
INTRODUCTION
Interleukin (IL)-24 is a novel tumor suppressor and a member of the IL-10 cytokine superfamily [1, 2]. Endogenous IL-24 protein expression is detectable in the peripheral blood mononuclear cells (PBMCs), T- and B-cells and in melanocytes [2, 3]. However, IL-24 protein expression is lost in a majority of cancer cells of human origin [1, 2–4]. Previous studies from our laboratory and others have demonstrated that IL-24 has anti-tumor, anti-metastatic, and anti-angiogenic activities [3–8]. Further, studies have also shown that IL-24 is a pro-inflammatory cytokine and stimulates the Th1-type immune response [2, 9], and is subject to post-translational modifications (PTMs), including phosphorylation, glycosylation, and ubiquitination [9–11]. IL-24 is reported to interact with protein kinase [12]. However, whether phosphorylation is required for IL-24-mediated antitumor activities is unknown.
In the present study, we investigated whether IL-24 phosphorylation is required for antitumor activities. The human IL-24 DNA sequence has five potential phosphorylation sites: Serine (Ser) 88, 101, and 161, and Threonine (Thr) 111 and 133. Using molecular techniques, we replaced all of the five phosphorylation sites, producing a mutant (IL-24mt). We compared IL-24mt with wild-type IL-24 (IL-24wt). New to science, our data show that IL-24 phosphorylation is required for IL-24-mediated anti-cancer activities.
The present study provides a platform for identifying the phosphorylation site(s) critical for IL-24 to function as an anti-cancer drug. Studies investigating the molecular mechanisms of IL-24 phosphorylation are also warranted.
RESULTS
IL-24wt and IL-24mt have different protein banding patterns and cellular localization
IL-24wt-expressing H1299 cells showed a typical expression pattern [3, 11] with multiple 17 Kd to 26 Kd bands, representing different post-translational modification and maturation stages of IL-24 protein (Figure 1A). However, IL-24mt-expressing cells showed a single 19–20 Kd protein band, suggesting that phosphorylation regulates IL-24 protein maturation.
We used immunoprecipitation to determine the phosphorylation status of IL-24wt and IL-24mt protein. We detected 26 Kd bands in sample lysates prepared from IL-24wt-expressing cells, reflecting IL-24wt protein (Figure 1B). No 26 Kd band was observed in lysates prepared from IL-24mt-expressing cells, indicating that IL-24mt protein is not phosphorylated.
Since exogenous IL-24wt protein expression is detectable in the cytoplasm, endoplasmic reticulum (ER), and secretory organelles [3, 13, 14], we investigated whether phosphorylation affected IL-24 cellular localization. Confocal microscopy and immunofluorescence revealed that IL-24wt protein localized to the cytoplasm, ER, and Golgi. However, IL-24mt protein was primarily localized to the perinuclear area and overlapped with ER (Figure 1C). These results suggest that phosphorylation of IL-24 protein is important for proper subcellular localization and trafficking in the cells.
To determine whether these results were unique to the stably-transfected H1299 single cell clone, we transiently transfected naïve H1299 cells with pcDNA3.1 plasmid DNA vector carrying IL-24wt and IL-24mt cDNA under the control of constitutively active cytomegalovirus (CMV) promoter (Supplementary Figure 1A). Western blotting showed that IL-24wt- and IL-24mt-expressing cells had different banding, irrespective of the time of analysis (Supplementary Figure 1B). We further tested IL-24 protein expression in a human melanoma (MeWo) cell line and compared to protein expression in H1299 cells by transient transfection using the pcDNA3.1-IL24wt or -IL24mt plasmids. MeWo and H1299 cells showed the same differences in the protein banding for IL-24wt and IL-24mt (Supplementary Figure 1C). Protein localization study results concurred with the results that were observed with the stable H1299 cell line (Supplementary Figure 1D). Thus, the banding pattern and organelle localization is not cell-line-specific, and protein phosphorylation is important for IL-24 protein maturation, localization, and secretion.
IL-24mt protein secretion is reduced in doxycycline-induced lung cancer cells
IL-24 protein is known to be secreted [6, 13, 14]. Phosphorylation is reported to regulate protein secretion and function [15, 16]. To determine whether phosphorylation modulates IL-24 protein secretion, we used ELISA to analyze IL-24 protein in cell culture supernatants from DOX-treated H1299-IL-24wt and H1299-IL-24mt cells. We detected IL-24 protein in supernatants collected from both cell types (Figure 1D). However, there was markedly less IL-24 protein in IL-24mt-expressing cells than in IL-24wt-expressing cells (Figure 1D). These results demonstrate that phosphorylation regulates IL-24 protein secretion. Modification of the phosphorylation sites resulted in intracellular retention of IL-24 protein, leading to reduced secretion. Our western blot data (Supplementary Figure 1C) also showed reduced IL-24 protein in the culture supernatant from H1299 and MeWo cells.
IL-24wt expression reduces cell viability and colony formation
Exogenous IL-24 expression was shown to inhibit tumor cell viability [3, 6, 9]. Therefore, we performed cell viability assays to determine whether modification of the phosphorylation sites in IL-24wt influences its ability to inhibit growth. IL-24wt-expressing H1299 cells were significantly less viable than IL-24mt-expressing cells (P < 0.05; Figure 1E). We also conducted colony formation assays on soft agar. IL-24wt expression inhibited cell growth, with fewer IL-24wt-expressing cell colonies than controls (25% reduction; Figure 1F). In contrast, IL-24mt-expressing H1299 cells had slightly more colonies than controls (Figure 1F).
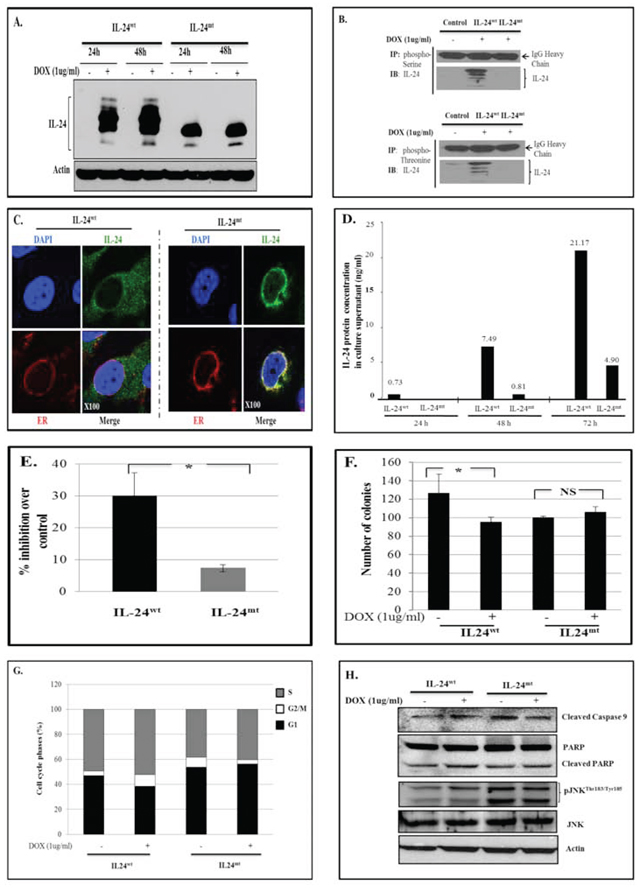
Figure 1: IL-24wt and IL-24mt have different protein banding patterns. A. Western blotting showed that IL-24wt and IL-24mt protein banding patterns differed following DOX treatment of H1299-IL-24wt and H1299-IL-24mt cells. Cells that did not receive DOX treatment served as controls. B. Cell lysates from DOX-treated H1299-IL24wt and H1299-IL24mt were immunoprecipitated (IP) with phosphorylated Serine or Threonine antibody and immunoblotted (IB) with human IL-24 antibody. IL-24 protein was detected in H1299-IL-24mt cell lysate, but not in H1299-IL-24mt cell lysate. This shows that only wild-type IL-24 protein is phosphorylated. IgG protein band served as internal protein loading control. C. Immunofluorescence studies showed that IL-24wt protein was uniformly distributed in the cytoplasm, with some localized in the endoplasmic reticulum (ER) of the cell. In contrast, IL-24mt protein was mostly localized in the ER, with little distributed in the cytoplasm of the cell. Magnification, x100. D. IL-24 protein level was markedly low in the culture supernatant collected from DOX-treated H1299-IL24mt cells compared with the IL-24 protein level in the supernatant from DOX-treated H1299-IL24wt cells, as determined by ELISA. Cell culture supernatant from untreated cells served as a negative control. The number above the bar indicates the protein concentration (ng/ml). E. Expression of IL-24wt following DOX treatment greatly reduced cell viability of H1299 cells, compared with cells expressing IL-24mt at 72 h. F. A colony formation assay on soft agar demonstrated that H1299-IL-24wt cells formed fewer colonies than H1299-IL-24mt when treated with DOX. G. Cell cycle analysis showed that only IL-24wt induced G2/M cell-cycle arrest at 48 h after DOX treatment. H. IL-24wt activated caspase-9, PARP and pJNKThr183/Tyr185 in H1299 cells at 48 h after DOX treatment, while IL-24mt did not. Beta actin was detected as protein loading control. *denotes P < 0.05. “NS” denotes Not Significant.
IL-24wt induces cell cycle arrest
Previous studies reported that IL-24wt expression in cancer cells results in cell cycle arrest, leading to apoptosis [3, 5, 17]. We therefore investigated whether IL-24wt and IL-24mt expression perturbed cell cycle progression in H1299 cells. IL-24wt expression increased the number of cells in the G2/M phase (9.3%) compared with controls (3.65%; Figure 1G). IL-24mt expression decreased the number of cells in the G2/M phase (3.4%) compared with controls (8.05%). These results demonstrate that phosphorylation is required for IL-24wt-mediated G2/M-phase cell-cycle inhibition.
IL-24wt activates caspase-9, PARP, and JNK
Our studies and others have demonstrated that adenovirus (Ad)-IL-24 induces apoptosis in cancer cells by activating caspase-9, poly (ADP ribose) polymerase (PARP), and JNK [3, 17–19]. In the present study, we determined the status of these proteins in IL-24wt- and IL-24mt-expressing H1299 cells. We observed activated caspase-9, PARP, and phosphorylated (p) JNKThr183/Tyr185 only in IL-24wt-expressing cells, indicating that phosphorylation is required for the pro-apoptotic activity of IL-24 (Figure 1H).
IL-24wt inhibits tumor cell migration and cell invasion
We previously demonstrated that lung cancer cells expressing exogenous wild-type IL-24 migrate and invade less [20]. Here, we performed a scratch assay to evaluate whether cell migration and invasion were altered by modification of the phosphorylation sites in IL-24wt cells.
IL-24wt-expressing cells migrated into 26.5% of the scratched area, whereas IL-24mt-expressing cells migrated into 94% of the scratched area (Figure 2A). To further confirm that lack of IL-24 phosphorylation abrogates the inhibition of cell migration, cell migration assays were performed. IL-24wt expression reduced tumor cell migration, compared with controls, at 24 h and 48 h (P = 0.01; Figure 2B). IL-24mt-expressing cells also migrated less than controls, although to a lesser degree than IL-24wt-expressing cells (Figure 2B).
We next performed a Matrigel assay, which mimics the invasive process of tumor cells travelling through basement membrane components. A significant reduction in cell invasion was observed only in IL-24wt-expressing cells (P = 0.001; Figure 2C). These results demonstrate that phosphorylation is required for IL-24-mediated inhibition of tumor cell migration and invasion.
IL-24wt inhibits the AKT-mTOR signaling pathway
We investigated the signaling mechanism by which IL-24 phosphorylation regulates its anti-tumor activity. Reverse phase protein array (RPPA) analysis [21, 22] on IL-24wt and IL-24mt cell lysates indicated different expression levels of proteins involved in the AKT-mammalian target of rapamycin (mTOR) pathway (data not shown). Thus, we investigated the regulation of the AKT-mTOR pathway by IL-24. Induction of IL-24wt in H1299-IL24wt cells significantly decreased pAKTS473 protein expression at 24 h and 48 h (Figure 3A). Expression of a critical downstream target of AKT, pPRAS40T246, was also reduced upon IL-24wt induction (Figure 3A). Furthermore, IL-24wt markedly reduced pmTORS2448 expression at 24 and 48 h (Figure 3A). The expression of IL-24mt protein in H1299-IL24 cells produced no change in pAKTS473 expression at 24 h (Figure 3A). However, we observed significantly increased expression at 48 h (P < 0.05; Figure 3A). pPRAS40T246 expression increased at 24 h and 48 h, while pmTORS2448 expression only increased at 24 h.
We further evaluated the effect of IL-24 phosphorylation on pAKTT308 expression and on AKT isoforms AKT1 and 2. AKT1 and 2 are ubiquitously expressed and individually contribute to cell survival, proliferation, and metastasis via different mechanisms [23]. In IL-24wt-expressing cells, we observed a marked reduction in pAKTT308, pAKT1S473, and pAKT2S474 expression at 48 h, compared with controls. IL-24mt expression increased pAKTT308 and pAKT2S474, with no change in pAKT1S473, suggesting that IL-24 phosphorylation is involved in the regulation of AKT isomers (Figure 3B).
IL-24 mediates anti-tumor activity by suppressing AKT and AKT-mediated PRAS40 phosphorylation
To evaluate whether IL-24 mediates its anti-tumor function by inhibiting AKT activation and phosphorylation is required for AKT inhibition, we knocked down AKT in H1299-IL-24 cells with si-AKT. pAKTS473 expression and cell migration were significantly lower in IL-24wt-expressing cells than controls (Figure 4A, 4B). Si-AKT knockdown mimicked the effect of IL-24wt expression on cell migration. We observed enhanced cell migration inhibition upon combining IL-24wt and si-AKT, compared with the individual treatments (Figure 4A, 4B). IL-24mt-expressing cells showed increased pAKTS473 expression that was, however, reduced in the presence of si-AKT to levels similar to those observed in H1299-IL-24wt cells (Figure 4A, 4B). IL-24mt, unlike IL-24wt, showed reduced cell migration inhibition compared with controls. The combination of treatment inhibited cell migration more than the individual treatments did. Together, our data show that IL-24wt mediates its anti-tumor activity by regulating AKT, and, importantly, that IL-24wt must be phosphorylated to regulate AKT.
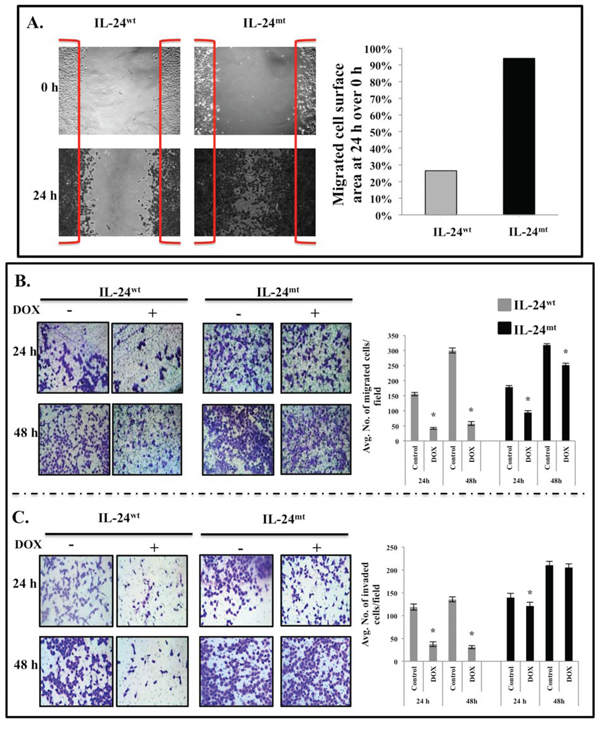
Figure 2: IL-24wt, but not IL-24mt, inhibits cell migration and invasion. A. Scratch assay showed that IL-24wt-expressing H1299 cells migrated less than IL-24mt-expressing H1299 cells. B. IL-24wt produced a greater inhibitory effect on cell migration than did IL-24mt at 24 h and 48 h after DOX treatment. C. Matrigel invasion assay showed that IL-24wt inhibited cell invasion to a greater extent than IL-24mt at 24 h and 48 h after DOX treatment. Cells that did not receive DOX treatment served as controls. Results shown are the means ± SD of three independent experiments. *P < 0.05.
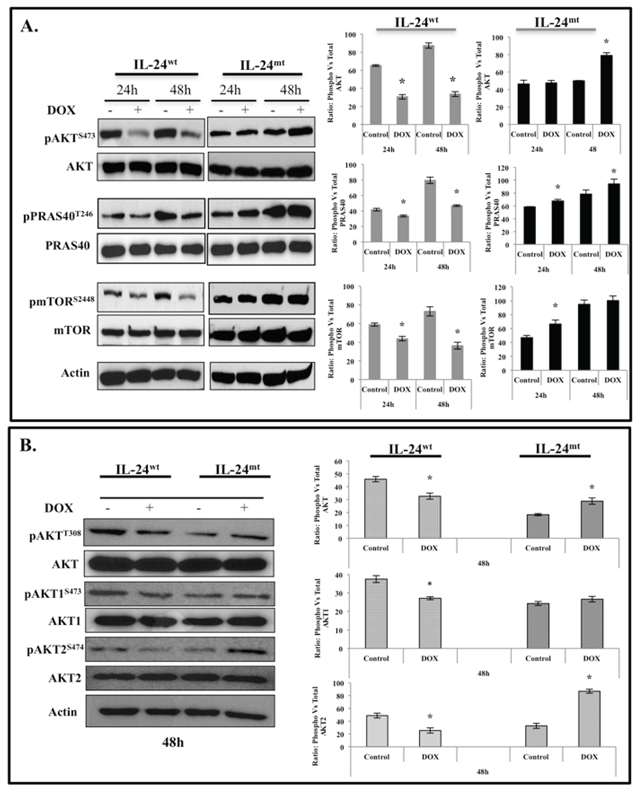
Figure 3: IL-24wt, but not IL-24mt, inhibits the AKT-mTOR signaling pathway. Cell lysates prepared from DOX-treated H1299-IL-24wt and H1299-IL-24mt cells were analyzed for proteins associated with the AKT signaling pathway and changes in protein expression quantified by Western blotting. A. IL-24wt, but not IL-24mt, significantly reduced the expression of phosphorylated (p) AKTS473, pPRAS40T246, and pmTORS2448 proteins at the two time points tested. B. Analysis for changes in the expression of AKT isoforms demonstrated that IL-24wt significantly reduced the expression of pAKTT308, pAKT1S473, and pAKT2S474. Increased expression of all of the AKT isoforms was observed in IL-24mt-expressing H1299 cells. Beta actin was detected as protein loading control. *denotes P < 0.05.
We also examined whether IL-24wt could mediate its tumor suppressor effects when AKT was overexpressed. Constitutive AKT expression was achieved by transfecting H1299-IL24wt cells with myr-AKT plasmid. We observed significantly increased pAKTS473, pPRAS40T246, and mTORS2448 expression in myr-AKT-expressing cells compared with vector controls (Figure 4C). However, we observed no noticeable IL-24wt-mediated inhibition of AKT or its downstream signaling proteins in myr- AKT-expressing cells. Furthermore, expression of myr-AKT abrogated the IL-24wt-mediated inhibition of cell migration compared with IL-24wt-mediated inhibition in the absence of myr-AKT (P < 0.05; Figure 4D). Our results confirm that the regulation of AKT signaling and phosphorylation are crucial for IL-24wt to produce anti-tumor effects.
AKT-mediated phosphorylation of PRAS40 at Threonine (T) 246 inactivates PRAS40 and influences mTOR activation [24]. Our myr-AKT study results concurred with this report. We used PRAS40mt plasmid to further test whether reduced PRAS40 expression in IL-24wt-expressing cells occurs via AKT. The plasmid expresses PRAS40 protein, in which the T is replaced with Alanine (A) at amino acid 246 (T246A), such that AKT cannot phosphorylate PRAS40, resulting in reduced pPRAS40 expression and mTOR activation. We anticipated that induction of IL-24wt, along with mutated PRAS40 expression, would show greater reduction in pPRAS40 expression. PPRAS40T246 expression was significantly reduced in mutant PRAS40T246A plasmid-transfected cells compared with controls (P < 0.05; Figure 4E). However, pPRAS40 expression was further reduced when combined with IL-24wt, comparable to our findings from cells expressing IL-24wt alone (Figure 4E). Our results show that IL-24wt inhibits AKT-mediated phosphorylation and PRAS40 inactivation to execute its anti-tumor activity.
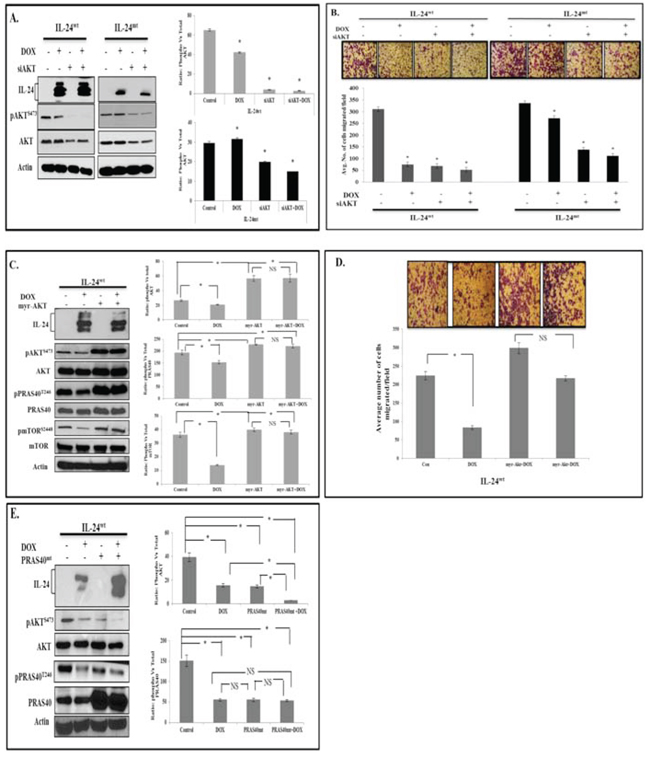
Figure 4: IL-24 mediates anti-tumor activity by suppressing AKT and AKT-mediated PRAS40 phosphorylation. H1299-IL-24wt and H1299-IL-24mt cells were transfected with AKT siRNA, or not transfected, followed by treatment with or without DOX (1 μg/ml). Then, cells were subjected to molecular analysis at 48 h after DOX treatment. A. Western blotting showed that IL-24wt plus AKT siRNA produced a greater inhibitory effect on pAKTS473 expression than IL-24mt plus siAKT. B. Cell migration assay results correlated with Western blotting and showed that IL-24wt plus siAKT produced inhibited cell migration more than IL-24mt plus siAKT. C. H1299- IL-24wt cells were either not transfected, or transfected with myr-AKT plasmid, followed by treatment with or without DOX (1 μg/ml) and subjected to molecular analysis at 48 h after treatment. Expression of myr-AKT abrogated the inhibitory effect of IL-24wt on AKT and its downstream targets, PRAS40 and mTOR. D. IL-24wt-mediated inhibition of cell migration was reduced when myr-AKT was expressed. E. Phosphorylation of PRAS40T246 and AKTS473 was significantly reduced in IL-24wt-expressing and PRAS40mt-expressing cells compared with controls. However, greater inhibition of pPRAS40T246 and pAKTS473 was observed when IL-24wt and PRAS40mt were co-expressed. Control cells did not receive any treatment. Beta actin was detected as protein loading control. *denotes P < 0.05. “NS” denotes Not Significant.
IL-24wt modulates PRAS40-Raptor interaction
AKT is known to inactivate PRAS40 via phosphorylation, thereby disrupting PRAS40-mTORC1 binding and removing PRAS40 inhibition of mTORC1 [24, 25]. PRAS40 binds to mTORC1 primarily through interactions with Raptor. Our results showed that IL-24wt expression reduced AKT-mediated PRAS40 inactivation. IL-24wt expression might, then, increase PRAS40 inhibition of mTORC1 by increasing the tight interaction between PRAS40 and Raptor. To test this, we treated H1299-IL24wt cells with DOX for 48 h. The cytosolic extracts were immunoprecipitated with PRAS40 and immunoblotted for Raptor. We observed a marked reduction in Raptor binding to PRAS40 upon IL-24wt induction (Figure 5A). Similar results were observed in H1299-IL24wt cells that were treated with mTORC1 inhibitor rapamycin (100 nM) for 48 h (Figure 5B), suggesting that IL-24wt functions similarly to rapamycin in inhibiting mTORC1.
IL-24wt alters the expression of downstream targets of AKT
We next determined the expression of proteins that are downstream of AKT in H1299 cells after induction of IL-24wt/mt protein expression. IL-24wt significantly reduced pGSK-3S21/9 and cyclin D1 and markedly increased pβ-cateninS33/37/T41 expression, compared with controls (Figure 6). Filamin A (FLNa) and p21-activated kinase 1 (PAK1) are also downstream targets of AKT. Expression of pFLNaS2152 and its downstream target pPAK1T423 were significantly reduced in IL-24wt-expressing cells (Figure 6). However, IL-24mt-expressing cells exhibited significant reduction of pβ- cateninS33/37/T41, and increased cyclin D1, pFLNaS2152, and pPAK1T423, with no change in pGSK-3S21/9 protein expression, compared with controls (Figure 6). Our results demonstrate that only IL-24wt effectively inhibits AKT and its downstream target proteins.
DISCUSSION
Protein phosphorylation is a common PTM in mammalian cells, playing a crucial role in regulating protein functions [16, 26]. Upon phosphorylation, proteins regulate critical cellular processes, such as the cell cycle, apoptosis, signal transduction, metabolism, survival, proliferation, and development [16, 26, 27]. In this study, we investigated whether phosphorylation of IL-24 is critical for anti-tumor activity. For this, we generated DOX-inducible H1299-IL24wt and H1299-IL24mt cells. We chose H1299 cells because these cells do not express endogenous IL-24 protein and lack IL-24 receptors [6, 13]. Thus, any anti-tumor activity observed against H1299 cells would be solely attributable to the exogenous IL-24 that is expressed intracellularly when induced by DOX and not confounded by secreted IL-24 [6, 13].
We demonstrated for the first time that lack of IL-24 phosphorylation affects its subcellular localization, secretion, and anti-tumor activity (Figures 1 and 2). Further, transient transfection studies revealed that our results were not cell-type-specific or attributable to the use of stable clones (Supplementary Figure 1). Other tumor suppressor proteins, including p53, LKB1, and von Hippel-Lindau proteins, require phosphorylation to function [28–31]. Similarly, phosphorylation of the herpes simplex virus type 1 ICP0 protein is necessary for its subcellular localization and efficient function [32]. Phosphorylation is also known to regulate protein secretion. Oh et al. [33] demonstrated that HMGB1phosphorylation is critical for HMGB1 protein secretion.
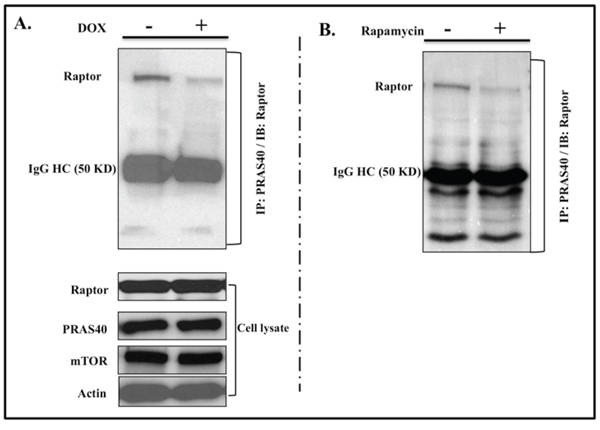
Figure 5: IL-24wt modulates PRAS40 and Raptor interaction. Immunoprecipitation studies showed that Raptor protein bound to PRAS40 was markedly reduced in A. IL-24wt-expressing cells and B. rapamycin-treated cells, compared with untreated controls. Total proteins for Raptor, PRAS40, and mTOR were detected in cell lysates to ensure that the reduced Raptor binding was not due to a reduction in total protein levels. Beta actin was detected as protein loading control.
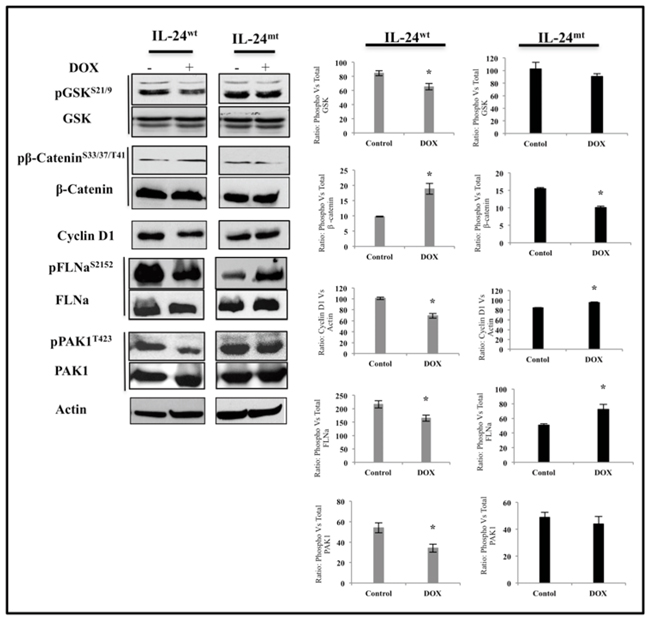
Figure 6: IL-24wt alters the expression of downstream targets of AKT. H1299-IL-24wt and H1299-IL-24mt cells treated with DOX (1 μg/ml) were subjected to molecular analysis at 48 h after treatment. Control cells did not receive any treatment. Only IL-24wt reduced the protein expression of pGSKS21/9, pFLNaS2152, pPAK1T423, and cyclin D1. Expression of pβ-cateninS33/37/T41, however, was increased in IL-24wt-, but not in IL-24mt-, expressing cells. Beta actin was detected as protein loading control. *denotes P-value < 0.05.
Functional studies showed that only IL-24wt significantly inhibited cell growth and colony formation, induced G2/M cell-cycle arrest, and activated caspase-9, PARP, and JNK, indicating that phosphorylation is required for IL-24 to exert anti-tumor effects (Figure 1). Zhao et al. [34] showed that AMPK protein phosphorylation was essential for inducing G1 cell cycle arrest and inhibition of cell proliferation and colony formation in nasopharyngeal carcinoma cells. Similarly, stress-activated protein kinases (SAPK)-mediated phosphorylation of p53 induces nuclear p53 accumulation and transcriptional activation of proteins involved in cell cycle arrest and apoptosis [35]. These findings are consistent with our results showing that subcellular localization, protein secretion, and the tumor-suppressive function of IL-24 is modulated by phosphorylation.
Tumor cell migration and invasion play important roles in lung cancer progression and metastasis [36]. We and others have shown that IL-24 inhibits metastasis by inhibiting cell migration and invasion [20]. Therefore, we focused on investigating the importance of IL-24 phosphorylation in the inhibition of migration and invasion of H1299-IL24 cells in vitro. Migration and invasion were significantly reduced only in IL-24wt-expressing cells (Figure 2). Consistent with our data, phosphorylation of the human estrogen receptor-beta at Serine 105 resulted in inhibition of breast cancer cell migration and invasion [37]. Our data reveal that IL-24 phosphorylation is important for its anti-metastatic function.
We next determined the molecular signaling mechanism by which IL-24mt and IL-24wt differed. The AKT/mTOR signaling pathway is associated with the development of human cancers, including lung cancer [38]. AKT is an essential downstream component of PI3K-mediated oncogenic signaling and provides critical cell survival signals for tumor progression by phosphorylating several proteins involved in cell cycle regulation and pro-apoptotic factors [38]. Studies showed that pharmacologic and biological inhibition of AKT/mTOR signaling suppressed cancer cell migration, invasion, and metastasis [38]. In parallel, we previously reported that IL-24 can negatively regulate the PI3K/AKT signaling pathway in Ad-IL24-transduced lung and breast cancer cells [39]. Consistent with these reports, only IL-24wt significantly inhibited the expression of AKT isoforms and their downstream signaling proteins (Figure 3). Studies using si-AKT and myr-AKT plasmid DNA demonstrated that IL-24wt specifically inhibits AKT (Figure 4A, 4B). This inhibition activated PRAS40, as evidenced by its reduced phosphorylation at Threonine 246 residue, and mTOR inhibition. AKT-mediated PRAS40 phosphorylation at Threonine 246 promotes PRAS40 binding to 14-3-3 protein complex, thereby weakening PRAS40 interaction with mTORC1, resulting in mTOR activation [25]. Further evidence for the involvement of PRAS40 in AKT/mTOR pathway inhibition was demonstrated using PRAS40T246A plasmid DNA (Figure 4E).
In the absence of nutrients, PRAS40 binds to Raptor and inhibits mTOR kinase activity by blocking TOR signaling (TOS)-mediated substrate access to Raptor [25, 40]. Since IL-24wt relieved PRAS40 from AKT inhibition, indicated by reduced pPRAS40T246 expression, we examined whether activated PRAS40 further interacted with Raptor to inhibit mTORC1. We observed less interaction between PRAS40 and Raptor in IL-24wt-expressing H1299 cells than in controls (Figure 5A). To determine whether our observation was common among the agents that disrupt mTOR, we tested rapamycin on H1299-IL24wt cells and observed reduced interaction between PRAS40 and Raptor in rapamycin-treated cells, compared with controls (Figure 5B). While we might speculate that IL-24wt likely disrupts the mTOR complex, additional studies investigating the molecular mechanism are warranted.
Finally, we investigated the expression of proteins that are molecular targets of and regulated by AKT. Only IL-24wt reduced the phosphorylation of GSK-3 and increased β-catenin phosphorylation (Figure 6). GSK-3 and β-catenin regulate cellular processes and are often deregulated in cancer [41]. The ability of IL-24wt to modulate GSK-3 and β-catenin concurs with our previous report demonstrating that Ad-mediated IL-24 expression in breast cancer cells reduced the expression of these proteins [39]. The expression of cyclin D1, a cell-cycle regulator and a target gene of β-catenin, is increased in lung cancer and correlated with increased risk of tumor progression and metastasis [42, 43]. In the present study, only IL-24wt reduced cyclin D1 expression (Figure 6). The ability of IL-24wt to markedly inhibit β-catenin and cyclin D1 expression and activate GSK-3, determined by its phosphorylation, makes IL-24-based cancer therapy attractive.
Recent studies have shown that cyclin D1 interacts with and regulates FLNa expression in migrating breast cancer cells [44]. FLNa is a widely expressed molecular scaffold protein that organizes filamentous actin into networks and stress fibers [45], is involved in cell motility and invasion [46], and is frequently overexpressed in cancers, including lung cancer [46]. SiRNA-mediated knock-down of cyclin D1 reduced phosphorylation of FLNa at Ser2152 and Ser1459, resulting in reduced cell migration and invasion [44].
FLNa is also regulated by PAK1 [47]. PAKs play an important role in cellular processes, including cytoskeletal rearrangements and growth and apoptotic signal transduction through mitogen-activated protein kinases [47]. PAK1 phosphorylation at Threonine 423 by PDK1 is associated with an increase in phosphorylation of PAK1 substrates, including FLNa at Serine 2152 [48]. Physical interaction of FLNa with PAK1 has been shown to enhance PAK1 kinase activity, which subsequently phosphorylates FLNa at Serine 2152 [47, 48]. This bidirectional PAK1/FLNa interaction has been demonstrated to influence actin cytoskeletal structures and enhance tumor cell migration. Finally, PAK1 has been reported to be regulated by AKT and regulate chemotaxis [48]. Our study indicated that only IL-24wt reduced pFLNaS2152 and pPAK1T423 expression, coinciding with reduced AKT and cyclin D1 (Figure 6). We did not, however, analyze for cyclin B1, which has also been shown to regulate FLNa [49]. To our knowledge, this is the first report demonstrating that IL-24 protein phosphorylation is critical for modulating the AKT/mTOR signaling axis and exerting its anti-tumor activities in lung cancer. Whether IL-24-mediated inhibition occurs via the AKT/mTOR pathway in human cancers of different origins is unknown and should be investigated. Additionally, all of the experiments in this study were performed in a receptor-negative H1299 lung cancer cell line. Thus, the role of phosphorylation in IL-24 cytokine function in immune cells remains unexplored. Finally, the contribution of protein phosphorylation on IL-24 protein stability is not known and is currently being investigated in the laboratory.
In conclusion, we demonstrated that IL-24 phosphorylation is essential for its anti-cancer properties. IL-24 is the only IL-10 cytokine family member whose functions are regulated by PTMs, analogous to classical tumor suppressors. Our results provide a platform for identifying the phosphorylation site(s) that are critical for IL-24 to function as an anti-cancer drug.
MATERIALS AND METHODS
Cell culture
We cultured human non-small cell lung cancer cells (H1299) in RPMI 1640 medium (GIBCO) as previously described [4, 6]. The cell line was authenticated at the Genetic Resource Core Resource Facility, Johns Hopkins University, Baltimore, MD. In all experiments, untreated cells served as controls.
IL-24 plasmid vectors and cell lines
MDA-7/IL-24 (GenBank accession NM_006850, variant 1) cDNA is 621 bp in size with 206 amino acids. Potential phosphorylation sites of MDA-7/IL-24 protein predicted by NIH NCBI ORF finder are S88, S101, T111, T133 and S161 where S stands for Serine; T stands for Threonine. The software predicted that phosphorylation in MDA-7/IL-24 are as following: amino acid 88 SAR, amino acid 133 TLK, amino acid 161 SIR are casein kinase II consensus sites; amino acid 101 SDAE, amino acid 111 TLLE, amino acid 161 SIRD are protein kinase C (PKC) consensus sites [50].
Human IL-24wt cDNA cloned in pLJ143 plasmid backbone was used as a template to create the IL-24 phosphorylation mutant (IL-24mt). Briefly, IL-24wt cDNA was retrieved by BamHI and XhoI restriction enzyme digestion and ligated back into to pBlueScript SK+ (Agilent). The sequence was verified by polymerase chain reaction (PCR) using primers MDA-7.F.BamHI (forward primer) and MDA7.R.XhoI (reverse primer; Table 1) and confirmed by DNA sequencing from T7 and M13R.
On the basis of wild-type IL-24wt cDNA sequence, IL-24 phosphorylation mutant (IL-24mt) plasmid was generated by replacing the three serine (S) and two threonine (T) sites using the forward and reverse PCR primers (Table 1) and the “Overlay PCR” method. The primers designed (Table 1) for creating the IL-24mt was such that the amino acid replacing the native amino acid in MDA-7/IL24 protein had equivalent size, structural similarity, charge and polarity. In the “Overlay PCR” method, the forward primer was designed such that the intended one or two or three nucleotides which are to be replaced were flanked on each side with approximately 20 oligonucleotide bases. The forward primer was used in conjunction with a complementing reverse primer. The forward primer for a mutant is paired with a next 3′ reverse primer for a high fidelity PCR reaction; the reverse primer is paired with previous 5′ forward primer in a high fidelity PCR reaction. Then the two segments of PCR products, which share approximately 40 bases together (so-called overlay), are used as templates for overlaying both segments together using previous 5′ forward primer and next 3′ reverse primer. The resulting PCR product with all five phosphorylation site replaced was subsequently TA cloned into TOPO plasmid vector (Invitrogen) and sequenced at the DNA sequencing core facility (MD Anderson Cancer Center, Houston, TX).
The IL-24wt and IL-24mt cDNA from the TOPO plasmid vector was sub-cloned into pcDNA3.1+ plasmid expression vector (Invitrogen). The resultant plasmids were verified by BamHI-XhoI restriction enzyme digestion, PCR, and by the inserted signature restriction enzyme digestion. All PCR reactions performed in the study followed the High Fidelity PCR system protocol (Roche). Restriction enzymes used in the study were purchased from New England Biolabs. The pcDNA3.1+ plasmid vector carrying IL-24wt and IL-24mt cDNA sequence were either used in transient transfection studies or used in creating doxycycline (DOX)-inducible plasmid vector as described below.
Table 1: PCR Primers used in “Overlay PCR” for generating IL-24mt
Prime Amino acid/Restriction E enzyme |
Primer sequences |
IL-24. F.BamHI |
5′GCGGATCCGAGGAAGGCCAGGAGGAACAC 3′ |
IL-24. R.XhoI |
5′ GCCTCGAGTGAAATGACACAGGGAACAAACCAGT 3′ |
IL-24- S88A.F.EagI |
5′ GCTCAGGATAACATCACGGCCGCCCGGCTGCTGCAGCAG 3′ |
IL-24- S88A.R.EagI |
5′ CTGCTGCAGCAGCCGGGCGGCCGTGATGTTATCCTGAGC 3′ |
IL-24- S101D.F.Hinc2 |
5′ GAGGTTCTGCAGAACGTCGACGATGCTGAGAGCTGTTAC 3′ |
IL-24- S101D.R.Hinc2 |
5′ GTAACAGCTCTCAGCATCGTCGACGTTCTGCAGAACCTC 3′ |
IL-24- T111Q.F.PvuII |
5′ GAGCTGTTACCTTGTCCACCAGCTGCTGGAGTTCTACTTG 3′ |
IL-24- T111Q.R.PvuII |
5′ CAAGTAGAACTCCAGCAGCTGGTGGACAAGGTAACAGCTC 3′ |
IL-24- T133Q.F.Pvu2 |
5′ GAACAGTTGAAGTCAGGCAGCTGAAGTCATTCTCTACTC 3′ |
IL-24- T133Q.R.Pvu2 |
5′ GAGTAGAGAATGACTTCAGCTGCCTGACTTCAACTGTTC 3′ |
IL-24- S161D.F.EcoRV |
5′ CAAGAAAATGAGATGTTTGATATCAGAGACAGTGCACACAGG 3′ |
IL-24- S161D.R.EcoRV |
5′ CCTGTGTGCACTGTCTCTGATATCAAACATCTCATTTTCTTG 3′ |
“F” and “R” preceding restriction enzyme denote forward and reverse primer respectively.
Tet-ON inducible IL-24 expression in H1299 lung cancer cell line
The Tet-ON inducible system was purchased from Clontech. The IL-24wt and IL-24mt cDNA sequence cloned in pcDNA3.1+ plasmid vector was retrieved by BamHI and PmeI restriction enzyme digestion and sub-cloned into BamHI and EcoRV enzyme digested pTRE-Tight plasmid (Clontech). The resulting pTre-IL24wt and pTre-IL24mt plasmids with appropriate inserts were verified by PCR, restriction enzyme digestions and signature enzyme digestions. The plasmids were subsequently used for creating Tet-ON inducible stable cell lines per manufacturer’s guidelines. Briefly, human H1299 lung cancer cell line was transfected with pTet-ON-adv plasmid (Clontech) using Fugene (Roche) and selected in Neomycin (800 μg/ml; Sigma) for fourteen days. Transfection studies were performed using 10% tetracycline free fetal bovine serum (Atlanta Biologicals). The surviving fraction of cells was replated and transfected with pTreT-IL-24wt or pTreT-IL-24mt plasmid followed by hygromycin (400 μg/ml; Sigma) for two weeks. The surviving fraction of cells were then expanded and screened for IL-24wt and IL-24mt expression by Western blotting after DOX (1 μg/ml) treatment for 24 h. The IL-24wt and IL-24mt expressing cells were then seeded in ninety-six-well plates and subjected to single-cell cloning. Each clonal population of cells was subsequently screened for IL-24 protein expression by Western blotting. The clone that demonstrated the highest IL-24wt and IL-24mt protein expression was expanded and labeled as H1299-IL24wt and H1299-IL24mt respectively and used in all of the studies described herein.
Cell viability assay
H1299-IL24wt and H1299-IL24mt cells (1 − 2 × 105 cells/well) seeded in six-well plates were treated with DOX (1 μg/ml). The number of viable cells at 72 h after DOX treatment was determined by the trypan-blue exclusion assay method as described previously [6, 17].
Colony-forming assay
H1299-IL24wt and H1299-IL24mt cells (2 × 104) were mixed with 0.7% low-melting agarose and seeded in six-well tissue culture plates previously coated with 0.7% agarose. The plates were incubated at 37°C for 24 h. Then, the cells were treated with DOX (1 μg/ml). Incubation continued for fourteen days at 37°C, with culture medium replenished twice a week. The number of surviving colonies was determined by staining with 0.005% crystal violet. Colonies were photographed and counted.
Cell cycle analysis
H1299-IL24wt and H1299-IL24mt cells (1 × 105) seeded in six-well plates were treated with DOX (1 μg/ml). Untreated cells served as controls. Cells were collected at 48 h after treatment and subject to flow-cytometric analysis as previously described [6, 17].
Western blotting assay
H1299-IL24wt and H1299-IL24mt cells (1 × 105/well) seeded in six-well plates were trypsinized and collected at 24 h and/or at 48 h after DOX (1 μg/ml). Cell lysates were prepared and protein samples subjected to western blotting [6, 11, 17]. Primary antibodies against IL-24 (Introgen Therapeutics), pAKTS473, pAKTT308, AKT, pAKT1S473, AKT1, pAKT2S474, AKT2, pPRAS40T246, PRAS40, pmTORS2448, mTOR, pGSK-3S21/9, GSK-3, pβ- catenin S33/37/T41, β-catenin, pFLNaS2152, FLNa, pPAK1T423, PAK1, pJNKThr183/Tyr185, JNK, cyclin D1, caspase-9, and PARP (Cell Signaling) were purchased and used. Beta-actin (Sigma) was used as an internal loading control. Proteins were detected using the appropriate secondary antibodies, as previously described [6, 11, 17, 39]. Protein expression levels were quantified using Image Quant (Syngene) software.
Immunoprecipitation assay
Cells (5 × 106) actively growing in 150-mm tissue culture plates were treated with DOX (1 μg/ml). Forty-eight hours after treatment, the cells were washed and trypsinized, and total cell lysates prepared. About 500 μg of total protein was mixed with 100 μl of appropriate antibody (phospho-Serine, phospho-Threonine, PRAS40; Cell Signaling) and incubated at 4°C overnight with constant mixing. Immune complexes were captured by incubation with 75–100 μl protein A/G-coated agarose beads for 1 h at 4°C. The immunoprecipitated proteins were recovered by boiling the agarose beads in 2X SDS sample buffer and western blotting. The membrane was subsequently probed using the appropriate primary antibody (IL-24, Raptor), and the protein bands detected as previously described [6, 17].
Immunofluorescence assay
H1299-IL24wt and H1299-IL24mt cells (1 × 103) seeded in two-well glass chamber slides were treated with DOX (1 μg/ml). Twenty-four hours after treatment, the cells were washed with 1.0 mL warm 1X phosphate-buffered saline (PBS) at 37°C and subsequently fixed in 4% paraformaldehyde in PBS for 10 min at room temperature (RT). After fixation, the cells were washed with PBS, incubated with 100% methanol for 10 min, and permeabilized with 0.5% TritonX-100 for 10 min. The cells were washed with PBS, followed by incubation in blocking buffer (3% BSA + 1% normal serum) in PBS for 1 h at RT. Then, we added a mixture of anti-human IL-24 antibody raised in goat (5.0 μl; Introgen Therapeutics) and mouse anti-PDI antibody (0.5 μl; Molecular Probes) in blocking buffer and incubated for 2 h at RT. The cells were subsequently washed with 0.5 ml of 1X blocking buffer, followed by the addition of a mixture of Alexa Fluor® 488 (anti-mouse green) and Alexa Fluor® 594 (anti-goat red) diluted in 1 ml of blocking buffer. The cells were incubated in the dark for 1 h at RT, then counter stained with DAPI (Sigma) for 1 min. The stained cells were washed with 1X PBS, mounted with Dako cytomation fluorescent mounting media (Dako), and then viewed using a laser confocal microscope equipped with appropriate fluorescein filters.
Enzyme-linked immunosorbent assay (ELISA)
H1299-IL24wt and H1299-IL24mt cells (1 × 104) seeded in six-well plates were incubated overnight at 37°C, 5% CO2. The next day, the culture medium was replaced with fresh RPMI-1640 medium containing 2% FBS. DOX (1 μg/ml) was added. Culture supernatants were collected at different times after treatment, centrifuged, and stored at −80°C. We detected IL-24 protein in the supernatant by sandwich ELISA (R&D Systems). Briefly, ninety-six well flat-bottomed plates were coated with 100 μl of anti-IL-24 capture antibody (2.0 μg/mL; R&D Systems) overnight at +4°C. The plates were washed with sterile PBS followed by incubation in 100 μl of blocking buffer containing 10% bovine serum albumin (BSA in PBS) for 1 h at RT. On completion of incubation, the blocking buffer was aspirated and 100 μl of culture supernatant collected from H1299-IL24wt and H1299-IL24mt cells with and without DOX treatment was diluted in PBS (1:2) and added in duplicate wells and incubated for 2 h at RT. The culture medium from the wells was subsequently aspirated and washed with sterile PBS. To the wells 100 μl of horse-radish peroxide (HRP)-conjugated anti-IL-24 detection antibody (R&D Systems) was added. After 1 h incubation, the wells were washed with sterile PBS followed by addition of streptavidin-horse radish peroxidase (100 μl/well) and incubation in the dark for 20 min at RT. The reaction was stopped by adding 50 μl of stop solution (R&D Systems). The plates were then read using a plate reader equipped with 450 nm excitation and 540 nm emission filter.
In vitro scratch assay
The in vitro scratch assay was performed using the CytoselectTM 24-well wound healing assay (Cell Biolabs). Briefly, H1299-IL24wt and H1299-IL24mt cells (2.5 × 104) were added to 24-well plates with inserts in place. After 24 h incubation, we removed the inserts to generate a 0.9 mm wound field, washed it with media, and incubated at 37°C. The cells were then treated with DOX (1 μg/ml). We photographed the scratch area 24 h later using a Nikon TiU microscope (Nikon). The distances between the two cell edges were measured per the manufacturer’s instructions.
Cell migration assay
H1299-IL24wt and H1299-IL24mt cells (5 × 104) suspended in 1 ml RPMI-1460 medium were seeded in the upper chambers (8 μm; BD Biosciences) and placed in a six-well plate filled with serum-free RPMI-1640 medium (lower chamber). After 24 h, the culture medium was replaced with fresh medium containing 20% tetracycline-free FBS (Atlanta Biologicals). The upper chamber was filled with 2% tetracycline-free FBS-containing medium, with or without DOX (1 μg/ml; Sigma). Following incubation for 24 h and 48 h, the inserts were removed and processed as previously described [20, 51, 52]. Four random fields were selected and we counted the migrated cells per field under an inverted bright-field microscope at 10X magnification. The results were expressed as the average number of cells of four fields.
Cell invasion assay
The cell invasion assay was performed using six-well Matrigel invasion chambers with 8-μm pore inserts (BD Biosciences). H1299-IL24wt and H1299-IL24mt (5 × 104) cells were seeded into the upper chambers. The lower chambers were filled with serum-free medium. After 24 h, the medium was replaced with 20% FBS-containing culture medium, and the upper chambers were filled with 2% FBS-containing culture medium, with or without DOX (1 μg/ml). After an additional 24 h and 48 h incubation, the filters were removed and processed as previously described [20, 51, 52]. Tumor cell invasiveness was determined by counting cells in four microscopic fields per well. The extent of invasion was expressed as an average number of cells per field.
RNA interference studies
H1299-IL24 (wt and mt) cells (1 × 105 cells/well) were seeded in serum-free medium in six-well plates and transfected with 100 nM AKT siRNA (Santa Cruz) using transfection reagent (Dharmacon), per the manufacturer’s instructions. Six hours later, the medium was replaced with RPMI-1460 containing 2% tetracycline-free serum, with or without 1 μg/ml DOX. Untransfected cells served as a control. After 48 h of incubation, the cells were harvested and total cell lysates prepared for western blot analysis.
For the cell migration assay, H1299-IL24 (wt and mt) cells (5 × 104) were seeded in the upper chamber of the inserts and transfected with AKT siRNA (100 nM), with or without 1 μg/ml DOX. The lower chamber was filled with 20% tetracycline-free FBS-containing medium. We counted the number of migrated cells 48 h after treatment.
AKT and PRAS40 overexpression studies
H1299-IL24wt cells (1 × 105 cells/well) were seeded in six-well plates and transfected with 0.5 μg myr-AKT or pPRAS40T246 DNA (Addgene) using Lipofectamine 2000 (Invitrogen), per the manufacturer’s instructions. Six hours after transfection in serum-free medium, the medium was replaced with RPMI-1460 containing 2% tetracycline-free serum, with or without 1 μg/ml DOX. Untransfected cells served as controls. After 48 h of incubation, the cells were harvested and total cells lysate subjected to western blotting.
For the cell migration assay, H1299-IL24wt cells (5 × 104) were seeded in the upper chamber of the inserts and were transfected with myr-AKT plasmid (0.5 μg), with or without 1 μg/ml DOX. The rest of the procedure followed the description under “cell migration assay”.
Statistical analysis
All experiments were performed at least twice. Unless otherwise stated, all data are shown as mean ± SD. Data were analyzed using two-tailed student’s t test or one-way analysis of variance (ANOVA) using SPSS 12.0 (SPSS, Inc., Chicago, IL). P < 0.05 was considered statistically significant.
ACKNOWLEDGMENTS
The study was supported in part by funds received from the Jim and Christy Everest Endowed Chair in Cancer Developmental Therapeutics, The University of Oklahoma Health Sciences Center. The authors thank Ms. Kathy Kyler at the office of Vice President of Research, OUHSC for editorial assistance. Rajagopal Ramesh is an Oklahoma TSET Research Scholar and holds the Jim and Christy Everest Endowed Chair in Cancer Developmental Therapeutics.
CONFLICTS OF INTEREST
The authors report no conflicts of interest in this work.
Authors’ contributions
JP, MS, JJ, CDB, RM: conducted the studies and collected data; JJ and MS: constructed and characterized the wild-type and phosphorylation mutant plasmids; and created the doxycycline-inducible stable cell lines; DZ: performed statistical analysis; JP, MS, JJ, CDB, RM, DZ, SC, AM, and RR: conceived and designed the studies; JP, SC, AM, and RR wrote the manuscript. JP, MS, JJ, CDB, RM, DZ, SC, AM and RR: critically analyzed and interpreted the data; JP, MS, JJ, CDB, RM, DZ, SC, AM and RR: critically reviewed, provided suggestions and edited the manuscript; RR supervised the project.
REFERENCES
1. Jiang H, Lin JJ, Su ZZ, Goldstein NI, Fisher PB. Subtraction hybridization identifies a novel melanoma differentiation associated gene, mda-7, modulated during human melanoma differentiation, growth and progression. Oncogene. 1995; 11:2477–2486.
2. Caudell EG, Mumm JB, Poindexter N, Ekmekcioglu S, Mhashilkar AM, Yang XH, Retter MW, Hill P, Chada S, Grimm EA. The protein product of the tumor suppressor gene, melanoma differentiation-associated gene 7, exhibits immunostimulatory activity and is designated IL-24. J Immunol. 2002; 168:6041–6046.
3. Mhashilkar AM, Schrock RD, Hindi M, Liao J, Sieger K, Kourouma F, Zou-Yang XH, Onishi E, Takh O, Vedvick TS, Fanger G, Stewart L, Watson GJ, et al. Melanoma differentiation associated gene-7 (mda-7): a novel anti-tumor gene for cancer gene therapy. Mol Med. 2001; 7:271–282.
4. Saeki T, Mhashilkar A, Swanson X, Zou-Yang XH, Sieger K, Kawabe S, Branch CD, Zumstein L, Meyn RE, Roth JA, Chada S, Ramesh R. Inhibition of human lung cancer growth following adenovirus-mediated mda-7 gene expression in vivo. Oncogene. 2002; 21:4558–4566.
5. Gopalan B, Shanker M, Chada S, Ramesh R. MDA-7/IL-24 suppresses human ovarian carcinoma growth in vitro and in vivo. Mol Cancer. 2007; 6:11.
6. Ramesh R, Mhashilkar AM, Tanaka F, Saito Y, Branch CD, Sieger K, Mumm JB, Stewart AL, Boquoi A, Dumoutier L, Grimm EA, Renauld JC, Kotenko S, Chada S. Melanoma differentiation-associated gene 7/interleukin (IL)-24 is a novel ligand that regulates angiogenesis via the IL-22 receptor. Cancer Res. 2003; 63:5105–5113.
7. Panneerselvam J, Munshi A, Ramesh R. Molecular targets and signaling pathways regulated by interleukin (IL)-24 in mediating its antitumor activities. J Mol Signal. 2013; 8:15.
8. Tong AW, Nemunaitis J, Su D, Zhang Y, Cunningham C, Senzer N, Netto G, Rich D, Mhashilkar A, Parker K, Coffee K, Ramesh R, Ekmekcioglu S, et al. Intratumoral injection of INGN 241, a nonreplicating adenovector expressing the melanoma-differentiation associated gene-7 (mda-7/IL24): biologic outcome in advanced cancer patients. Mol Ther. 2005; 11:160–172.
9. Inoue S, Shanker M, Miyahara R, Gopalan B, Patel S, Oida Y, Branch CD, Munshi A, Meyn RE, Andreeff M, Tanaka F, Mhashilkar AM, Chada S, Ramesh R. MDA-7/IL-24-based cancer gene therapy: translation from the laboratory to the clinic. Curr Gene Ther. 2006; 6:73–91.
10. Fuson KL, Zheng M, Craxton M, Pataer A, Ramesh R, Chada S, Sutton RB. Structural mapping of post-translational modifications in human interleukin-24: role of N-linked glycosylation and disulfide bonds in secretion and activity. J Biol Chem. 2009; 284:30526–30533.
11. Gopalan B, Shanker M, Scott A, Branch CD, Chada S, Ramesh R. MDA-7/IL-24, a novel tumor suppressor/cytokine is ubiquitinated and regulated by the ubiquitin-proteasome system, and inhibition of MDA-7/IL-24 degradation enhances the antitumor activity. Cancer Gene Ther. 2008; 15:1–8.
12. Pataer A, Vorburger SA, Chada S, Balachandran S, Barber GN, Roth JA, Hunt KK, Swisher SG. Melanoma differentiation-associated gene-7 protein physically associates with the double-stranded RNA-activated protein kinase PKR. Mol Ther. 2005; 11:717–723.
13. Sieger KA, Mhashilkar AM, Stewart A, Sutton RB, Strube RW, Chen SY, Pataer A, Swisher SG, Grimm EA, Ramesh R, Chada S. The tumor suppressor activity of MDA-7/IL-24 is mediated by intracellular protein expression in NSCLC cells. Mol Ther. 2004; 9:355–367.
14. Sauane M, Lebedeva IV, Su ZZ, Choo HT, Randolph A, Valerie K, Dent P, Gopalkrishnan RV, Fisher PB. Melanoma differentiation associated gene-7/interleukin-24 promotes tumor cell-specific apoptosis through both secretory and nonsecretory pathways. Cancer Res. 2004; 64:2988–2993.
15. Jones JI, D’Ercole AJ, Camacho-Hubner C, Clemmons DR. Phosphorylation of insulin-like growth factor (IGF)-binding protein 1 in cell culture and in vivo: effects on affinity for IGF-I. Proc Natl Acad Sci USA. 1991; 88:7481–7485.
16. Johnson LN. The regulation of protein phosphorylation. Biochem Soc Trans. 2009; 37:627–641.
17. Saeki T, Mhashilkar A, Chada S, Branch C, Roth JA, Ramesh R. Tumor-suppressive effects by adenovirus-mediated mda-7 gene transfer in non-small cell lung cancer cell in vitro. Gene Ther. 2000; 7:2051–2057.
18. Yacoub A, Mitchell C, Lebedeva IV, Sarkar D, Su ZZ, McKinstry R, Gopalkrishnan RV, Grant S, Fisher PB, Dent P. mda-7 (IL-24) Inhibits growth and enhances radiosensitivity of glioma cells in vitro via JNK signaling. Cancer Biol Ther. 2003; 2:347–353.
19. Kawabe S, Nishikawa T, Munshi A, Roth JA, Chada S, Meyn RE. Adenovirus-mediated mda-7 gene expression radiosensitizes non-small cell lung cancer cells via TP53-independent mechanisms. Mol Ther. 2002; 6:637–644.
20. Ramesh R, Ito I, Gopalan B, Saito Y, Mhashilkar AM, Chada S. Ectopic production of MDA-7/IL-24 inhibits invasion and migration of human lung cancer cells. Mol Ther. 2004; 9:510–518.
21. Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discover. 2005; 4:988–1004.
22. Tibes R, Qiu Y, Lu Y, Hennessy B, Andreeff M, Mills GB, Kornblau SM. Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol Cancer Ther. 2006; 5:2512–2521.
23. Chin YR, Toker A. Function of Akt/PKB signaling to cell motility, invasion and the tumor stroma in cancer. Cell Signal. 2009; 21:470–476.
24. Nascimento EB, Ouwens DM. PRAS40: target or modulator of mTORC1 signalling and insulin action? Arch Physiol Biochem. 2009; 115:163–175.
25. Wang L, Harris TE, Roth RA, Lawrence JC Jr. PRAS40 regulates mTORC1 kinase activity by functioning as a direct inhibitor of substrate binding. J Biol Chem. 2007; 282:20036–20044.
26. Lannigan DA. Estrogen receptor phosphorylation. Steroids. 2003; 68:1–9.
27. Karve TM, Cheema AK. Small changes huge impact: the role of protein posttranslational modifications in cellular homeostasis and disease. J Amino Acids. 2011; 2011:207691.
28. Ashcroft M, Kubbutat MH, Vousden KH. Regulation of p53 function and stability by phosphorylation. Mol Cell Biol. 1999; 19:1751–1758.
29. Ostermeyer AG, Runko E, Winkfield B, Ahn B, Moll UM. Cytoplasmically sequestered wild-type p53 protein in neuroblastoma is relocated to the nucleus by a C-terminal peptide. Proc Natl Acad Sci USA. 1996; 93:15190–15194.
30. Zhan YY, Chen Y, Zhang Q, Zhuang JJ, Tian M, Chen HZ, Zhang LR, Zhang HK, He JP, Wang WJ, Wu R, Wang Y, Shi C, et al. The orphan nuclear receptor Nur77 regulates LKB1 localization and activates AMPK. Nature Chem Biol. 2012; 8:897–904.
31. Lee S, Chen DY, Humphrey JS, Gnarra JR, Linehan WM, Klausner RD. Nuclear/cytoplasmic localization of the von Hippel-Lindau tumor suppressor gene product is determined by cell density. Proc Natl Acad Sci USA. 1996; 93:1770–1775.
32. Davido DJ, von Zagorski WF, Lane WS, Schaffer PA. Phosphorylation site mutations affect herpes simplex virus type 1 ICP0 function. J Virol. 2005; 79:1232–1243.
33. Oh YJ, Youn JH, Ji Y, Lee SE, Lim KJ, Choi JE, Shin JS. HMGB1 is phosphorylated by classical protein kinase C and is secreted by a calcium-dependent mechanism. J Immunol. 2009; 182:5800–5809.
34. Zhao L, Wen ZH, Jia CH, Li M, Luo SQ, Bai XC. Metformin induces G1 cell cycle arrest and inhibits cell proliferation in nasopharyngeal carcinoma cells. Anat Rec. 2011; 294:1337–1343.
35. Alarcon-Vargas D, Ronai Z. p53-Mdm2—the affair that never ends. Carcinogenesis. 2002; 23:541–547.
36. Bulk E, Sargin B, Krug U, Hascher A, Jun Y, Knop M, Kerkhoff C, Gerke V, Liersch R, Mesters RM, Hotfilder M, Marra A, Koschmieder S, et al. S100A2 induces metastasis in non-small cell lung cancer. Clin Cancer Res. 2009; 15:22–29.
37. Lam HM, Suresh Babu CV, Wang J, Yuan Y, Lam YW, Ho SM, Leung YK. Phosphorylation of human estrogen receptor-beta at serine 105 inhibits breast cancer cell migration and invasion. Mol Cell Endocrinol. 2012; 358:27–35.
38. Yap TA, Garrett MD, Walton MI, Raynaud F, de Bono JS, Workman P. Targeting the PI3K-AKT-mTOR pathway: progress, pitfalls, and promises. Curr Opin Pharmacol. 2008; 8:393–412.
39. Mhashilkar AM, Stewart AL, Sieger K, Yang HY, Khimani AH, Ito I, Saito Y, Hunt KK, Grimm EA, Roth JA, Meyn RE, Ramesh R, Chada S. MDA-7 negatively regulates the beta-catenin and PI3K signaling pathways in breast and lung tumor cells. Mol Ther. 2003; 8:207–219.
40. Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, Carr SA, Sabatini DM. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007; 25:903–915.
41. Li VS, Ng SS, Boersema PJ, Low TY, Karthaus WR, Gerlach JP, Mohammed S, Heck AJ, Maurice MM, Mahmoudi T, Clevers H. Wnt signaling through inhibition of beta-catenin degradation in an intact Axin1 complex. Cell. 2012; 149:1245–1256.
42. Ratschiller D, Heighway J, Gugger M, Kappeler A, Pirnia F, Schmid RA, Borner MM, Betticher DC. Cyclin D1 overexpression in bronchial epithelia of patients with lung cancer is associated with smoking and predicts survival. J Clin Oncol. 2003; 21:2085–2093.
43. Dobashi Y, Goto A, Fukayama M, Abe A, Ooi A. Overexpression of cdk4/cyclin D1, a possible mediator of apoptosis and an indicator of prognosis in human primary lung carcinoma. Int J Cancer. 2004; 110:532–541.
44. Zhong Z, Yeow WS, Zou C, Wassell R, Wang C, Pestell RG, Quong JN, Quong AA. Cyclin D1/cyclin-dependent kinase 4 interacts with filamin A and affects the migration and invasion potential of breast cancer cells. Cancer Res. 2010; 70:2105–2114.
45. Stossel TP, Condeelis J, Cooley L, Hartwig JH, Noegel A, Schleicher M, Shapiro SS. Filamins as integrators of cell mechanics and signalling. Nat Rev Mol Cell Biol. 2001; 2:138–145.
46. Zhu TN, He HJ, Kole S, D’Souza T, Agarwal R, Morin PJ, Bernier M. Filamin A-mediated down-regulation of the exchange factor Ras-GRF1 correlates with decreased matrix metalloproteinase-9 expression in human melanoma cells. J Biol Chem. 2007; 282:14816–14826.
47. Vadlamudi RK, Li F, Adam L, Nguyen D, Ohta Y, Stossel TP, Kumar R. Filamin is essential in actin cytoskeletal assembly mediated by p21-activated kinase 1. Nat Cell Biol. 2002; 4:681–690.
48. King CC, Gardiner EM, Zenke FT, Bohl BP, Newton AC, Hemmings BA, Bokoch GM. p21-activated kinase (PAK1) is phosphorylated and activated by 3-phosphoinositide-dependent kinase-1 (PDK1). J Biol Chem. 2000; 275:41201–41209.
49. Cukier IH, Li Y, Lee JM. Cyclin B1/Cdk1 binds and phosphorylates Filamin A and regulates its ability to cross-link actin. FEBS Lett. 2007; 581:1661–1672.
50. Gupta P, Su ZZ, Lebedeva IV, Sarkar D, Sauane M, Emdad L, Bachelor MA, Grant S, Curiel DT, Dent P, Fisher PB. mda-7/IL-24: multifunctional cancer-specific apoptosis-inducing cytokine. Pharmacol Ther. 2006; 111:596–628.
51. Stewart AL, Mhashilkar AM, Yang XH, Ekmekcioglu S, Saito Y, Sieger K, Schrock R, Onishi E, Swanson X, Mumm JB, Zumstein L, Watson GJ, Snary D, et al. PI3 kinase blockade by Ad-PTEN inhibits invasion and induces apoptosis in RGP and metastatic melanoma cells. Mol Med. 2002; 8:451–461.
52. Panneerselvam J, Jin J, Shanker M, Lauderdale J, Bates J, Wang Q, Zhao YD, Archibald SJ, Hubin TJ, Ramesh R. Disruption of the CXCR-4/SDF axis by interleukin (IL)-24 reduces tumor cell migration and invasion in lung cancer cells. PLoS One. 10:e0122439; 2015.