
Introduction
Ovarian cancer is a heterogeneous disease composed of several histotypes including mucinous, clear cell, endometrioid, low-grade and high-grade serous. In the United States, 23,000 women will be diagnosed and 14,000 will succumb to these diseases each year [1]. High-grade serous carcinoma (HGSC) is the most common and lethal histotype and is suggested to arise from the fallopian tube fimbria. Putative fallopian precursor, lesions are thought to arise from the mutation of p53, followed by subsequent oncogenic events that confer expansion into serous tubal intraepithelial carcinoma (STIC) and ultimately into HGSC [2]. HGSC are primarily diagnosed at the metastatic stage (75% of cases) and are treated with debulking surgery and a combination of paclitaxel and carboplatin, but patients often develop chemoresistance [3]. Defining the molecular events leading to HGSC carcinogenesis and progression from the fallopian tube is essential for identification of targets for early detection and personalized drug therapy.
Murine models of HGSC originating from the fallopian tube equivalent, the oviductal epithelium, reveal that these cells can give rise to tumors. These murine models utilize the PAX8-TetOn-Cre promoter to drive the deletion of PTEN, BRCA1 or 2, and mutation of p53 in combination, as well as deletion of PTEN together with deletion of p53 [4]. These models produced tumors that were highly metastatic, colonized the ovaries, and expressed oviductal markers. Interestingly, another murine model with floxed alleles of PTEN and KRAS developed high-grade endometrioid carcinoma when exposed to adenovirus encoding for Cre-recombinase regardless of the tissue type targeted (ovarian surface or oviductal epithelium [5]). A further model utilizing the MISRII promoter driving Cre-recombinase expression to delete PTEN alone or in combination with DICER in oviductal stromal cells forces a stromal-to-epithelial derived high-grade tumor type [6]. Unfortunately, despite the advantages of using the Cre-Lox system to generate murine models, the limited viral infectivity of the oviduct epithelium and the inability to target the oviduct specifically with promotor driven Cre-recombinase without impacting additional cell types could mask or confuse the question of cellular origin [7]. Furthermore, although p53 is mutated in 96-100% of HGSC tumors and is thought to be one of the earliest molecular changes in fallopian derived HGSC, animals with mutation in p53 alone have not yet been characterized. Additionally, mutation in p53 has not been examined with the loss of PTEN, in the absence of BRCA mutation, which is of interest as ~70-80% of HGSC tumors are spontaneous and not the result of mutation or methylation in BRCA (11). Moreover, human fallopian cell models have demonstrated the importance of KRAS and c-myc in generating HGSC and mucinous histotypes from fallopian tube epithelium [8-10]. In these human models, p53 is either silenced or inactivated by SV40T, which hinders the ability to study the impact of gain-of-function p53 mutations, which represent the vast majority of mutations in HGSC.
The Cancer Genome Atlas Network demonstrated that several key pathways are altered in HGSC, yet many of them are not altered strictly through mutation [11]. For example, PTEN is rarely mutated in HGSC but instead tumors demonstrate a loss of heterozygosity. Similarly, tissue microarray studies demonstrate AKT activation through phosphorylation occurs early in tumorigenesis and is independent of PTEN, AKT, and PIK3Ca mutation [12, 13]. KRAS is also rarely mutated, but instead is amplified in many tumors. As many HGSC tumors are studied in a late stage, metastatic setting, it is unclear whether altered pathways are initiators of disease or if they are compounded genetic alterations.
The purpose of this study was to engineer clonogenic murine oviductal epithelial (MOE) models with individual and multiple pathway modifications to mimic changes detected in human HGSC tumors. The ability to study mutant p53 alone and in combination with several other events allows for clarification of its role and its ability to modify other pathways. Similarly, models were constructed with PTEN silenced, but not completely deleted, similar to loss of heterozygosity in HGSC and with AKT constitutively activated by myristolation. Lastly, the G12V mutation was utilized to activate KRAS function. These unique MOE cell models indicate that loss of PTEN alone is sufficient to drive tumorigenesis and widely spread metastasis in the peritoneal cavity. Mutation of p53 did not enhance this process, while mutation of KRAS significantly decreased survival time. These models indicate that specific mutation in oviductal epithelium can produce tumors with unique morphologies and specific secondary sites of tumor spread, including the ovary suggesting a role for this tissue in the dissemination of disease. Finally this study validates that novel MOE models can be utilized in syngeneic allografts allowing for preclinical therapeutic development in an immune-competent in vivo environment.
Results
MOE cells stably expressing targets altered in HGSC patients
To examine the effects of genetic alterations similar to those observed in HGSC patients, MOE cells were stably transfected with mutation in p53 R273H (p53R273H), silenced PTEN via shRNA (PTENshRNA), activation of AKT via myristolation (AKTMYR) and activating mutation in KRAS G12V (KRASG12V) (Suppl. Table 1). In addition cell lines harboring multiple genetic modifications were created, as often multiple mutations are required for oncogenic transformation. These cells were validated to express targets of interest via western blotting (Suppl. Fig 1A) compared to parental wildtype and scrambled shRNA control MOE cell lines. Signaling downstream of genetic manipulations was also altered as expected, demonstrating that both the targets themselves and their signaling pathways were functionally impacted (Suppl. Fig 1B).
Silencing PTEN increases MOE cell proliferation and addition of p53R273H, AKTMYR, or KRASG12V promotes in vitro transformation
Stably modified MOE cells were characterized for phenotypic changes in vitro, specifically enhanced proliferation or anchorage independent growth. SRB assay determined PTENshRNA MOE had enhanced cell growth at 5 days (9.2±0.4 fold, p = 0.01) when compared with wildtype parental MOE cells (6.2±0.3 fold) or scrambled shRNA control MOE cells (7.1±0.5 fold, Figure 1A). MOE cells harboring PTENshRNA in combination with either p53R273H or KRASG12V also displayed enhanced proliferation (10.6±1.1 fold, p = 0.02 and 10.8±0.7 fold, p = 0.01 respectively). The expression of p53R273H,KRASG12V, or AKTMYR alone in MOE cells did not alter proliferation (Figure 1B) [14]. Addition of AKTMYR to generate PTENshRNA/AKTMYR MOE abrogated the enhanced growth seen with PTENshRNA alone. Further, MOE cells harboring triple manipulation; PTENshRNA/AKTMYR/KRASG12V exhibited reduced cell growth (4.9±0.7 fold increase, p = 0.02).
In vitro transformative potential was investigated using a soft agar assay for anchorage independent growth. Wildtype and scrambled shRNA control MOE cells did not form colonies in soft agar (Figure 1C). Similarly, MOE cells with single genetic alterations were unable to form colonies [14]. However, MOE cells with PTEN knockdown in combination with another alteration did form colonies in soft agar (Figure 1D); PTENshRNA/p53R273H (5-fold increase compared to scrambled shRNA control), PTENshRNA/AKTMYR (3-fold increase), and PTENshRNA/KRASG12V (22-fold increase). MOE cells stably transfected with three genetic alterations; PTENshRNA/AKTMYR/ KRASG12V, were also transformed in vitro, with a 7-fold increase in colony formation.
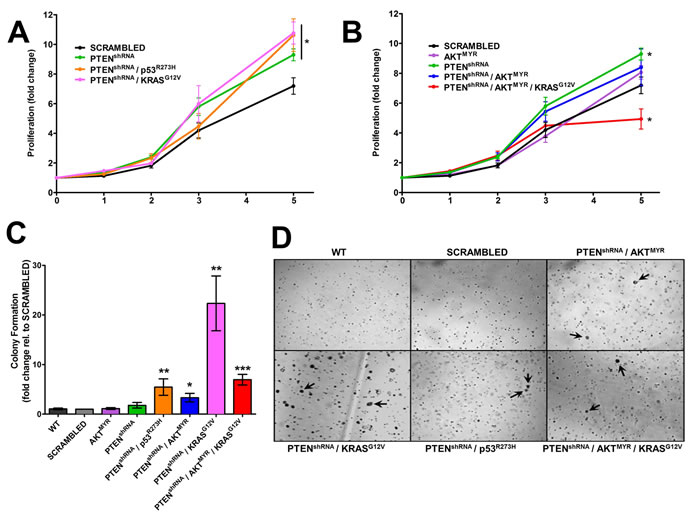
Figure 1: Genetically engineered MOE have tumorigenic characteristics in vitro. A. MOE cells stably altered with PTENshRNA, PTENshRNA/p53R273H, and PTENshRNA/KRASG12V were significantly more proliferative than scrambled shRNA transfected and parental MOE cells (data not shown) after 5 days in culture (n = 8). B. Proliferation was not altered in cells harboring AKTMYR or PTENshRNA/AKTMYR. PTENshRNA/ AKTMYR/KRASG12V MOE cells had a reduction in proliferation compared to scrambled shRNA control (n = 5). C and D. Anchorage independent growth occurred in several engineered MOE cells as demonstrated by colony formation in vitro, specifically cells with PTENshRNA in combination with other genetic alterations (denoted with arrows). Parental MOE cell (WT), SCR, and MOE cells harboring single genetic alterations did not form colonies. *p ≤ 0.05, **p ≤ 0.01 and ***p ≤ 0.001 as determined by t-test.
Transformed PTENshRNA MOE cell grafts mimic HGSC tumors in patients
To test tumorigenic potential of MOE cells in vivo, genetically engineered MOE cell lines and parental wildtype and scrambled shRNA controls were allografted into athymic mice subcutaneously (s.c) and intraperitoneally (i.p.) and tumorigenesis was assessed over a 6 month period (Figure 2A). Mice grafted with wildtype or scrambled shRNA control MOE had no evidence of tumor burden. MOE with single genetic changes to either p53R273H or KRASG12V also did not form disease in athymic mice [14]. However, several MOE models generated tumors. Subcutaneous tumor growth was tracked via weekly caliper measurement (Figure 2B). When tumor burden reached a moribund stage animals were sacrificed, s.c. tumors weighed and i.p. disease assessed (Table 1).
Table 1: Summary of MOE allografted animal survival and tumor formation.
MOE Cell Type |
Survival (days) |
S.C. Tumor Weight (mg) |
Peritoneal Disease |
Wildtype (WT) |
no mortality |
N/A |
N/A |
Scrambled shRNA (SCR) |
no mortality |
N/A |
N/A |
PTENshRNA |
147 ± 21 |
376.8 ± 107.1 |
100% (4/4) |
PTENshRNA / p53R273H |
130 ± 8 |
771.1 ± 198.1 |
60% (3/5) |
PTENshRNA / AKTMYR |
no mortality |
69.93 ± 14.1 |
40% (2/5) |
PTENshRNA / KRASG12V |
45 ± 1 |
520.1 ± 38.9 |
100% (6/6) |
PTENshRNA / AKTMYR / KRASG12V |
164 ± 8 |
347.5 ± 88.7 |
67% (4/6) |
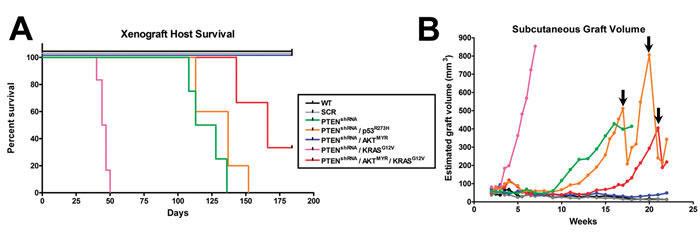
Figure 2: MOE cells form subcutaneous (s.c) and intraperitoneal tumors in an allograft model. A. Several pathway modified MOE models were capable of tumorigenesis when grafted into athymic mice and had markedly reduced survival after 6 months in vivo (n = 4-6 mice per group). B. S.C. tumor growth was tracked via weekly caliper measurements. No growth was seen in parental (WT) or scrambled shRNA (SCR) control cell lines whereas pathway modified MOE cells formed large s.c. tumors. Arrows denote where animals were humanely sacrificed due to tumor burden, which modified the average tumor size.
PTENshRNA MOE cells formed large s.c. tumors (376.8±107.1 mg, Figure 3A) and were sacrificed at 147±21 days post grafting. Further, PTENshRNA MOE formed tumor explants throughout the peritoneal cavity (4/4 animals, 100%, Figure 3B), including spread to the reproductive tract (4/4 animals), G.I./omentum (4/4 animals), and the diaphragm (3/4 animals, Table 2) Further, kidney, liver and lung were relatively disease free, a disease pattern that is common in patients, demonstrating that malignancy of oviductal epithelial origin mimics HGSC peritoneal colonization. In addition to the analogous disease spread, one of the four PTENshRNA MOE host animals (25%) also developed ascites fluid, another hallmark of HGSC tumorigenesis in women. When host reproductive tracts were excised, metastasis into the bursa and spread to the ovaries was noted (Figure 3C).
Table 2: Immunohistochemical analysis on MOE-derived tumors.
PTENshRNA |
PTENshRNA / p53R273H |
PTENshRNA / KRASG12V |
PTENshRNA / AKTMYR |
PTENshRNA / AKTMYR/KRASG12V |
|
CK8 |
+ |
+ |
+ |
- |
+ |
PAX8 |
+ |
+ |
+ |
+ |
+ |
WT1 |
+ |
+/- |
+ |
- |
+/- |
Ki67 |
50-60% |
40-50% |
60-70% |
20-30% |
30-40% |
p53 |
1+ / 2+ |
1+ |
1+ |
- |
- |
Immunohistochemical analysis of PTENshRNA MOE derived tumors and subsequent pathological analysis confirmed the tumors were moderately differentiated high-grade carcinoma of oviductal epithelial origin. This was determined by moderate nuclear atypia seen in H&E staining (Figure 3D). Tumors consisted of epithelioid cells, primarily solid and nesting growth patterns with areas of poorly formed glandular structure. Cytokeratin 8 (CK8) and PAX8 staining (Figure 3E-3F) confirmed the tumors to be of oviductal epithelial origin. PTENshRNA MOE derived tumors also expressed the serous marker WT1 (Figure 3G), were highly proliferative (50-60% Ki-67 positive, Figure 3H) and demonstrated stabilized p53 protein expression via strong and diffuse immunoreactivity (Figure 3I).
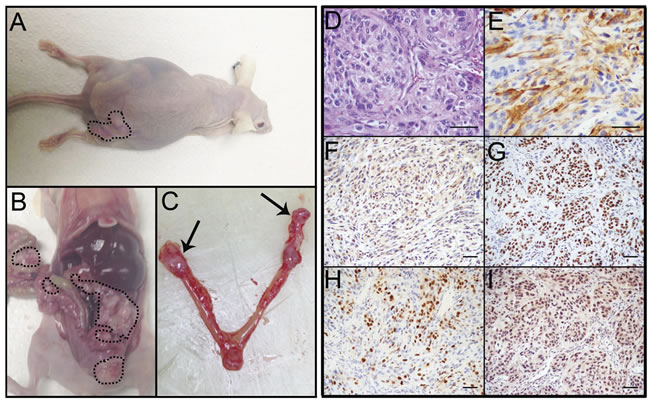
Figure 3: PTENshRNA MOE form high-grade carcinoma in vivo. A. PTENshRNA MOE grafted mice formed large s.c. tumors (dashed outline) and B. disseminated i.p. disease (dashed outline). C. Notably, MOE tumors spread to the ovaries of grafted animals (arrows). Immunohistochemical analysis of PTENshRNA MOE via D. H&E, E. CK8, F. PAX8, G. WT1, H. p53, and I. Ki67 staining determined the tumors to be moderately differentiated high-grade carcinomas of oviductal epithelial origin. Scale bars equal 50 microns.
Mutation in p53 does not enhance PTENshRNA MOE phenotype
Animals grafted with PTENshRNA/p53R273H MOE cells had similar s.c. disease formation (Figure 4A) and survival rates to that of PTENshRNA MOE grafted animals (130±8 days post grafting). However, peritoneal tumor burden was reduced, as not all PTENshRNA/p53R273H allograft mice presented with peritoneal disease (3 of 5 animals (60%), Figure 4B). Of the mice that developed i.p. disease, fewer malignant explants were found. Despite fewer sites of dissemination, animals with peritoneal disease had tumors in their ovaries (Figure 4C-4D), with infiltration noted via CK8 and PAX8 positive tumor cells engulfing the ovaries and leaving follicular structures surrounded by MOE-derived tumors.
PTENshRNA/p53R273H MOE derived tumors were pathologically assessed and determined to be poorly differentiated, high-grade tumors with features of sarcomatoid carcinoma. Tumor cells had high nuclear atypia with little to no glandular structure (Figure 4E). Cells were of MOE origin, characterized by their diffuse immunoreactivity for CK8 and PAX8 (Figure 4F-4G), serous marker WT1 expression was variable (Figure 4H). PTENshRNA/p53R273H tumors were highly proliferative (40-50% of cells staining positively for Ki67, Figure 4I) and expressed stabilized p53 (Figure 4J).
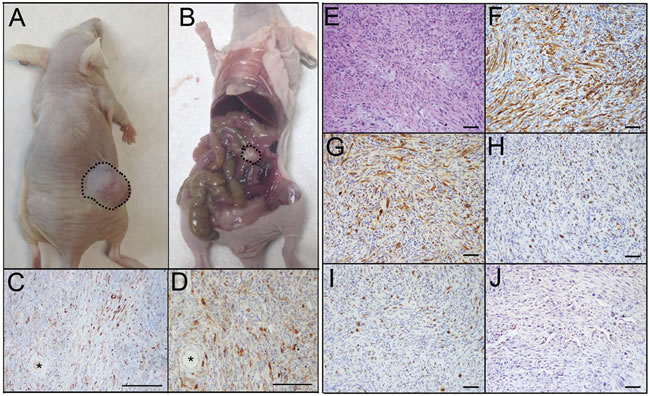
Figure 4: PTENshRNA/p53R273H MOE form high-grade carcinomas with reduced i.p. spread compared to PTENshRNA MOE. A. As with PTENshRNA-alone MOE, PTENshRNA/p53R273H MOE grafted mice formed large s.c. tumors (dashed outline) but B. had reduced i.p. disease (dashed outline). C. Despite the reduction in i.p. tumor explants, PTENshRNA/p53R273H MOE, staining for CK8 and D. PAX8 preferentially formed disease on the ovaries, with tumor invasion engulfing oocytes (*). Immunohistochemical analysis of PTENshRNA/p53R273H MOE via E. H&E, F. CK8, G. PAX8, H. WT1, I. Ki67, and J. p53 staining determined the tumors to be poorly differentiated high-grade sarcomatoid carcinoma. Scale bars equal 200 microns (C-D) and 50 microns (E-J).
Activation of KRAS enhances PTENshRNA phenotype
The addition of a mutation in KRAS (G12V) to PTENshRNA (PTENshRNA/ KRASG12V) enhanced MOE cell carcinogenesis, with an average survival of 45±1 days. Large s.c. tumors developed quickly (520.1±38.9 mg, Figure 5A). Ascites fluid (1/6 animals (17%)), vast peritoneal disease spread (6/6 animals, 100%, Figure 5B), and reproductive explants (6/6 animals, 100%, Figure 5C) were found in PTENshRNA/KRASG12V grafted hosts. Tumor explants were noted in the ovaries in the majority of animals with i.p. disease, while oviducts remained disease free in MOE grafted mice (Suppl. Fig. 2).
Immunohistochemical analysis of PTENshRNA/KRASG12V MOE derived tumors determined them to be high-grade carcinomas of oviductal epithelial origin. Tumors had solid and sheath growth patterns with high-grade and pleomorphic nuclear atypia, and brisk of mitosis (Figure 5F). As with previous MOE grafts, PTENshRNA/KRASG12V MOE derived tumors had strong and diffuse immunoreactivity for CK8, PAX8, and WT1, consistent with oviductal secretory cell origin (Figure 5G-5I). PTENshRNA/KRASG12V tumors were highly proliferative (60-70% Ki67 staining, Figure 5J), which likely contributed to enhanced aggression of the disease. p53 was enhanced but varied from case to case (Figure 5K).
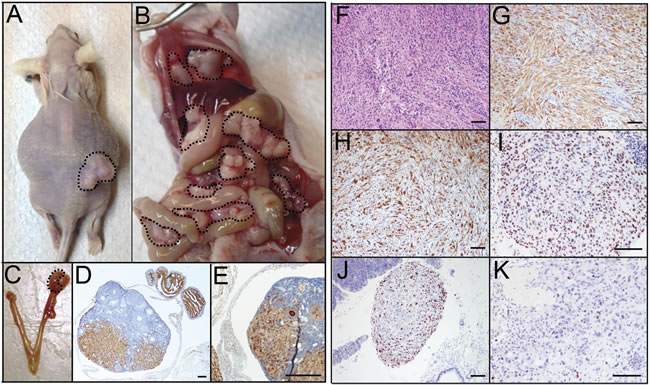
Figure 5: PTENshRNA/KRASG12V MOE give rise to highly-aggressive carcinomas with advanced i.p. spread. A. PTENshRNA/KRASG12V MOE grafted mice quickly developed large s.c. tumors (dashed outline) and some hosts developed ascites fluid. B. i.p. explants were dispersed throughout the peritoneal cavity including the G.I. tract and diaphragm (dashed outline). C. As with PTENshRNA and PTENshrNA/p53R273H grafted animals ovarian tumors were noted staining positively for D. CK8 and E. PAX8. Immunohistochemical analysis of PTENshRNA/KRASG12V MOE via F. H&E, G. CK8, H. PAX8, I. WT1, J. p53, and K. Ki67 staining determined the tumors to be undifferentiated high-grade carcinomas of oviductal epithelial origin. Scale bars equal 200 microns (D-E) and 50 microns (F-K).
Myristolation of AKT diminishes MOE tumor phenotype
Interestingly, the addition of AKTMYR to PTENshRNA (PTENshRNA/AKTMYR) or in combination with PTENshRNA and KRASG12V (PTENshRNA/AKTMYR/KRASG12V) resulted in reduced tumor burden. PTENshRNA/AKTMYR grafts were not lethal after six months, although s.c. tumors 5-fold smaller than those found in PTENshRNA MOE mice, were recovered at the time of sacrifice (69.93±14.1 mg). Two of five animals (40%) had singular metastatic explants, a drastic reduction in tumor spread compared to PTENshRNA grafted mice. Pathological assessment determined PTENshRNA/AKTMYR tumors were poorly differentiated high-grade sarcomatoid carcinoma. H&E staining revealed extensive tumor necrosis and apoptosis (Figure 6A). Tumors were hypercellular with high nuclear to cytoplasmic ratio, and features of ‘small-cell like’ morphology. Further, tumor cells had reduced CK8 and low PAX8 expression (Figure 6B-6C). The serous marker WT1 was also lost (Figure 6D). PTENshRNA/AKTMYR s.c. tumors displayed low mitotic indices (20-30% Ki67 staining, Figure 6E), and no stabilized p53 expression (Figure 6F).
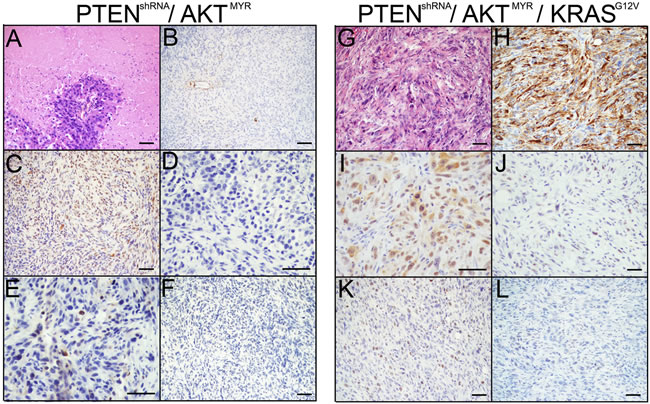
Figure 6: MOE with AKTMYR had reduced tumor phenotypes. The addition of AKTMYR to PTENshRNA in PTENshRNA/AKTMYR MOE grafted mice generated a less aggressive phenotype. Immunohistochemical analysis of PTENshRNA/AKTMYR MOE via A. H&E, B. CK8, C. PAX8, D. WT1, E. p53, and F. Ki67 staining determined the tumors to be full of necrotic and regressing cells as well as proliferating cells and reduction of epithelial markers, resulting in poorly differentiated high-grade mesenchymal-like carcinomas. Similarly, PTENshRNA/AKTMYR/KRASG12V MOE had a reduced phenotype compared to either PTENshRNA or PTENshRNA/KRASG12V MOE cells. Pathological evaluation of tumors stained for via G. H&E, H. CK8, I. PAX8, J. WT1, K. p53, and L. Ki67 revealed the PTENshRNA/AKTMYR/KRASG12V derived tumors were poorly differentiated high-grade epithelioid sarcomas. Scale bars equal 50 microns.
PTENshRNA/AKTMYR/KRASG12V grafts also produced a diminished tumor phenotype when compared to either PTENshRNA or PTENshRNA/ KRASG12V grafted animals, identified as poorly differentiated high-grade sarcomatoid carcinoma, with an over three-fold increase in survival compared with PTENshRNA/ KRASG12V grafted mice (164±8 days). Only four of six (67%) PTENshRNA/AKTMYR/KRASG12V grafted mice were moribund prior to the 6 month experimental timepoint. Resulting s.c. tumors were comparable to other lethal MOE allografts (347.5±88.7 mg). Four of six (67%) PTENshRNA/AKTMYR/KRASG12V grafted mice formed peritoneal explants; however disease was reduced compared to PTENshRNA or PTENshRNA/ KRASG12V, with no animal harboring more than two peritoneal explants. Both the reproductive tract and the pancreas remained preferential sites of tumor colonization (Table 3).
Table 3: Sites of MOE peritoneal explant colonization.
PTENshRNA |
PTENshRNA/ p53R273H |
PTENshRNA/ KRASG12V |
PTENshRNA/ AKTMYR |
PTENshRNA/ AKTMYR/ KRASG12V |
PTENshRNA/ KRASG12V SYNGENEIC |
|
Reproductive Tract |
4 / 4 |
3 / 5 |
6 / 6 |
0 / 5 |
3 / 6 |
0 / 5 |
G.I. Tract / Omentum |
4 / 4 |
3 / 5 |
6 / 6 |
1 / 5 |
1 / 6 |
3 / 5 |
Diaphragm |
3 / 4 |
1 / 5 |
4 / 6 |
0 / 5 |
0 / 6 |
2 / 5 |
Pancreas |
4 / 4 |
2 / 5 |
4 / 6 |
2 / 5 |
4 / 6 |
4 / 5 |
Kidney |
0 / 4 |
0 / 5 |
1 / 6 |
0 / 5 |
0 / 6 |
0 / 5 |
Liver/Lung |
1 / 4 |
0 / 5 |
1 / 6 |
0 / 5 |
0 / 6 |
1 / 5 |
Ascites |
1 / 4 |
0 / 5 |
1 / 6 |
0 / 5 |
0 / 6 |
5 / 5 |
As with PTENshRNA/AKTMYR, tumors were high-grade carcinoma with notable nuclear atypia (Figure 6G). PTENshRNA/AKTMYR/KRASG12V tumors were immunoreactive for CK8 and PAX8 although expression was diminished compared to lethal MOE models (Figure 6H-6I). WT1 expression and proliferation were reduced (30-40% proliferation, Figure 6J-6K). Moreover, p53 expression was not stabilized (Figure 6L). These data demonstrate that although enhanced phosphorylation of AKT is seen in HGSC tumors, enhanced AKT signaling does not phenocopy loss of PTEN, with notable alterations in tumor spread, p53 stabilization, and expression of serous markers such as WT1.
Therapeutic targeting and syngeneic allografts of PTENshRNA/KRASG12V MOE cells
PTENshRNA/KRASG12V MOE cells could be utilized to test novel targeted therapeutics prior to human clinical trials. To highlight potential pathways for therapeutic intervention, PTENshRNA/KRASG12V MOE cells were treated with specific chemical inhibitors downstream of genetic alterations and subjected to soft agar assay. MK2206 (10µM), an allosteric AKT inhibitor, was utilized to diminish the effects of PTEN silencing, and U0126 (10µM), a MEK1/2 inhibitor, was utilized to inhibit uncontrolled signaling downstream of KRAS mutation (Figure 7A). U0126 reduced colony formation by 60%, MK2206 by 78% and combined inhibitor treatment by 84%, demonstrating both pathways are required for colony formation , inhibition of which is able to suppress in vitro transformation.
Additionally, PTENshRNA/KRASG12V cells were utilized to demonstrate the potential for MOE cells as a syngeneic model, allowing for preclinical investigation of novel therapeutics in the presence of an immune system. FVB mice were i.p. allografted with PTENshRNA/KRASG12V MOE and developed high-grade carcinomas. Interestingly, immno-competent FVB host animals had reduced survival (28±0.2 vs 45±1 days, Figure 7B). This was likely due to increased occurrence of ascites fluid build-up, seen in all syngeneic grafted animals. Tumor explants were of a similar morphology and disease pattern to that seen previously in athymic allografted animals (Figure 7C-7F, Table 3). MOE models could be utilized for therapeutic development in the presence of an immune-competent in vivo environment.
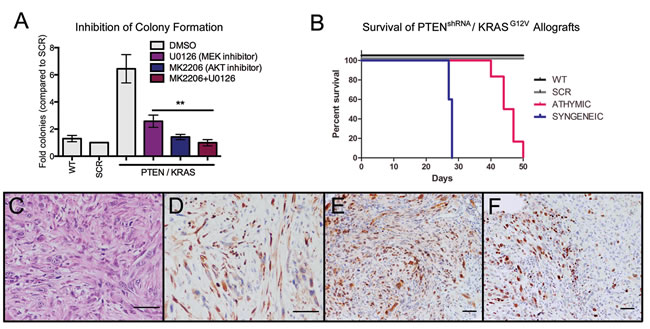
Figure 7: MOE cells as preclinical models for therapeutic development. A. Treatment with the AKT inhibitor (MK2006 (10 µM), counteracts PTEN suppression) or with the MEK inhibitor (U0126 (10 µM), inhibits signaling downstream of KRAS) represses colony formation in soft agar compared to vehicle-treated (DMSO) control (n = 6). B. Syngeneic PTENshRNA/KRASG12V allografts have drastically reduced survival time compared to athymic PTENshRNA/KRASG!2V allografts. C. Tumors from syngeneic PTENshRNA/KRASG!2V allografted mice were similar in morphology to those of athymic hosts as determined via H&E staining and expression of D. CK8, E. PAX8, and F. Ki67 staining. Scale bars equal 50 microns.
Discussion
To address origin and initiation factors leading to HGSC tumorigenesis, murine oviductal epithelial cells (MOE) were modified to stably express alterations in key pathways affected in HGCS patients. This study demonstrated that oviductal epithelium can give rise to tumors similar to those identified in HGSC patients from characterized genetic drivers of disease. In vitro characterization of MOE cells demonstrated specific genetic alterations, namely PTENshRNA alone and in combination with KRASG12V, p53R273H, and AKTMYR induced phenotypic changes in proliferation and anchorage independent growth. In vivo characterization in allografted athymic and subsequent syngeneic murine model produced MOE derived high-grade carcinomas, which resembled HGSC found in women both in pathological staining and pattern of disease spread. These data support the fallopian tube/oviduct as a potential origin for HGSC, expand upon previous studies of key genetic regulators in human and murine models, and provide a novel immune-competent model for biomarker and therapeutic development.
Silencing PTEN alone initiated tumor formation from MOE cells. Studies using PAX8 to drive Cre-recombinase mediated deletion of PTEN did so only in combination with other genes such as BRCA1/2 or mutant p53 R270H [4]. Importantly, recent evidence indicates that PTEN loss is more common than initially thought in HGSC when epithelium are isolated away from stromal contaminants [15]. This is the first publication to demonstrate that PTEN silencing alone in a murine model of oviductal epithelium can generate tumors. PTEN loss also drives tumor formation from the endometrium, thyroid, and prostate epithelium indicating that multiple types of hormone responsive epithelium are dependent on expression of PTEN to block tumor formation [16]. Another Cre-recombinase murine model driven by MISRII to delete PTEN alone did not demonstrate a phenotype, but this was likely due to Cre-mediated excision in the oviductal stroma and ovarian surface epithelium rather than the oviductal epithelium and suggests that PTEN governs different pathways in these cell types [6, 17]. In agreement with this hypothesis, findings from our lab indicated that PTEN silencing in murine OSE was unable to generate soft agar colonies [18]. Moreover, a publication recently attempted to intraoviductally inject adenovirus into MUC1/KRAS/PTEN floxed mice, which resulted in high-grade endometrioid carcinoma; however, intraoviductal injections can be leaky, often infecting the ovarian surface, and as previous reports suggest, oviductal infectivity is limited in vivo and in 3D in vitro cultures [5, 7].
The incorporation of mutant p53 into PTENshRNA cells did not enhance proliferation or tumor forming potential observed with PTENshRNA alone. These data are consistent with studies performed in human samples that indicate that mutation in p53 does not correlate with changes in tumor progression or survival [19]. In fact, mutation of p53 reduced the extent of tumor spread in mice with PTENshRNA suggesting that p53 mutation functions in a very context specific manner. These results are similar to the PAX8-Cre models where the addition of mutant p53 to PTEN and BRCA did not dramatically impact survival [4]. Previous reports suggest that mutation of p53 can slow migration as compared to wildtype [20] and that wildtype p53 in OSE derived tumors enhances survival [21]. However, mutation in p53 enhances tumor formation in the APC/PTEN model of OSE derived endometrioid cancer providing evidence that both the mutation landscape and the cell of origin impact the functional context of p53 mutation and resulting tumor histotype [22]. The current study focused on one DNA mutation of p53, R273H, the most common mutation in HGSC. However, many other mutations are seen in HGSC, making it difficult to decipher a general role for p53 and how it impacts tumorigenesis from the oviduct.
Data from this study highlight the importance of context in the generation of disease; namely the cell of origin, the genetic landscape/specific mutations, and the site of tumor development, especially when allografted. This is highlighted by PTEN silencing alone in the OSE, which does not drive tumor formation [23], whereas in the oviductal epithelium PTEN loss is sufficient for tumorigenesis. We speculate, this variation in susceptibility may be due in part to disparity between the two cell types; oviductal epithelium, which may be more representative of a true epithelium and is highly similar to that of other PTEN susceptible epithelial populations, such as endometrial epithelium [15], as compared to the OSE, which is developmentally less differentiated and more characteristic of a mixed mesothelium [24]. The molecular signaling landscape also impacts tumor formation, as evidenced by PTENshRNA alone and in combination with KRAS mutation developing more aggressive peritoneal disease as compared to PTENshRNA in combination with p53 mutation or activation of AKT. Furthermore, tumor histology was influenced by the site of grafting as PTENshRNA tumors appeared more epithelioid when allografted into the peritoneal cavity, a more orthotopic model of HGSC as compared to the subcutaneous space. The cell of origin from which a tumor is derived further impacts the spectrum of spread. The ovary was a common site of infiltration of MOE models that formed peritoneal explants. In contrast, despite cell models being derived from the oviduct, colonization in the oviduct was not seen. In addition to the ovary, the gynecologic tract in general was a site of tumor adhesion and growth suggesting that the ovaries and uterus express receptors or growth factors that help to stimulate colonization. As the rodent ovary is surrounded by a bursa, identification of MOE derived ovarian tumors indicates metastasis through the bursa or through the lymphatic or hematopoietic systems to penetrate the ovarian cortex after peritoneal injection. Taken together these data highlight the context of disease, in particular the cells impacted, the specific pathways altered, and the site of tumor growth will influence the phenotype and therefore the future use of models for preclinical testing.
Overall these findings indicate that the secretory epithelium of the oviduct can form tumors that resemble the human disease through genetic manipulation of pathways that are frequently modified in HGSC. PTEN silencing alone was sufficient to drive widespread disease and was further enhanced with activation of KRAS but not mutation of p53. KRAS mutation enhanced PTENshRNA phenotype, while AKTMYR blunted the peritoneal spread seen in PTENshRNA and PTENshRNA/KRASG12V MOE cells. These data are intriguing as they suggest PTENshRNA does not phenocopy AKTMYR and may have functionality independent of PI3K/AKT signaling. This is supported by previous studies which demonstrated PTEN loss results in reduction of p53 expression in tissues and cells [25]. Further, it is postulated p53 transcriptional activity is modulated by direct PTEN interaction with p300 and p53 in the nucleus, a mechanism independent of the AKT pathway [26]. However, the difference between PTENshRNA and AKTMYR in the present study may also be the result of artificial activation of AKT via myristolation, which via lipid modification, forces AKT to be presented at the cell membrane and thus constitutively activated. As a result, it is possible actions of AKT away from the cell membrane, such as in the nucleus where it is postulated to interact with FOXO family transcription factors may be suppressed in this model, thereby creating the differences seen between PTEN suppression and AKT activation [27]. Further, the construct utilized by this study activates AKT1, and although it is thought that AKT2 and 3 isoforms can compensate, evidence suggests they play distinct roles in cancer progression [28, 29]. Thus it may be beneficial to utilize a cell line incorporating all AKT isoforms, such as PI3K activation, to further clarify possible phosphatase-independent mechanisms by which PTEN promotes tumorigenesis. Despite these differences, colony formation was disrupted with an AKT small molecule inhibitor in cells with absent PTEN. This was also demonstrated with the MEK inhibitor U0126 downstream of KRAS function. Recent reports demonstrate FOXO3 is differentially expressed in p53 signatures and STICs and correlated with changes in KRAS and PTEN signaling that may underlie alterations from a benign to a malignant phenotype [30].
The novel MOE cell model created in this study has demonstrated that context of carcinogenesis is extremely impactful on the resulting disease, in particular the cell of origin and genetic alterations leading to tumorigenesis. These models can be used to investigate secreted factors that might serve as biomarkers, to directly compare against similar OSE models [31] and can be allografted sygeneically, thereby allowing for the study of tumorigenesis in the presence of immune function and immune-modulating therapies. Clarifying the mechanisms contributing to HGSC formation will inform the development of targeted screening, prevention and therapy, thereby impacting HGSC patient prognosis and survival.
materials and Methods
Cell culture
MOE were isolated from the oviducts of cycling FVB/N mice in accordance with the guidelines of the Canadian Council on Animal Care and with institutional approval by the University of Ottawa. Cells were clonogenically grown from single cells and confirmed as oviductal epithelium by expression of PAX2, PAX8, and OVGP1 proteins. MOE were cultured as previously described [18].
Stable cell development
Parental MOE cells were transfected via TransIT LT1 (Mirus Bio, Madison, WI) according to the manufacturer’s instructions with plasmids containing targets of interest (Suppl. Table 1). Cells were clonogenically isolated via antibiotic selection (Suppl. Table 1) and validated via qRT-PCR and western blotting.
Western blotting
Cell lysates (30μg) were run on 12% SDS-PAGE gels and transferred to nitrocellulose membranes (Fisher Scientific, Pittsburgh, PA). Membranes were incubated with antibodies targeting proteins of interest overnight at 4° C (Suppl. Table 2) prior to detection via horseradish peroxidase-conjugated secondary antibody (1:1000, Cell Signaling, Cambridge, MA) and SuperSignal West Femto substrate (Thermo Scientific, Rockford, IL, USA).
Proliferation assay
Sulforhodamine B (SRB) assay was utilized to determine cell density as previously described [32]. Briefly, MOE were plated at 1,000 cells per well in a 96-well plate and fixed at varying time points as described. Plates were probed with SRB and cell density determined by comparison to a cell-matched control plate fixed 2 hours post plating.
Soft agar colony formation assay
Anchorage independent growth was assessed via soft agar assay as previously described [18]. MOE cells were seeded at a density of 1.5x105 cells per well in 0.35% agarose (Sigma Aldrich, St. Louis, MO) / DMEM (Life Technologies, Carlsbad, CA) in a 24-well plate. Cells were incubated for 2 weeks, with media changed every 4 days (DMEM, 1x penicillin/streptomycin (Gibco, Carlsbad, CA), 4% FBS (Life Technologies)). Data was blinded prior to quantification via ImageJ software (NIH, Bethesda, MD). Subsequent assays utilizing inhibitors (MK2206 (10μM) and U0126 (10μM)) with DMSO as vehicle control, treatments were refreshed every 4 days with overlying media.
Study approval
All animals were treated in accordance with the NIH guidelines for Laboratory Animals and established Institutional Animal Use and Care protocol at the University of Illinois at Chicago.
Allografting
Athymic mice were acquired from (Taconic, Germantown, NY) and allografted with MOE cells including the parental/wildtype (WT) and scrambled shRNA (SCR) control lines 2x106 cells in 300μL PBS:matrigel (1:1 vol/vol) subcutaneously (s.c) and intraperitoneally (i.p.) 1x107 cells in 300μL PBS. Animal weight and s.c. tumor growth (via caliper measurement) were tracked weekly and animals sacrificed when tumor burden or general health was determined to be moribund. Syngeneic grafts were performed in FVB/N mice via i.p. injection of 1x107 cells in 300μL PBS. Subcutaneous grafts were not performed in immune competent hosts.
Tissue collection and analysis
At the time of sacrifice, s.c. tumors were dissected and weighed, evidence of i.p. disease noted and collected along with reproductive and gastrointestinal tracts for further investigation. Tissues were fixed in 4% paraformaldehyde before dehydration in ethanol and xylene prior to paraffin embedding. Immunohistochemistry was performed as previously described [33]. Briefly, tissues were sectioned and rehydrated in a gradient of ethanol prior to antigen retrieval and peroxidase block. Sections were incubated in primary antibody overnight at 4° C (Suppl. Table 2) before detection via biotinylated secondary antibody (1:200, Vector Laboratories (Burlingame, CA) and ABC peroxidase (Vector Laboratories). Targets were visualized via 3,3’-diaminobenzidine (DAB, Vector Laboratories) and counterstained with hematoxylin.
Statistics
All data are displayed as mean ± standard error. Significance for in vitro proliferation and anchorage independent growth assays was determined via paired t-test utilizing Prism software (GraphPad, La Jolla, CA). All data sets were analyzed for significant outliers by Grubbs’ test of deviation.
Acknowledgments
We would like to thank Patricia Corona for her assistance with optimization of in vitro assays, and Rosemarie Tagare and David Davis for their assistance with microscopy. This research has been generously funded by the Department of Defense (OC110133) and the American Cancer Society RSG-12-230-01-TBG.
Conflicts of Interest Statement
The authors have declared that no conflict of interest exists.
Author Contributions
SLE and JEB drafted the manuscript. SLE, SMQ, EOH, GMH, and DDL performed experiments. JJW performed pathological analysis. BCV supplied parental MOE cells. All authors assisted in manuscript preparation.
References
1. Siegel R, Ma J, Zou Z and Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014; 64:9-29.
2. Nik NN, Vang R, Shih Ie M and Kurman RJ. Origin and pathogenesis of pelvic (ovarian, tubal, and primary peritoneal) serous carcinoma. Annu Rev Pathol. 2014; 9:27-45.
3. Schmid BC and Oehler MK. New perspectives in ovarian cancer treatment. Maturitas. 2014; 77:128-136.
4. Perets R, Wyant GA, Muto KW, Bijron JG, Poole BB, Chin KT, Chen JY, Ohman AW, Stepule CD, Kwak S, Karst AM, Hirsch MS, Setlur SR, Crum CP, Dinulescu DM and Drapkin R. Transformation of the fallopian tube secretory epithelium leads to high-grade serous ovarian cancer in Brca;Tp53;Pten models. Cancer Cell. 2013; 24:751-765.
5. Tirodkar TS, Budiu RA, Elishaev E, Zhang L, Mony JT, Brozick J, Edwards RP and Vlad AM. MUC1 Positive, Kras and Pten Driven Mouse Gynecologic Tumors Replicate Human Tumors and Vary in Survival and Nuclear Grade Based on Anatomical Location. PLoS One. 2014; 9:e102409.
6. Kim J, Coffey DM, Creighton CJ, Yu Z, Hawkins SM and Matzuk MM. High-grade serous ovarian cancer arises from fallopian tube in a mouse model. Proc Natl Acad Sci U S A. 2012; 109:3921-3926.
7. Quartuccio SM, Lantvit DD, Bosland MC and Burdette JE. Conditional Inactivation of p53 in Mouse Ovarian Surface Epithelium Does Not Alter MIS Driven Smad2-Dominant Negative Epithelium-Lined Inclusion Cysts or Teratomas. PLoS One. 2013; 8:e65067.
8. Karst AM, Levanon K and Drapkin R. Modeling high-grade serous ovarian carcinogenesis from the fallopian tube. Proc Natl Acad Sci U S A. 2011; 108:7547-7552.
9. Jazaeri AA, Bryant JL, Park H, Li H, Dahiya N, Stoler MH, Ferriss JS and Dutta A. Molecular requirements for transformation of fallopian tube epithelial cells into serous carcinoma. Neoplasia. 2011; 13:899-911.
10. Shan W, Mercado-Uribe I, Zhang J, Rosen D, Zhang S, Wei J and Liu J. Mucinous adenocarcinoma developed from human fallopian tube epithelial cells through defined genetic modifications. Cell Cycle. 2012; 11:2107-2113.
11. Integrated genomic analyses of ovarian carcinoma. Nature. 2011; 474:609-615.
12. Pal SK, Reckamp K, Yu H and Figlin RA. Akt inhibitors in clinical development for the treatment of cancer. Expert opinion on investigational drugs. 2010; 19:1355-1366.
13. Zhang HY, Zhang PN and Sun H. Aberration of the PI3K/AKT/mTOR signaling in epithelial ovarian cancer and its implication in cisplatin-based chemotherapy. Eur J Obstet Gynecol Reprod Biol. 2009; 146:81-86.
14. Quartuccio SM, Karthikeyan S, Eddie SL, Lantvit DD, hAinmhire EO, Modi DA, Wei JJ and Burdette JE. Mutant p53 expression in fallopian tube epithelium drives cell migration. Int J Cancer. 2015.
15. Martins FC, Santiago I, Trinh A, Xian J, Guo A, Sayal K, Jimenez-Linan M, Deen S, Driver K, Mack M, Aslop J, Pharoah PD, Markowetz F and Brenton JD. Combined image and genomic analysis of high-grade serous ovarian cancer reveals PTEN loss as a common driver event and prognostic classifier. Genome biology. 2014; 15:526.
16. Mirantes C, Eritja N, Dosil MA, Santacana M, Pallares J, Gatius S, Bergada L, Maiques O, Matias-Guiu X and Dolcet X. An inducible knockout mouse to model the cell-autonomous role of PTEN in initiating endometrial, prostate and thyroid neoplasias. Disease models & mechanisms. 2013; 6:710-720.
17. Fan HY, Liu Z, Paquet M, Wang J, Lydon JP, DeMayo FJ and Richards JS. Cell type-specific targeted mutations of Kras and Pten document proliferation arrest in granulosa cells versus oncogenic insult to ovarian surface epithelial cells. Cancer Res. 2009; 69:6463-6472.
18. King SM, Quartuccio SM, Vanderhyden BC and Burdette JE. Early transformative changes in normal ovarian surface epithelium induced by oxidative stress require Akt upregulation, DNA damage and epithelial-stromal interaction. Carcinogenesis. 2013; 34:1125-1133.
19. Darcy KM, Brady WE, McBroom JW, Bell JG, Young RC, McGuire WP, Linnoila RI, Hendricks D, Bonome T, Farley JH and Gynecologic Oncology G. Associations between p53 overexpression and multiple measures of clinical outcome in high-risk, early stage or suboptimally-resected, advanced stage epithelial ovarian cancers A Gynecologic Oncology Group study. Gynecologic oncology. 2008; 111:487-495.
20. O hAinmhire E, Quartuccio SM, Cheng W, Ahmed RA, King SM and Burdette JE. Mutation or loss of p53 differentially modifies TGFbeta action in ovarian cancer. PLoS One. 2014; 9:e89553.
21. Mullany LK, Fan HY, Liu Z, White LD, Marshall A, Gunaratne P, Anderson ML, Creighton CJ, Xin L, Deavers M, Wong KK and Richards JS. Molecular and functional characteristics of ovarian surface epithelial cells transformed by KrasG12D and loss of Pten in a mouse model in vivo. Oncogene. 2011; 30:3522-3536.
22. Wu R, Baker SJ, Hu TC, Norman KM, Fearon ER and Cho KR. Type I to type II ovarian carcinoma progression: mutant Trp53 or Pik3ca confers a more aggressive tumor phenotype in a mouse model of ovarian cancer. Am J Pathol. 2013; 182:1391-1399.
23. Wu R, Hendrix-Lucas N, Kuick R, Zhai Y, Schwartz DR, Akyol A, Hanash S, Misek DE, Katabuchi H, Williams BO, Fearon ER and Cho KR. Mouse model of human ovarian endometrioid adenocarcinoma based on somatic defects in the Wnt/beta-catenin and PI3K/Pten signaling pathways. Cancer Cell. 2007; 11:321-333.
24. Auersperg N, Wong AS, Choi KC, Kang SK and Leung PC. Ovarian surface epithelium: biology, endocrinology, and pathology. Endocr Rev. 2001; 22:255-288.
25. Freeman DJ, Li AG, Wei G, Li HH, Kertesz N, Lesche R, Whale AD, Martinez-Diaz H, Rozengurt N, Cardiff RD, Liu X and Wu H. PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms. Cancer cell. 2003; 3:117-130.
26. Li AG, Piluso LG, Cai X, Wei G, Sellers WR and Liu X. Mechanistic insights into maintenance of high p53 acetylation by PTEN. Mol Cell. 2006; 23:575-587.
27. Tzivion G and Hay N. PI3K-AKT-FoxO axis in cancer and aging. Biochim Biophys Acta. 2011; 1813:1925.
28. Irie HY, Pearline RV, Grueneberg D, Hsia M, Ravichandran P, Kothari N, Natesan S and Brugge JS. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition. The Journal of cell biology. 2005; 171:1023-1034.
29. Heron-Milhavet L, Franckhauser C, Rana V, Berthenet C, Fisher D, Hemmings BA, Fernandez A and Lamb NJ. Only Akt1 is required for proliferation, while Akt2 promotes cell cycle exit through p21 binding. Molecular and cellular biology. 2006; 26:8267-8280.
30. Levanon K, Sapoznik S, Bahar-Shany K, Brand H, Shapira-Frommer R, Korach J, Hirsch MS, Roh MH, Miron A, Liu JF, Vena N, Ligon AH, Fotheringham S, Bailey D, Flavin RJ, Birrer MJ, et al. FOXO3a loss is a frequent early event in high-grade pelvic serous carcinogenesis. Oncogene. 2014; 33:4424-4432.
31. Xing D and Orsulic S. A genetically defined mouse ovarian carcinoma model for the molecular characterization of pathway-targeted therapy and tumor resistance. Proc Natl Acad Sci U S A. 2005; 102:6936-6941.
32. Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S and Boyd MR. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst. 1990; 82:1107-1112.
33. King SM, Hilliard TS, Wu LY, Jaffe RC, Fazleabas AT and Burdette JE. The impact of ovulation on fallopian tube epithelial cells: evaluating three hypotheses connecting ovulation and serous ovarian cancer. Endocr Relat Cancer. 2011; 18:627-642.