |
3 |
3 |
- |
0.00427 |
RNA binding, RNA degradation |
FAM166B |
family with sequence similarity 166, member B |
2 (16%) |
1 |
- |
1* |
0.01062 |
unknown |
RPL17 |
ribosomal protein L17 |
2 (16%) |
2 |
1 |
1 |
0.01405 |
RNA binding, translation |
CMYA5 |
cardiomyopathy associated 5 |
1 (8%) |
7 |
7 |
- |
0.02032 |
protein binding |
UNC80 |
unc-80 homolog |
4 (33%) |
4 |
4 |
- |
0.02762 |
ion transmembrane transport |
SCN9A |
sodium channel, voltage-gated, type IX, alpha subunit |
3 (25%) |
3 |
3 |
- |
0.03008 |
ion transmembrane transport |
ZNF598 |
zinc finger protein 598 |
3 (25%) |
1 |
- |
1* |
0.03008 |
translation repressor |
KIF2B |
kinesin family member 2B |
2 (16%) |
2 |
1 |
1 |
0.09976 |
chromosome segregation, genome stability |
Note: *Identical recurrent variants found in unpaired pPCL samples annotated in both dbSNP and COSMIC catalogue and confirmed somatic in COSMIC website.
Table 3: Pathways significantly enriched in mutated genes by ToppGene analysis
Pathway |
Source |
p-value |
FDR q-value |
No. Damaging/Total variants |
Genes |
Cadherin signaling pathway |
PantherDB (ID P00012) |
1.26E-07 |
3.07E-04 |
24/48 |
PCDH15*, FZD6, PCDH7*, PCDH20*, CDH20*, DCHS1*, CDH17*, CDH4*, CDH9*, CDH23*, CTNNA2, PCDHGB1*, PCDHGC5*, PCDHGC4*, PCDHGA2*, PCDHGA1*, PCDHA7*, PCDHA13*, PCDHAC1*, PCDHB2*, PCDHB3*, PCDHB7*, PCDHB8*, PCDHA2*, PCDHA1*, PCDHA3*, CELSR3*, FAT1*, FAT2*, FER, FZD10, CDHR2*, FAT3*, YES1, PCDH11X* |
ECM-receptor interaction |
BioSystems: KEGG (ID 83068) |
2.01E-06 |
2.45E-03 |
9/26 |
CD36, SV2B, COL1A2†, COL1A1†, COL5A1†, COL6A3†, COL4A2†, DAG1†, COL6A6†, RELN†, TNN†, FN1†, THBS3, TNXB†, HMMR, HSPG2†, ITGA1, LAMB1†, LAMA4†, LAMA5†, LAMA2†, LAMA3† |
Cell Cycle G2/M Checkpoint |
MSigDB C2: BioCarta (ID M8560) |
1.25E-05 |
1.02E-02 |
13/20‡ |
ATM, ATR, BRCA1, CDC25A, CDKN1A, PRKDC, EP300, CHEK2, RPS6KA1, TP53 |
Wnt signaling pathway |
PantherDB (ID P00057) |
2.17E-05 |
1.18E-02 |
36/66‡ |
MYH13, PCDH15*, FZD6, PCDH7*, MYH7, PCDH20*, CDH20*, DCHS1*, CDH17*, CDH4*, CDH9*, CDH23*, PRKCZ, PPP2R5E, PPP3R2, CREBBP, CTNNA2, PLCB4, PCDHGB1*, PCDHGC5*, PCDHGC4*, PCDHGA2*, PCDHGA1*, DVL3, PCDHA7*, PCDHA13*, PCDHAC1*, PCDHB2*, PCDHB3*, PCDHB7*, PCDHB8*, PCDHA2*, PCDHA1*, PCDHA3*, EP300, CELSR3*, FAT1*, FAT2*, SRCAP, FZD10, INO80, TP53, CDHR2*, FAT3*, PCDH11X*, KREMEN1, ITPR2, MYH14, PLCB1 |
Extracellular matrix organization |
BioSystems: REACTOME (ID 576262) |
2.50E-05 |
1.18E-02 |
22/51 |
ACTN1, ACAN†, COL6A5†, DDR2, LTBP4†, ADAMTS9†, COL1A2†, COL1A1†, COL15A1†, COL17A1†, COL9A3†, PDGFB, COL9A1†, COL5A1†, COL6A3†, COL4A2†, PLEC, VCAN†, DAG1†, ADAMTS16†, ADAMTS18†, COL6A6†, BCAN†, PSEN1, DSPP†, TNN†, PTPRS, FBN2†, FGA, FN1†, ADAMTS5†, TNXB†, ADAM18, LEPREL1†, HSPG2†, ICAM4, ITGA1, LOXL4, LAMB1†, LAMA4†, LAMA5†, LAMA2†, LAMA3†, FBN3† |
Note: *30 genes showing a cadherin domain according to significant enrichment by ToppGene analysis (InterPro IPR002126 domain, q-value 3.24E-08, FDR correction). †34 genes annotated as proteinaceous extracellular matrix (ECM) cellular components according to significant enrichment by ToppGene analysis (GO Cellular Component GO:0005578, q-value 4.62E-04, FDR correction). ‡Significant enrichment for damaging variants (q-value < 0.05, Bonferroni correction).
Gene expression profiles of mutated genes in MMs and pPCLs
A proprietary dataset of 55 MM and 21 pPCL cases (comprising the 12 in this study) and four bone marrow healthy donors profiled on Affymetrix Gene 1.0 array [12] was exploited to assess the expression profiles of the mutated genes (1, 598 of which included in the chip array). A hierarchical clustering based on the 132 mutated genes found among the most variably expressed genes (2AVEFC) of our dataset revealed that sample grouping was principally driven by the main cytogenetic translocations (Figure 5). Specifically, we found a good separation of the two t(4;14) (TC4) and t(14;16)/t(14;20) (TC5) MM-PCL sample groups (15/15 and 14/14, respectively; P < 10−4), whereas t(11;14) cases (TC1) were divided in two sub-branches, clearly separating MM from PCL samples (11/12 MMs and 7/8 PCLs; P < 10−4). Additionally, three distinct clusters were identified for TC2 (12/13; P < 3 × 10−4) and TC3 MM cases (7/12; P < 10−4, and 4/12; P < 9.4 × 10−3). Notably, functional enrichment in regulation of cellular developmental process and morphogenesis, phosphatase activity, ECM/cytoskeleton organization, cell adhesion and endocytosis processes was observed in these 132 genes (Suppl Table S5). Interestingly, the probability of replicating the same sample clustering using a comparable number of genes randomly extracted from the whole 2AVEFC list (regardless of mutational state) was calculated as P < 0.004. This finding indicated that these 132 genes, representing the 11% of the 2AVEFC list, had peculiar clustering abilities and strongly suggested that genes found mutated in pPCL may also have relevant biological roles in the context of the different molecular and clinical types of MM.
Finally, a two-class supervised analysis comparing pPCL to MM samples on the 1, 598 mutated genes identified 102 differentially expressed genes (DEGs), of which 42 up- and 60 down-regulated in pPCL versus MM group (Suppl Table S6). Specifically, up-regulated genes were functionally enriched in regulation of transcriptional processes, while several down-regulated genes were involved in cell death regulation. Interestingly, concerning genes included in the five enriched functional pathways aforementioned, we found ATM and CHEK2 (G2/M cell cycle checkpoint) as well as CD36, HMMR and RELN (ECM-receptor interaction pathway) as down-regulated in pPCL versus MM, whereas EP300 (Wnt signaling pathway) resulted up-regulated in pPCL as compared to MM samples. Notably, 33 out of the 102 mutated DEGs (32%) were in common with our published 503-gene transcriptional signature distinguishing pPCL and MM [12].
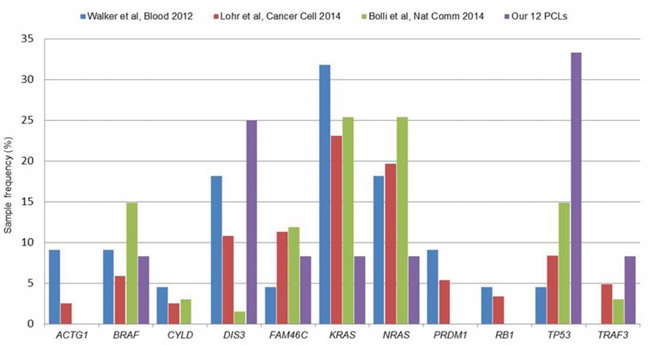
Figure 4: Recurrence of the 11 multiple myeloma genes in our pPCL series with respect to MM published datasets. Recently published datasets for multiple myeloma include: Walker et al., Blood 2012 (22 cases, [20]), Lohr et al., Cancer Cell 2014 (203 cases, [17]) and Bolli et al., Nat. Commun. 2014 (67 cases, [16]). Recurrence rates were calculated as the percentage of positive samples on the total number of cases included in each study.
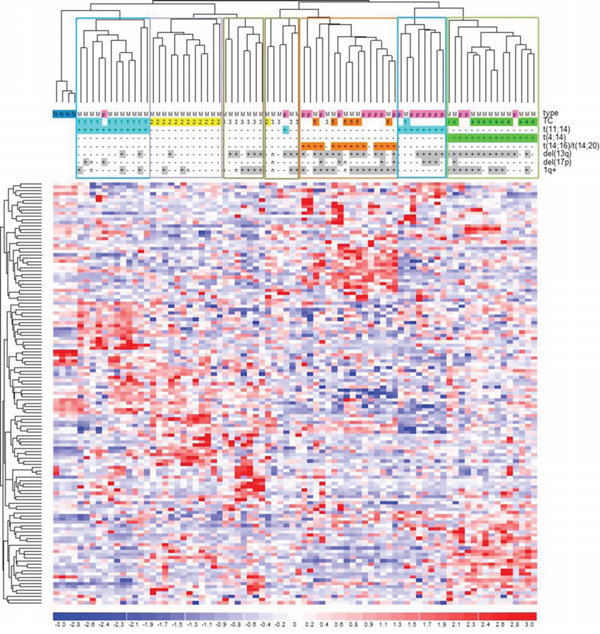
Figure 5: Hierarchical clustering of the 55 MM-21pPCL-4N dataset based on the expression levels of the 132 most variable and mutated genes. Data were previously generated on GeneChip Human Gene 1.0 ST array platform [12]. Samples were labelled according to tumor type (MM, multiple myeloma; p, primary plasma cell leukemia), translocation/cyclin D (TC) classification and the main associated cytogenetic alterations. Significant sample clusters, according to major IgH translocations, are highlighted.
DISCUSSION
Over the last few years, NGS technology has extended our knowledge of the genetic alterations associated with transformation and progression in MM. Notably, these studies showed a considerable number of somatic variants per patient, few recurrently mutated genes with pathogenetic significance, and an heterogeneous sub-clonal structure at presentation [16, 17, 20, 26]. Furthermore, although performed on only two PCL cases, Walker et al. [15] recently demonstrated that number and pattern of somatic variants at PCL stage are higher and more complex than in MM, smoldering multiple myeloma (SMM) and monoclonal gammopathy of undetermined significance (MGUS). In the present study, we aimed to provide novel and more comprehensive information concerning the mutational profile of pPCL by performing WES of highly purified malignant PCs from a prospective panel of pPCL patients already extensively characterized at clinical and molecular levels in our previousreports [10–13].
Our WES analysis in pPCL confirmed and extended the remarkable genetic heterogeneity associated with MM. Based on our results, 1, 928 coding somatic non-silent variants distributed on 1, 643 genes were identified in the whole dataset, with a mean of 166 variants per sample. However, as already observed for MM, the recurrence of identical variants did not appear a frequent event in pPCL, where only 1.9% of variants were recurrent in two or more samples, even if affecting genes with pathogenetic relevance already reported in COSMIC and MM [14]. Though we could have slightly over-estimated the number of identified variants due to the availability of paired samples only in five patients, our study represents so far the most comprehensive overview of the mutational patterns of primary PCL.
In our study on pPCL, we observed that, among those genes found recurrently mutated in MM, KRAS and NRAS were three-fold less frequent, while DIS3 and TP53 were till to two-fold more recurrent in pPCL than MM considered as a whole [16, 17, 20]. These differences in mutational frequencies could be partially explained by the composition of our pPCL series in terms of cytogenetic abnormalities, not strictly comparable to that generally observed for MM. This mainly concerns TP53 and DIS3, whose mutations have been reported in several MM series as preferentially associated, respectively, with chr 17p deletion [16, 27] and non-hyperdiploid status [17]. Both these conditions are over-represented in our pPCL dataset, in line with previous reports in other pPCL series [3, 9, 28]. In particular, as regards TP53 mutations, it should be noted, as aforementioned, that all the four TP53-mutated cases also carried del(17p), and the frequent co-occurrence of these two genetic alterations is compatible with the demonstrated association of TP53 inactivation with advanced forms of malignancy (also substantiated by its nearly ubiquity in MM cell lines) [29]. Three of the four TP53 mutations we found (I195T, R273C, P278L) were deleterious missense variants targeting the DNA binding domain and frequently reported as mutated (IARC TP53 Database, http://p53.iarc.fr/), also in MM patients [16, 17]; the fourth variant was a deletion of ten nucleotides, of which eight at the 3′-end of intron 9 and two at the 5′-end of the following exon 10, introducing a reading frameshift and consequently a premature stop codon in the first part of the oligomerization domain (I332Pfs*4). As regards NRAS and KRAS, a slight enrichment of mutated cases among hyperdiploid MM patients can be observed in both the main WES studies for MM: both NRAS and KRAS were reported mutated in 18.5% of non-hyperdiploid (NHD) and 30% of hyperdiploid (HD) patients in the dataset from Bolli et al. [16], while in the cohort by Lohr et al. [17] NRAS mutations were detected in 16.3% of NHD and 22.4% of HD patients, and KRAS mutations in 18.6% of NHD and 26.7% of HD cases (although association did not reach statistical significance). Based on these data, the low occurrence of NRAS and KRAS mutations in our 12 pPCLs could be partly expected. Concerning DIS3, although its role in the pathogenesis of the disease remains to be elucidated, this gene was characterized as potential tumor suppressor in MM. This suggestion is based on the loss of enzymatic activity caused by the MM-associated DIS3 mutations that have been functionally studied [30], and on the observation of frequent loss of heterozygosity involving DIS3 (also occurring in our dataset) due to the combination of gene mutation and chr 13 deletion. Anyhow, further investigations in considerably larger cohorts will be definitely useful to confirm the involvement of these genes in pPCL and their actual recurrence with respect to MM.
Interestingly, we identified significantly enriched pathways associated with mutated genes in pPCL, namely cadherin/Wnt signaling, ECM-receptor interaction and organization, and G2/M cell cycle checkpoint. Notably, both Wnt signaling and G2/M cell cycle checkpoint pathways resulted enriched also in damaging variants, thus enforcing the hypothesis of their crucial involvement in pPCL biology. Finally, several of the candidate driver genes found in our study were included or strictly correlated with these pathways.
Cadherins play a major role in cell-cell adhesion and tissue integrity and homeostasis maintenance and their alterations are associated with tumorigenesis and metastasis; in particular, several protocadherins were recognized as candidate TSGs, mainly silenced by methylation in different solid tumors [31]. Furthermore, cadherins are functionally associated with cytoplasmic members of the catenin family, including β-catenin which represents a crucial player in Wnt signaling [32]. Notably, two genes of the cadherin/Wnt cascade, PPP2R5E and EP300, resulted as potential drivers for pPCL (though with q-value slightly above significance threshold, probably due to the small size of our dataset). PPP2R5E, involved in apoptosis control, was proposed as TSG in acute myeloid leukemia [33]. EP300 encodes a histone acetyltransferase, whose altered transcriptional and epigenetic functions were associated to leukemia and other cancers [34]. Notably, EP300/β-catenin interaction is critical in Wnt pathway regulation and particularly in mediating neoplastic transformation [35]. Moreover, among the mutated cadherin genes interacting with Wnt pathway, FAT1 (FAT atypical cadherin 1) was found mutated in solid tumors [36] and chronic lymphocytic leukemia [37]. When inactivated, FAT1 is unable to sequester β-catenin at the cell membrane, which in turn promotes the activation of Wnt cascade and tumor growth [38]. Notably, other FAT genes frequently mutated in solid tumors [38] resulted affected in our series, such as FAT2 (one case) and FAT3 (three cases), in a mutually exclusive fashion. Interestingly, FAT3 was found recurrently mutated in a recently published MM series [16].
ECM is part of the bone marrow (BM) microenvironment and includes many components as fibronectin, laminins, collagens and proteoglycans. ECM can provide a protective environment against drug effects, thus favoring the emergence of chemoresistant cancer cells [39]; for instance, fibronectin was found directly involved in the mechanisms of cell adhesion-mediated drug resistance of MM cells [40], whereas collagen degradation in the BM ECM by matrix metalloproteinases may contribute to MM progression [41]. Recent evidences suggested that ECM components expressed by myeloma patients differ from those of healthy individuals, involving the progressive up-regulation of specific ECM proteins in the transition from MGUS to MM [42]. Here, we found several mutated ECM components, including proteoglycans and proteases, involved in ECM homeostasis and remodeling, as well as collagen and laminin genes, which can interact with surrounding cells through integrin membrane receptors and participate in differentiation, adhesion and migration [43]. Finally, enrichment in ECM organization and cell adhesion functions was found in the 132 most variably expressed among the mutated genes in our analyses, further supporting the hypothesis that they could play important pathogenetic roles in these PC disorders. Overall, based on these considerations, alterations of both these pathways may be strongly involved in the mechanisms of extramedullary spread associated with pPCL.
The G2/M cell cycle checkpoint pathway represents a complex process interconnected with several other cascades, first of all with the DNA damage response (DDR) mechanisms. MM and in particular PCL are disorders characterized by high genetic instability; the expression of genes involved in distinct DNA repair mechanisms has recently been associated with a disease-specific prognostic relevance in MM [44]. Here, we evidenced the marked involvement of TP53 gene. Although referring to a relatively small series, its occurrence was higher than that reported for MM in previous NGS studies [16, 17, 20]. The increased frequency of TP53 deletions found in our pPCLs with respect to MM is confirmed in all published pPCL series [3, 6, 7, 9, 28] and can be partially explained by the different composition in terms of cytogenetic alterations between pPCL and MM. Furthermore, we reported the recurrent involvement of ATM and ATR genes, which encode the two checkpoint kinases required for cell cycle arrest and DNA damage repair activation. In particular, the PI3/PI4-kinase ATR, carrying four missense variants in three pPCLs, resulted a potential driver gene in our series. Recently, ATR inhibitors showed to sensitize tumor cells to topoisomerase inhibitors already used in clinical trials, thus suggesting the possibility of a combined strategy to enhance cancer treatment efficacy [45]. The co-occurrence of mutations in different members of the DDR cascade downstream to ATM or ATR (CHEK2, CDC25A, TP53, CDKN1A, BRCA1) may suggest a potential synergic role in deregulated DNA repair function in pPCL. This is of particular relevance since our study involved patients that did not receive any previous treatment, suggesting that alterations of this function may represent a specific hallmark of tumorigenesis in pPCL.
Overall, our results suggested a peculiar involvement for a variegated set of genes and pathways in pPCL biology and evidenced the importance of their comprehensive characterization to extend the knowledge of molecular mechanisms responsible for this disease. Moreover, since pPCL is a highly aggressive form of PC dyscrasia, understanding its molecular processes might also shed new light and help to unravel the genetic complexity of high-risk MM. Candidate genes identified in these pathways might be useful as actionable targets for therapeutic approaches, thus opening new perspectives or improving the efficacy of already existing strategies.
MATERIALS AND METHODS
Patients and sample preparation
Pathological specimens were collected from 12 untreated pPCL patients, all but one (PCL-038) included in a multicenter Italian clinical trial (RV-PCL-PI-350, EudraCT N° 2008-003246-28), an open label, exploratory, single arm, two-stage study aimed at evaluating safety and anti-tumor activity of combined lenalidomide and dexamethasone as first-line treatment for previously untreated pPCLs [11]. All cases have been characterized by fluorescent in situ hybridization (FISH) for the main MM genomic aberrations (Suppl Table S7) and profiled for whole-genome DNA copy number and gene and miRNA expression by microarray technology [10, 12, 13]. Data are available at NCBI GEO repository (Accession numbers GSE39383, GSE39925 and GSE37053).
Highly purified (≥ 90%) bone marrow PCs were obtained as previously reported [46]. CD138-negative bone marrow population (CD138+ < 0.5%, as assessed by FACS analysis) was obtained for five of the 12 patients and used as normal counterpart for WES.
Whole-exome capture and sequencing
Whole-exome capture for the 12 pPCLs and five control samples was performed using the TruSeq Exome Enrichment kit (62 Mb) (Illumina, San Diego, CA, USA). Libraries were sequenced on an Illumina GAIIx platform, in paired-end 85-cycle runs (see Supplementary Methods for details). Raw data have been deposited in NCBI Sequence Read Archive (SRA), under Study Accession Number SRP051153.
WES data analysis
Since matched controls were not available in all pPCL cases, traditional somatic callers could not be used in our study. Therefore, to analyze all the 12 samples using the same method, we implemented a custom pipeline based on GATK UnifiedGenotyper caller and used at first public databases to filter out known polymorphisms and then the pool of control samples to further remove germline variants. Briefly, reads were mapped to the NCBI human reference genome (GRCh37) using BWA [47]. Variant calling for SNVs and indels was performed using GATK software [48], and functional annotation was carried out using Annovar [49], according to Ensembl GenCode v67 gene model. By applying a stringent custom filtering pipeline, any variant already annotated as polymorphism in public databases (dbSNP, 1000 Genomes, ESP) was discarded, unless present also in COSMIC (v67) or NCBI ClinVar database given their potential relevance for tumor or clinical associations. Finally, control samples were used to remove germline background and to select coding somatic non-silent variants, including non-synonymous SNVs and indels. Full description of bioinformatics pipeline and filtering steps is provided in Supplementary Methods.
Confirmation of WES variants was performed by PCR amplification and Sanger or Roche Junior sequencing. Calculation of nucleotide contexts for identification of mutational signatures was performed according to Alexandrov et al. [50].
Since control samples were not available for all the investigated cases, there was the possibility that calls in unpaired samples still included private polymorphisms, not removed because absent in the control pool. To evaluate the possible carry-over of germline background, our pipeline was applied on the five paired pPCLs by analyzing them first in “paired” manner (subtracting to each tumor its matched control, as described in Supplementary Methods) and then in “unpaired” manner (subtracting to each tumor the pool of the other not matched controls). Results showed that the private background was ultimately marginal thanks to the adopted filtering steps (Suppl Table S8). Identical recurrent variants found in two or more samples and annotated in both dbSNP and COSMIC catalogue were manually checked in COSMIC website for somatic status. The few variants (all occurring in unpaired pPCLs) whose somatic status was not confirmed in COSMIC website were flagged for further analyses as they could be residual germline background (Suppl Table S9).
Copy number analysis and integration
Genome-wide analysis of somatic DNA copy number alterations (CNAs) was performed on WES data using the read count-based EXCAVATOR software [51], with default parameters and setting c = 1 (Supplementary Methods). To assess the CN state of mutated genes, genes mapping in CNA regions were retrieved using UCSC RefSeq transcript annotation track. In case of multiple transcripts for the same gene, the largest RefSeq transcript was chosen. Then, this CNA-gene list was intersected with WES mutated gene list, to evaluate the CN state of affected genes across the whole dataset.
Identification of driver genes
Specifically implemented for the analysis of small tumor datasets, DOTS-Finder tool [52] was applied with default settings to identify significant recurrently affected genes with potential driver role in our series, based on functional and frequentist approaches taking into account gene length, dataset size and number of non-synonymous/synonymous variants found in our samples with respect to COSMIC and The Cancer Genome Atlas (TCGA) cancer databases. Identical recurrent variants found in unpaired pPCL samples and annotated in both dbSNP and COSMIC catalogue but not confirmed as somatic in COSMIC website were considered just once to not over-score corresponding genes. Annovar output was used to produce the initial marf file and a q-value < 0.1 (Benjamini-Hochberg correction) was set to define statistically significant driver genes.
Pathway analysis
Functional enrichment analysis of the mutated genes was performed using ToppGene Suite [53]. Pathways with q-value < 0.05 (FDR Benjamini-Hochberg correction) were defined as significantly enriched. Before running ToppGene, a list of potentially spurious genes frequently found mutated in cancer but not necessarily relevant to tumor biology was compiled according to Lawrence et al. [21]. Twenty-three of them resulted as present in our list of 1, 643 mutated genes (flagged in Suppl File S2) and were excluded from this analysis.
Moreover, mutated genes were catalogued according to a selection of eight pathways chosen on the basis of the existing knowledge about MM and lymphoid tumor malignancies [23, 24]. KEGG database (http://www.genome.jp/kegg/) was used to retrieve full lists of genes composing each pathway (Supplementary Methods).
Gene expression profiles of mutated genes
A panel of highly purified bone marrow PCs from 55 MM and 21 pPCL patients (12 of whom investigated in this study) and from four healthy donors were previously profiled by us on GeneChip Human Gene 1.0 ST array (Affymetrix, Santa Clara, CA, USA) [12]. These data were used to investigate gene expression levels of mutated genes and to perform hierarchical clustering and differential expression analysis (Supplementary Methods). To assess the clustering abilities of mutated genes, the biological homogeneity index (BHI) [54] provided in R package clValid was used (Supplementary Methods).
ACKNOWLEDGMENTS
This work was supported by grants from the Associazione Italiana Ricerca sul Cancro (IG 10136) (A.N.), the Italian Ministry of Health Finalized Research for Young Researchers (CUP project E66110000230001) (K.T., P.M. and A.N.) (GR-2011-02352026) (I.C.) and the MIUR FIRB for Young Researchers (RBFR10OS4G) (I.C.).
CONFLICTS OF INTEREST
The authors declare no competing financial interests.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
REFERENCES
1. Kyle RA, Maldonado JE, Bayrd ED. Plasma cell leukemia. Report on 17 cases Arch Intern Med. 1974; 133:813–818.
2. Bladé J, Kyle RA. Nonsecretory myeloma, immunoglobulin D myeloma, and plasma cell leukemia. Hematology/oncology clinics of North America. 1999; 13:1259–72.
3. Tiedemann RE, Gonzalez-Paz N, Kyle RA, Santana-Davila R, Price-Troska T, Van Wier SA, Chng WJ, Ketterling RP, Gertz MA, Henderson K, Greipp PR, Dispenzieri A, Lacy MQ, et al. Genetic aberrations and survival in plasma cell leukemia. Leukemia. 2008; 22:1044–1052.
4. Fernandez de Larrea C, Kyle RA, Durie BG, Ludwig H, Usmani S, Vesole DH, Hajek R, San Miguel JF, Sezer O, Sonneveld P, Kumar SK, Mahindra A, Comenzo R, et al. Plasma cell leukemia: consensus statement on diagnostic requirements, response criteria and treatment recommendations by the International Myeloma Working Group. Leukemia. 2013; 27:780–791.
5. Albarracin F, Fonseca R. Plasma cell leukemia. Blood Rev. 2011; 25:107–112.
6. Avet-Loiseau H, Daviet A, Brigaudeau C, Callet-Bauchu E, Terré C, Lafage-Pochitaloff M, Désangles F, Ramond S, Talmant P, Bataille R. Cytogenetic, interphase, and multicolor fluorescence in situ hybridization analyses in primary plasma cell leukemia: a study of 40 patients at diagnosis, on behalf of the Intergroupe Francophone du Myélome and the Groupe Français de Cytogénétique Hématolo. Blood. 2001; 97:822–5.
7. Chang H, Qi X, Yeung J, Reece D, Xu W, Patterson B. Genetic aberrations including chromosome 1 abnormalities and clinical features of plasma cell leukemia. Leukemia research. 2009; 33:259–62.
8. Chang H, Sloan S, Li D, Patterson B. Genomic aberrations in plasma cell leukemia shown by interphase fluorescence in situ hybridization. Cancer Genetics and Cytogenetics. 2005; 156:150–153.
9. Chiecchio L, Dagrada GP, White HE, Towsend MR, Protheroe RKM, Kan LC, Stockley DM, Orchard KH, Cross NCP, Harrison CJ, Ross FM. Frequent upregulation of MYC in plasma cell leukemia. Genes Chromosomes and Cancer. 2009; 48:624–636.
10. Mosca L, Musto P, Todoerti K, Barbieri M, Agnelli L, Fabris S, Tuana G, Lionetti M, Bonaparte E, Sirchia SM, Grieco V, Bianchino G, D’Auria F, et al. Genome-wide analysis of primary plasma cell leukemia identifies recurrent imbalances associated with changes in transcriptional profiles. Am J Hematol. 2013; 88:16–23.
11. Musto P, Simeon V, Martorelli MC, Petrucci MT, Cascavilla N, Di Raimondo F, Caravita T, Morabito F, Offidani M, Olivieri A, Benevolo G, Mina R, Guariglia R, et al. Lenalidomide and low-dose dexamethasone for newly diagnosed primary plasma cell leukemia. Leukemia. 2014; 28:222–225.
12. Todoerti K, Agnelli L, Fabris S, Lionetti M, Tuana G, Mosca L, Lombardi L, Grieco V, Bianchino G, D’Auria F, Statuto T, Mazzoccoli C, De Luca L, et al. Transcriptional characterization of a prospective series of primary plasma cell leukemia revealed signatures associated with tumor progression and poorer outcome. Clin Cancer Res. 2013; 19:3247–58.
13. Lionetti M, Musto P, Di Martino MT, Fabris S, Agnelli L, Todoerti K, Tuana G, Mosca L, Gallo Cantafio ME, Grieco V, Bianchino G, D’Auria F, Statuto T, et al. Biological and clinical relevance of miRNA expression signatures in primary plasma cell leukemia. Clin Cancer Res. 2013; 19:3130–3142.
14. Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, Harview CL, Brunet JP, Ahmann GJ, Adli M, Anderson KC, Ardlie KG, Auclair D, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011; 471:467–472.
15. Walker BA, Wardell CP, Melchor L, Brioli A, Johnson DC, Kaiser MF, Mirabella F, Lopez-Corral L, Humphray S, Murray L, Ross M, Bentley D, Gutiérrez NC, et al. Intraclonal heterogeneity is a critical early event in the development of myeloma and precedes the development of clinical symptoms. Leukemia. 2014; 28:384–90.
16. Bolli N, Avet-Loiseau H, Wedge DC, Van Loo P, Alexandrov LB, Martincorena I, Dawson KJ, Iorio F, Nik-Zainal S, Bignell GR, Hinton JW, Li Y, Tubio JM, et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat Commun. 2014; 5:2997.
17. Lohr JG, Stojanov P, Carter SL, Cruz-Gordillo P, Lawrence MS, Auclair D, Sougnez C, Knoechel B, Gould J, Saksena G, Cibulskis K, McKenna A, Chapman MA, et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell. 2014; 25:91–101.
18. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, Boyault S, Burkhardt B, Butler AP, et al. Signatures of mutational processes in human cancer. Nature. 2013; 500:415–21.
19. Walker BA, Wardell CP, Murison A, Boyle EM, Begum DB, Dahir NM, Proszek PZ, Melchor L, Pawlyn C, Kaiser MF, Johnson DC, Qiang YW, Jones JR, et al. APOBEC family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Nature communications. 2015; 6:6997.
20. Walker BA, Wardell CP, Melchor L, Hulkki S, Potter NE, Johnson DC, Fenwick K, Kozarewa I, Gonzalez D, Lord CJ, Ashworth A, Davies FE, Morgan GJ. Intraclonal heterogeneity and distinct molecular mechanisms characterize the development of t(4, 14) and t(11;14) myeloma. Blood. 2012; 120:1077–86.
21. Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, Kiezun A, Hammerman PS, McKenna A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013; 499:214–8.
22. Wu Q, Maniatis T. A Striking Organization of a Large Family of Human Neural Cadherin-like Cell Adhesion Genes. Cell. 1999; 97:779–790.
23. Abdi J, Chen G, Chang H. Drug resistance in multiple myeloma: latest findings and new concepts on molecular mechanisms. Oncotarget. 2013; 4:2186–207.
24. Bommert K, Bargou RC, Stühmer T. Signalling and survival pathways in multiple myeloma. European journal of cancer. 2006; 42:1574–80.
25. Ocio EM, Richardson PG, Rajkumar SV, Palumbo A, Mateos MV, Orlowski R, Kumar S, Usmani S, Roodman D, Niesvizky R, Einsele H, Anderson KC, Dimopoulos MA, et al. New drugs and novel mechanisms of action in multiple myeloma in 2013. a report from the International Myeloma Working Group (IMWG). Leukemia. 2014; 28:525–42.
26. Egan JB, Shi CX, Tembe W, Christoforides A, Kurdoglu A, Sinari S, Middha S, Asmann Y, Schmidt J, Braggio E, Keats JJ, Fonseca R, Bergsagel PL, et al. Whole-genome sequencing of multiple myeloma from diagnosis to plasma cell leukemia reveals genomic initiating events, evolution, and clonal tides. Blood. 2012; 120:1060–6.
27. Lodé L, Eveillard M, Trichet V, Soussi T, Wuillème S, Richebourg S, Magrangeas F, Ifrah N, Campion L, Traullé C, Guilhot F, Caillot D, Marit G, et al. Mutations in TP53 are exclusively associated with del(17p) in multiple myeloma. Haematologica. 2010; 95:1973–6.
28. Avet-Loiseau H, Roussel M, Campion L, Leleu X, Marit G, Jardel H, Dib M, Decaux O, Lamy T, Tiab M, Cony-Makhoul P, Banos A, Lepeu G, et al. Cytogenetic and therapeutic characterization of primary plasma cell leukemia: the IFM experience. Leukemia. 2012; 26:158–9.
29. Neri A, Baldini L, Trecca D, Cro L, Polli E, Maiolo AT. p53 gene mutations in multiple myeloma are associated with advanced forms of malignancy. Blood. 1993; 81:128–35.
30. Tomecki R, Drazkowska K, Kucinski I, Stodus K, Szczesny RJ, Gruchota J, Owczarek EP, Kalisiak K, Dziembowski A. Multiple myeloma-associated hDIS3 mutations cause perturbations in cellular RNA metabolism and suggest hDIS3 PIN domain as a potential drug target. Nucleic acids research. 2014; 42:1270–90.
31. Imoto I, Izumi H, Yokoi S, Hosoda H, Shibata T, Hosoda F, Ohki M, Hirohashi S, Inazawa J. Frequent silencing of the candidate tumor suppressor PCDH20 by epigenetic mechanism in non-small-cell lung cancers. Cancer research. 2006; 66:4617–26.
32. Schambony A, Kunz M, Gradl D. Cross-regulation of Wnt signaling and cell adhesion. Differentiation. 2004; 72:307–18.
33. Cristóbal I, Cirauqui C, Castello-Cros R, Garcia-Orti L, Calasanz MJ, Odero MD. Downregulation of PPP2R5E is a common event in acute myeloid leukemia that affects the oncogenic potential of leukemic cells. Haematologica. 2013; 98:e103–4.
34. Wang F, Marshall CB, Ikura M. Transcriptional/epigenetic regulator CBP/p300 in tumorigenesis: structural and functional versatility in target recognition. Cellular and molecular life sciences: CMLS. 2013; 70:3989–4008.
35. Sun Y, Kolligs FT, Hottiger MO, Mosavin R, Fearon ER, Nabel GJ. Regulation of beta -catenin transformation by the p300 transcriptional coactivator. Proceedings of the National Academy of Sciences of the United States of America. 2000; 97:12613–8.
36. Morris LGT, Kaufman AM, Gong Y, Ramaswami D, Walsh LA, Turcan Ş, Eng S, Kannan K, Zou Y, Peng L, Banuchi VE, Paty P, Zeng Z, et al. Recurrent somatic mutation of FAT1 in multiple human cancers leads to aberrant Wnt activation. Nature genetics. 2013; 45:253–61.
37. Messina M, Del Giudice I, Khiabanian H, Rossi D, Chiaretti S, Rasi S, Spina V, Holmes AB, Marinelli M, Fabbri G, Piciocchi A, Mauro FR, Guarini A, et al. Genetic lesions associated with chronic lymphocytic leukemia chemo-refractoriness. Blood. 2014; 123:2378–88.
38. Morris LGT, Ramaswami D, Chan TA. The FAT epidemic: a gene family frequently mutated across multiple human cancer types. Cell cycle. Georgetown, Tex: 2013; 12:1011–2.
39. Vincent T, Mechti N. Extracellular matrix in bone marrow can mediate drug resistance in myeloma. Leukemia & lymphoma. 2005; 46:803–11.
40. Fei M, Hang Q, Hou S, Ruan C. Cell adhesion to fibronectin down-regulates the expression of Spy1 and contributes to drug resistance in multiple myeloma cells. International journal of hematology. 2013; 98:446–55.
41. Vacca A, Ribatti D, Ria R, Pellegrino A, Bruno M, Merchionne F, Dammacco F. Proteolytic activity of human lymphoid tumor cells. Correlation with tumor progression. Developmental immunology. 2000; 7:77–88.
42. Slany A, Haudek-Prinz V, Meshcheryakova A, Bileck A, Lamm W, Zielinski C, Gerner C, Drach J. Extracellular matrix remodeling by bone marrow fibroblast-like cells correlates with disease progression in multiple myeloma. Journal of proteome research. 2014; 13:844–54.
43. Colognato H, Yurchenco PD. Form and function: the laminin family of heterotrimers. Developmental dynamics: an official publication of the American Association of Anatomists. 2000; 218:213–34.
44. Kassambara A, Gourzones-Dmitriev C, Sahota S, Rème T, Moreaux J, Goldschmidt H, Constantinou A, Pasero P, Hose D, Klein B. A DNA repair pathway score predicts survival in human multiple myeloma: the potential for therapeutic strategy. Oncotarget. 2014; 5:2487–98.
45. Josse R, Martin SE, Guha R, Ormanoglu P, Pfister TD, Reaper PM, Barnes CS, Jones J, Charlton PA, Pollard JR, Morris J, Doroshow JH, Pommier Y. The ATR inhibitors VE-821 and VX-970 sensitize cancer cells to topoisomerase I inhibitors by disabling DNA replication initiation and fork elongation responses. Cancer Res. 2014; 74:6968–79.
46. Mattioli M, Agnelli L, Fabris S, Baldini L, Morabito F, Bicciato S, Verdelli D, Intini D, Nobili L, Cro L, Pruneri G, Callea V, Stelitano C, et al. Gene expression profiling of plasma cell dyscrasias reveals molecular patterns associated with distinct IGH translocations in multiple myeloma. Oncogene. 2005; 24:2461–73.
47. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–60.
48. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011; 43:491–498.
49. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research. 2010; 38:e164.
50. Alexandrov LB, Nik-Zainal S, Wedge DC, Campbell PJ, Stratton MR. Deciphering signatures of mutational processes operative in human cancer. Cell reports. 2013; 3:246–59.
51. Magi A, Tattini L, Cifola I, D’Aurizio R, Benelli M, Mangano E, Battaglia C, Bonora E, Kurg A, Seri M, Magini P, Giusti B, Romeo G, et al. EXCAVATOR: detecting copy number variants from whole-exome sequencing data. Genome Biol. 2013; 14:R120.
52. Melloni GE, Ogier AG, de Pretis S, Mazzarella L, Pelizzola M, Pelicci P, Riva L. DOTS-Finder: a comprehensive tool for assessing driver genes in cancer genomes. Genome Medicine. 2014; 6:44.
53. Chen J, Bardes EE, Aronow BJ, Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic acids research. 2009; 37:W305–11.
54. Datta S, Datta S. Methods for evaluating clustering algorithms for gene expression data using a reference set of functional classes. BMC bioinformatics. 2006; 7:397.