
INTRODUCTION
The androgen receptor (AR), a ligand-dependent nuclear receptor, plays a critical role in prenatal development of the prostate [1–3]. For example, work by wilkins and colleagues in 1950 showed that individuals with complete androgen insensitivity, caused by AR inactivating mutations, do not develop a prostate gland. Likewise, AR knockout mice show a lack of prostate gland development [4]. Even in adult males, the AR continues to support the survival of secretory prostate epithelia, the primary cell type argued to be transformed in prostate adenocarcinoma [5, 6]. Prostate cancer is the second leading cause of cancer related death amongst men in the united states, with some 233,000 individuals diagnosed with prostate cancer in 2014 alone, of which ~30,000 died as a consequence of the disease [7]. Many of the patients who develop prostate cancer receive AR antagonists, to inhibit the actions of androgens on proliferation of prostate epithelia [8, 9]. These therapies although effective in initial stages, quickly loose their benefit, as most patients eventually develop a castrate resistant prostate cancer (CRPC), a devastating though poorly understood disease state [10–13]. Studies using xenograft prostate tumors that recur following androgen ablation therapy show that although CRPC is insensitive to further androgen depletion treatment, the tissue still expresses AR regulated genes, suggesting that AR signaling pathways are still intact in CRPC [14, 15].
Kinase-signaling pathways have been implicated in the regulation and modulation of nuclear receptor activity [16, 17], [18–20]. However, the signaling mechanisms through which kinases modulate AR function are not well understood. Stress kinase signaling is known to regulate AR phosphorylation, increasing its rate of nuclear export and proteasome-mediated degradation [21]. In contrast, Protein Phosphatase 1C (PP1C) dephosphorylates AR, leading to an increase in AR nuclear retention and increased gene transcription [22]. Furthermore, the HER2/ERBB3 kinase signal has been shown to stabilize AR protein levels and optimize binding of the AR to promoter regions of androgen-regulated genes [23]. Clearly, dysregulation of kinase signaling pathways would impact heavily on AR activity in CRPC [24].
Recent Genome Wide Association Studies (GWAS) of patients with prostate cancer have identified a genetic variant of Lemur Tyrosine Kinase 2 (LMTK2) (also called BREK/KPI-2/CRPK/AATYK2), a membrane associated kinase (Fig. 1A) [25, 26], [27, 28] to be strongly associated with prostate cancer (P < 0.0001) [29–32]. Furthermore, this genetic variant of LMTK2 has a Single Nucleotide Polymorphism (SNP) in intron 9, causing decrease in LMTK2 mRNA levels [29]. These studies suggest that LMTK2 might be involved in the development and/or maintenance of prostate gland tumors. However, due to limited understanding of LMTK2 function [33, 34], its role in prostate cancer still remains unknown. Recently, LMTK2 has been reported to interact with PP1C (Fig. 1B) and inhibit its activity in CNS [35, 36]. Since, PP1C plays an important role in nuclear retention of AR by dephosphorylating AR, it is likely that decreased LMTK2 protein and/or activity would result in an increase in AR activity and sensitivity to androgens, events precisely observed in CRPC.
In summary, the results in this manuscript argue that LMTK2 interacts directly with AR and negatively regulates its activity. Furthermore, a decrease in LMTK2 protein expression, as proposed in prostate cancer, not only results in an increase in androgen mediated AR activity but also increases the androgen-independent activity of AR. Moreover, LMTK2-Knock Down (KD) in prostate cancer cells results in an increase in cell viability and tumorogenecity in the presence and absence of androgen. As such, our study takes observations made in genomic studies and reveals LMTK2 as a novel regulator of AR in prostate epithelium.
RESULTS
LMTK2 expression and localization
Given GWAS linking LMTK2 expression levels with prostate cancer, we initially determined if LMTK2 was expressed in prostate epithelia. We used a model cell line HEK293 as well as prostate cancer cell lines i.e. PTN1A, PC3 and LNCaP for the same. As predicted, immunoblot analysis showed robust endogenous expression of LMTK2 in prostate epithelial and HEK293 cells, which appeared as a single dominant band of ~210 kDa (Fig. 2A), consistent with previously published data [26]. In addition, we confirmed that the observation were not an artifact of cell lines by studying Lmtk2 expression in mouse primary prostate epithelial cells. Mouse primary prostate epithelial cells not only showed robust expression of Cytokeratin 5/8 (prostate epithelial cell marker) and AR as expected, but also Lmtk2 (Fig. 2B).
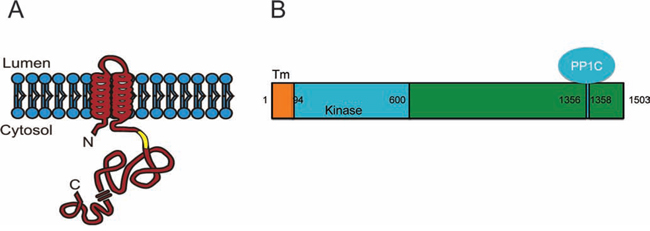
Figure 1: Predicted structure of Lemur Tyrosine Kinase 2 (LMTK2). A. Topology of LMTK2 in endosomal membrane is shown with N-terminal and C-terminal either in cytosol or within an endosomal lumen [26]. B. LMTK2 is 1503 amino acid long protein with predicted transmembrane (TM) helices located between 11–29 and 46–63 amino acids while kinase domain is predicted to lie between 94–600 amino acid as shown in yellow. LMTK2 interacts with Protein Phosphatase 1 C (PP1C) via its VTF motif (1356–1358 amino acids) [25].
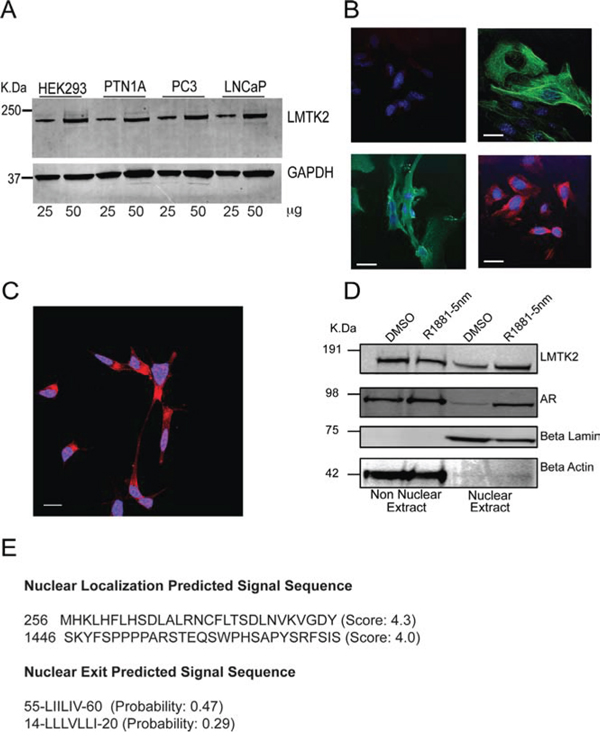
Figure 2: Expression and localization of LMTK2 in prostate epithelial cells. A. Immunoblot showing expression of LMTK2 in Human Embryonic Kidney Cell (HEK293), Human prostate epithelial cell (PTN1A), Prostate Cancer cell (PC3) & Prostate adenocarcinoma cell (LNCaP). Two concentrations for each cell type were loaded B. Top-left panel shows the mice prostate epithelial cell stained with secondary antibody only; top-right panel shows mice prostate epithelial cell marker cytokeratin 5/8 stained with alexa 488 (Green). LMTK2 is stained with alexa 488(Green) as shown in bottom left panel and bottom right panel shows the AR stained with CY-5 (Red). Nuclei stained in DAPI, appears blue. Bar = 0.25 μm. C. LMTK2 stained with Cy5 (red) and nuclei are stained with DAPI (blue), colocalization between LMTK2 and nuclei appears magenta. Bar = 0.5 μm. D. LNCaP cells grown in androgen deprived media were treated with DMSO or 2.5 nm R1881 (synthetic androgen) for 24 hours; cytoplasmic extract and nuclear extract were immunobloted to measure relative level of LMTK2 and AR. ß-lamin and ß-actin were used as nuclear and cytoplasmic protein control, respectively. E. Predicted Multiple Putative NLS and NES by “cNLS Mapper” & NetNES 1.1 (http://www.cbs.dtu.dk/services/NetNES/) and ValidNESs (http://validness.ym.edu.tw/).
Furthermore, several studies have showed LMTK2 to be an endosome membrane-anchored protein [26, 34]. Hence, a reasonable expectation was that LMTK2 would be localized in the extra-nuclear membrane fraction of prostate cancer cells. Surprisingly, our confocal images showed both nuclear as well as non-nuclear staining for LMTK2 in prostate cancer cells (Fig. 2C). We further confirmed this finding using subcellular fractionation, to enrich a nuclear fraction, which too showed presence of LMTK2 in nuclear and non-nuclear compartment of prostate cancer cells, irrespective of its androgen exposure (Fig. 2D). AR translocation, as reported in previous studies [37] was also seen in the fractionation analysis.
LMTK2 is down regulated in human prostate cancer
Previous studies have suggested that reduced LMTK2 mRNA levels are associated with prostate cancer, however whether this translates to altered protein levels has not been determined. Immunostaining analysis of a human prostate tissue array (US Biomax) containing prostate cancer (n = 48), prostate hyperplasia (n = 8) and normal prostate tissue (n = 14) from a total of 20 individual patients, revealed a marked difference in LMTK2 protein expression levels (Supplementary Table 2). LMTK2 intensity was determined using Image-J software and assigned arbitrary unit, which was binned as no (0), low (0–20), medium (20–40), high (40–80) and very high (80–170). A majority, >65% of normal prostate tissue had very high expression of LMTK2, around 13% and 25% of hyperplasia tissue had respectively very high or high expression of LMTK2. In contrast, >67% of tumor tissue contained undetectable or low levels of LMTK2 (Fig. 3A, 3C and 3D). The statistical significance of apparent differences in LMTK2 expression between normal, and prostate cancer was investigated by Mann-Whitney-U analysis for pairwise comparison, which revealed a strong association (P ≤ 0.001) between a decrease in LMTK2 protein expression and prostate cancer (Fig. 3B).
LMTK2 and AR interact in prostate cancer epithelial cells and co-localizes in human prostate tissue
We had initially hypothesized that LMTK2 might negatively regulate AR-dependent transcriptional activity. Hence, we asked whether LMTK2 and AR are binding partners in prostate epithelial cells. Protein complexes immunoprecipitated with AR antibody from whole cell lysate of LNCaP cells indicated the presence of LMTK2 (Fig. 4C). The specificity of LMTK2 and AR interaction was shown by absence of LMTK2 when AR antibody was replaced by IgG (Fig. 4C). AR pull down was confirmed by blotting with separate AR antibody (AR-N20) (Fig. 4C). However, in the reciprocal Co-IP experiment, LMTK2 antibody failed to pull down AR, this might be due to LMTK2 antibody interfering with the binding site to the AR.
In addition, a Proximity Ligation Assay (PLA), a means of assessing protein-protein interaction in situ, was used to verify CO-IP findings [38]. LMTK2/AR complexes were labeled using antibodies directed against LMTK2 and AR in LNCaP cells deprived of androgen or grown in the presence of synthetic androgen (R1881, 2.5 nM). Significantly higher numbers of LMTK2/AR protein complexes were detected in prostate epithelial cells in comparison to negative control (BAX/CFTR complexes), two known non-interacting protein (Fig. 4A and 4B). While under androgen deprivation the distribution of the LMTK2/AR complexes was predominantly extra-nuclear but in presence of androgens, they also localized within the nuclear space.
To rule out cell line artifact, we further explored possible interaction of the AR and LMTK2 in human prostate tissue using co-localization analysis. 16 normal prostate tissue samples were tested using immunostaining for possible co-localization between AR and LMTK2. Fig. 4D shows representative figure where AR and LMTK2 appear to co-localize in the glandular prostate epithelial cells. Results obtained from human prostate cancer cell and tissue model together shows that AR and LMTK2 are binding partners suggesting a functional significance of LMTK2 in AR-axis.
LMTK2 inhibits AR transcriptional activity
Our study shows that a decrease in LMTK2 expression is associated with human prostate cancer and that LMTK2 and AR are binding partners in prostate epithelial cells. These results prompted us to examine the potential role for LMTK2 in AR-mediated signaling. For this purpose, we created HEK293 cells stably expressing either shRNA against LMTK2, control shRNA (scrambled sequence) or an LMTK2 over-expression plasmid. We also created stable LNCaP cells expressing either shRNA against LMTK2 (LNCaP-KD cells) or control shRNA (LNCaP-Control cells). Knock down or overexpression of LMTK2 in these cell lines was confirmed using immunoblot analysis (Fig. 5A & 5B). To measure the affects of manipulating LMTK2 level on the androgen receptor activity we used a dual luciferase assay. Overexpression of LMTK2 in HEK293 cells expressing AR, decreased androgen-dependent activation of a luciferase reporter gene by two fold compared to parental cells expressing AR; no activation was observed in HEK293 cells without AR. In contrast, knock down of LMTK2 in HEK293 cells expressing AR (analogous to prostate cancer cells) enhanced androgen-dependent activation of reporter gene by three fold in comparison to parental cells and by six fold in comparison to cells overexpressing LMTK2 (Fig. 5C). Constitutively active renillia luciferase was used as transfection control.
To study the affects of LMTK2 on the expression of endogeneous AR regulated genes, we measured the expression of PSA, a prototypical AR regulated gene [39]. Consistent with the luciferase assay in HEK293 cells, there was a dramatic increase in activation of endogenous AR in LNCaP-KD cells in comparison to LNCaP-Control as measured by secreted and total PSA (Fig. 5D).
Decrease in LMTK2 expression increases basal AR activity
Since, the AR is active in CRPC and is able to transcribe AR-dependent genes either in the presence of low levels of androgens or absence of androgens [40], we determined if a decrease in LMTK2 is involved in activating AR in androgen deprived prostate cancer cells. Thus, we compared the expression of AR responsive genes between LNCaP-KD and LNCaP-Control cells deprived of androgens for 72 hours. LMTK2-KD cells showed a significant increase in mRNA expression of the AR dependent genes-KLK2, S100P, TMPRS22 and PSA (Fig. 5E) in comparison with LNCaP-control cells, no significant difference in mRNA levels of AR was found. No significant differences were observed in the initial experiments using GAPDH or Tubulin as internal control; hence experiments were conducted using GAPDH as internal control. In addition, we also compared protein expression of FKBP51, an AR regulated gene, between LNCaP-KD and LNCaP-control cells grown in absence of androgen for 72 hours followed by androgen (1 nm R1881) stimulation for times shown in the Fig. 5F. We found that LMTK2-KD significantly increased FKBP51 protein levels in comparison to LNCaP-control cells, with the biggest difference of about 5 fold was observed at time zero i.e. during the androgen deprivation stage (Fig. 5F and 5G).
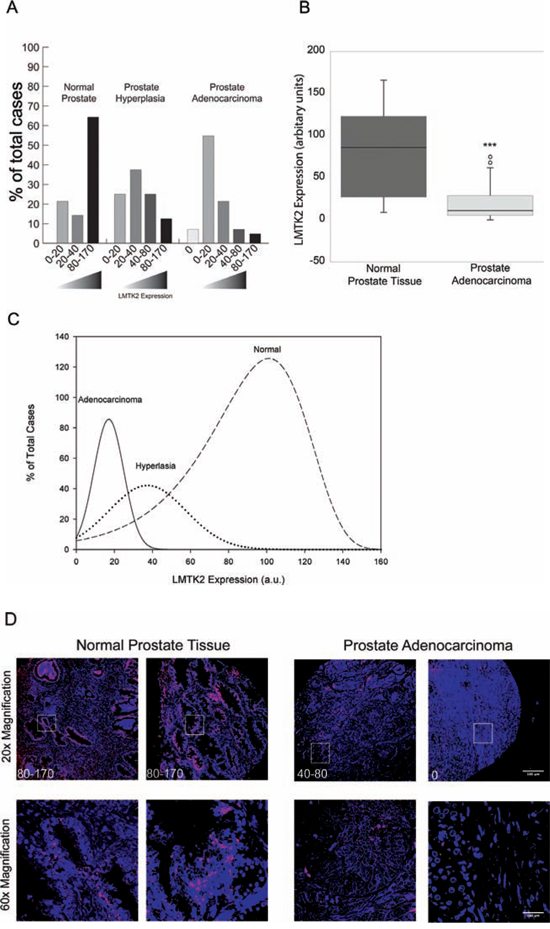
Figure 3: LMTK2 is down regulated in human prostate cancer. A. Expression levels of LMTK2 protein in 48 prostate cancer, 8 prostate hyperplasia and 14 normal prostate tissue samples measured by tissue array. LMTK2 protein levels were detected by immunofluorescence staining. Intensity of staining was classified as no (0), low (0–20), medium (20–40), high (40–80) and very high (80–170). B. This box plot gives the cancer status (Normal or prostate adenocarcinoma) on the X-axis, and the protein expression of LMTK2 on the Y-axis. Error bars represent the interquartile range (IQR) of the measurements. The level of significance, P ≤ 0.001, was determined by Mann-Whitney-U analysis for pairwise comparison and circles indicate outliers. C. To generate curves, data were fit using a Best-Fit Gamma distribution (SigmaPlot, Systat, San Jose, CA). D. Representative Immunofluorescence images, LMTK2 stained with secondary antibody linked to Cy-5 (red) and nuclei stained in DAPI. Top panel shows low magnification image, the boxed region is magnified in the bottom panel. Scale bars = 100 μm.
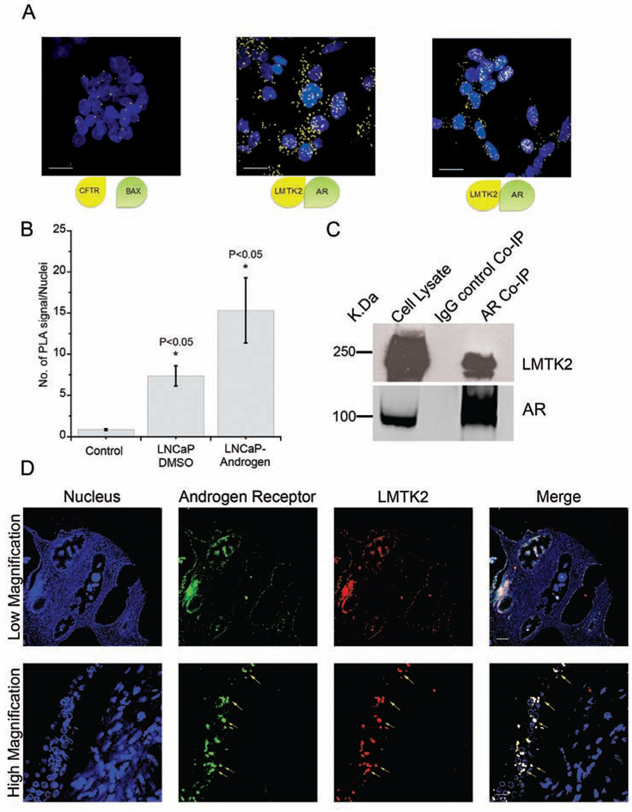
Figure 4: LMTK2 and AR interact in prostate cancer epithelial cells and co-localizes in human prostate tissue. A. Protein within interacting distance i.e. <40 nm is detected using proximity ligation assay (PLA). Cartoon shows two interacting protein being detected by fluorescent signal. PLA between non-interacting proteins, CFTR and BAX was used as negative control. PLA between LMTK2 and AR in LNCaP cells grown in DMSO or 2.5 nM R1881 (androgen) shows PLA signal (Yellow dots), representing interaction. B. LMTK2-AR PLA signals per nucleus in LNCaP cells are counted using Image J software & compared with CFTR-BAX (non interacting proteins). Error bars indicate SD, which is given for mean of n ≥ 30 cells per condition. *P < 0.05 for difference from CFTR-BAX by student-unpaired t-test. C. Lysates from LNCaP cells was precipitated using mouse anti-AR Ab or control mouse Ab and blotted for LMTK2 or AR. 10% of lysate was loaded in the first lane as lysate control. D. Representative figure for co-localization between LMTK2 (red) and AR (green) in normal human prostate tissue is shown. Nuclei were stained with DAPI (blue). Arrows in the merge image shows some, but not all areas of co-localization (yellow) between LMTK2 and AR. Top panel shows low magnification image, the boxed region is magnified in the bottom panel.
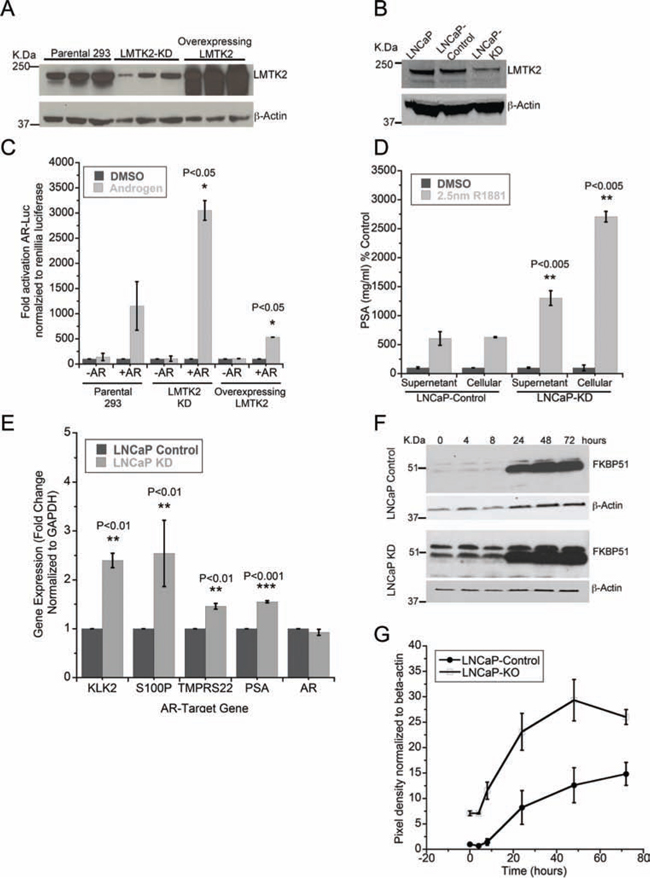
Figure 5: LMTK2 inhibits AR transcriptional activity. A. Immunoblot comparing LMTK2 expression between HEK293 cells stably transfected with control shRNA (Parental 293), LMTK2 shRNA (LMTK2-KD) or PCI-LMTK2 (Overexpressing LMTK2). β-actin was used as endogenous loading control. B. Immunoblot comparing LMTK2 expression between LNCaP cells and LNCaP cell stably transfected with either control shRNA (LNCaP-control) or LMTK2 shRNA (LNCaP-KD). β-actin was used as endogenous loading control. C. Modulation of AR responsive gene activity by LMTK2 (mean ± SD, n = 3) *p < 0.05 for difference from endogenous LMTK2 levels in HEK293 cells. D. ELISA was performed in duplicates to analyze the concentrations of PSA protein in culture supernatant and cell lysate from LNCaP sublines cultured in androgen deprived medium for 3 days and treated with either DMSO or 2.5 nM synthetic androgen (R1881) for 16 hours (R1881) for 24 hours. Data is presented as % change in relation to the control (DMSO treated), (mean ± SD, n = 3) **p < 0.005 for difference from DMSO treatment group. E. Real-time PCR comparing transcript level of AR and genes regulated by AR between LNCaP sublines grown under androgen deprivation for 3 days was performed in duplicates (KLK2, S100P, TMPRS22, PSA), (mean ± SD, n = 3) **p < 0.01, ***p < 0.001 value for difference from endogenous LMTK2 levels in LNCaP cells are denoted on the graph. F, G. Immunobloting of FKBP51 in control and knock down cells grown under androgen deprivation for 3 days and treated with 1 nm R1881 for time points shown in figure. Quantitative analysis of normalized protein levels of FKBP51 between control and knock down LNCaP cells is shown in panel G.
LMTK2 down-regulation promotes tumor forming capacity and proliferation in LNCaP cells
In order to better understand the physiological role of LMTK2 in prostate cancer, we investigated the effect of LMTK2 on the tumor forming capacity and cell viability of LNCaP cells, using a tumorsphere assay [41] and a cell viability assay. Tumorspheres are enriched in cancer stem cells [42], which are argued to be tumor-initiating cells and are believed to play an enabling role in development of CRPC [43]. LNCaP-KD cells showed significantly higher colony-forming capacity by forming ~5 times more clones compared to LNCaP control cells in tumorsphere assay (Fig. 6A & 6B). We further tested the effects of a decrease in LMTK2 expression on prostate cancer cell viability using the ATP-Glo Bioluminometric cell viability assay (Biotium, CA). LNCaP-KD cells, as expected, showed ~5 times higher cell viability under androgen starvation and ~2.5 times in presence of androgen when compared to LNCaP-control cells (Fig. 6C). It is important to note that while LNCaP-control cells treated with DMSO after 3 days of androgen starvation showed decrease in cell viability, there was no such decrease observed in LNCaP-KD cells.
DISCUSSION
Recently, several Genome Wide Association Studies (GWAS) involving prostate cancer patients of Caucasian and East Asian descent identified a genetic variant (SNP rs6465657) of Lemur Tyrosine Kinase 2 (LMTK2), leading to decrease in its mRNA expression, to be associated with prostate cancer [29–32]. However, no information is available in terms of protein expression or function of LMTK2 in prostate epithelial cells. In this report, we compared protein expression of LMTK2 in human prostate cancer tissue specimens with normal, hyperplasia and prostate tissue specimens. Our data shows that loss of LMTK2 protein is strongly associated with prostate cancer and prostate hyperplasia, disease states marked by dysregulation of AR. Since, we did not genotype prostate cancer tissues it remains to be determined if SNP rs6465657 associated with prostate cancer results in decrease in LMTK2 protein expression. Furthermore, we also show a high-level expression of LMTK2 in normal prostate epithelial cells (PNT1A), androgen-independent metastatic prostate cancer cells (PC3), androgen-dependent prostate cancer cells (LNCaP) and mouse primary prostate epithelial cells. Since, LMTK2 was expressed in androgen dependent as well as androgen independent prostate cancer epithelial cells, it is likely that function of LMTK2 is not just limited to AR axis and certainly previous studies have shown LMTK2 to be an important binding partner of myosin VI involved in endocytic trafficking pathways in prostate cancer cells [33]. Furthermore, this report proposes a novel function for LMTK2, as a negative regulator of AR transcription.
It was surprising to find LMTK2 to be localized in a nuclear fraction of prostate cancer cells as previous studies have suggested LMTK2 to be an endosomal-membrane associated kinase [26, 34] though none looked specifically for nuclear localization. Using a Nuclear Localization Signal (NLS) prediction tool [44–46] and Nuclear Exit Signal (NES) prediction tool [47], we predict 2 possible bipartite NLS signal and a N-terminal NES signal in LMTK2 (Fig. 2E), however these sites remain to be confirmed experimentally. Hence, we speculate that LMTK2 might be involved in retrograde endocytic transport of proteins i.e. from cytoplasmic fraction to nuclear fraction. Furthermore, LMTK2 immunostaining and duolink data in this report indicates a nuclear translocation of LMTK2/AR complexes in response to androgen treatment of prostate cancer cells. Together, these data suggests that LMTK2 may be involved in the activation and translocation of AR. This phenomenon of an endosomal membrane protein localized in nuclear fractions is not unique to LMTK2. For example, FAM21, a WASH complex subunit residing in early endosome membrane like LMTK2 has been shown to undergo nuclear translocation and participate in NF-kB-depenent gene regulation in pancreatic cancer cells [48, 49]. Similarly, tachykinin NK3 receptor (NK3R), a multi-pass membrane protein belonging to G-protein receptor-1 class undergoes nuclear translocation via importin pathway [50, 51].
The majority of prostate cancers (PCa) and Castrate Resistant Prostate Cancers (CRPC) have functional AR, which continues to drive the expression of AR-dependent cell proliferative genes and hence the tumor growth despite of low level of systemic androgens [52, 53]. Our report shows that LMTK2 is an essential negative regulator of AR transcriptional activity and knocking down its expression in prostate cancer cells leads to significant increase in the AR activity and also an increase in tumorogenecity and cell viability. In contrast, overexpressing LMTK2 leads to repression of AR activity. Hence, it can be deduced that a decrease in LMTK2 expression observed in prostate cancer patient promotes tumor cells proliferation by enhancing AR transcriptional activity.
One of the most important outcomes from this report was the role of LMTK2 in CRPC. Kinases play an important role in driving AR activity in CRPC either by direct activation of AR or by increasing its sensitivity to low level of androgens. FKBP51, an AR-dependent gene, which is also a positive regulator of AR activity, is expressed two fold higher in CRPC compared with primary tumors [54]. Data in this paper shows that decreasing LMTK2 expression in prostate cancer cells deprived of androgen results in significantly higher levels of FKBP51 protein as well as increased mRNA levels of AR-dependent genes (KLK2, S100P, TMPRS22 and PSA). These results provided strong circumstantial evidence of a role for LMTK2 in pathogenesis and progression of prostate cancer to castrate resistant stage. In addition, cell viability data showed the strongest role of decrease in LMTK2 in regards to androgen-independent growth in prostate cancer cells, androgen-dependent growth was also affected, although to a lesser degree.
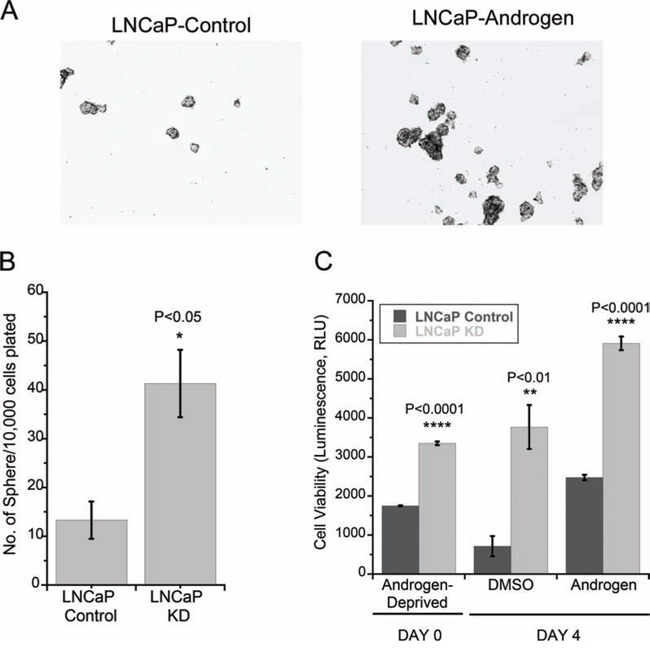
Figure 6: LMTK2 down-regulation promotes tumor forming capacity and proliferation in LNCaP cells. A. Panel showing affects of endogenous LMTK2 levels on LNCaP tumor forming capacity. Representative colonies are shown in the figure. Quantitative analysis of colony numbers is shown in the panel B. (mean ± SD, n = 3) *p < 0.05 for difference from endogenous LMTK2 in LNCaP cells. C. Androgen-independent and androgen-stimulated cell viability of LNCaP-control and LNCaP-KD cell lines measured by the ATP-Glo™ Bioluminometric cell viability assay, (mean ± SD, n = 3) ****p < 0001, **p < 0.01, ****p < 0.0001 value for difference from endogenous LMTK2 in LNCaP cells.
An important question that arises from our study is the mechanism by which LMTK2 might be regulating AR transcription and cell proliferation in the absence of exogenous androgen in prostate cancer cells. There are several possible mechanisms that might be playing a role. Firstly, LMTK2 has been shown to inactivate catalytic activity of PP1, which plays an important role in nuclear retention of AR by dephosphorylating at Ser-650. Hence a decrease in LMTK2 protein express might result in increase in PP1 activity leading to increased nuclear retention and hence increased transcription activity. Secondly, LMTK2 might be directly phosphorylating AR and resulting in increased transcription activity in absence or presence of exogenous androgen. Initial evidence supporting this possibility comes from our study where we show LMTK2 and AR to be binding partners in prostate cancer cells. However, further experiments are needed to support this hypothesis. And lastly, FKBP51, an AR regulated gene is also known to positively regulate AR transcriptional activity. Our study showed increase in FKBP51 protein expression in LMTK2-KD prostate cancer cells. Argument can be made that LMTK2 regulates AR activity through FKBP51. Further experiments are clearly needed to identify possible mechanism through which LMTK2 augments AR transcription and cell proliferation in prostate cancer cells.
In conclusion, our findings are the first evidence that LMTK2 negatively regulates AR activity in prostate cancer cells possibly by directly interacting with AR. Furthermore, loss of LMTK2, associated with prostate cancer can enhance AR transcriptional activity in absence of androgen, suggesting role of LMTK2 in development of CRPC. LMTK2 can now be considered negative regulator of androgen-induced AR-mediated cell growth and transcription. In terms of potential therapeutic target, small molecules that enhance the activity of LMTK2 can decrease AR-proliferative activity in patients with prostate cancer and more importantly with castrate resistant prostate cancer.
MATERIALS AND METHODS
Cell culture
The human cell lines, PNT1A (normal prostate epithelial cells, Sigma-Aldrich, St. Louis, Mo) and LNCaP (androgen sensitive human prostate adenocarcinoma cells, ATCC, Manassas, VA) were cultured in RPMI 1640 medium while PC3 (metastatic prostate cancer cells isolated from bones, ATCC) were maintained in F-12K medium. HEK293 (Female-Human Embryonic Kidney Epithelial cells, ATCC) were cultured in Advanced Dulbecco’s Medium. All culture medium were supplemented with 10% FBS and 1% Pen/Strep (Invitrogen, Carlsbad, CA) unless specified.
Plasmids and transfections
Wild type LMTK2 cloned in PCI vector was provided as a gift by Dr. Takeshi Inoue, University of Tokyo. Halo tagged Androgen Receptor construct was obtained from Promega (Madison, WI). Plasmids expressing and shRNA against human LMTK2 or control shRNA were obtained from DNA 2.0 (Menlo Park, CA). Transfections of cells were performed by 4D nucleofector (Lonza Group Ltd, Basel, Switzerland) according to the manufacturer’s instruction.
Prostate primary cell isolation
Male mice were euthanized at 8–12 week of age and primary prostate epithelial cells were isolated as described [55]. Cell type was verified by staining for Cytokeratin 5/8 (SC-32328, Santa Cruz Biotechnology, Santa Cruz, CA), widely used marker for prostate epithelial cells [56].
Immunofluorescence
Cells plated onto poly-L-lysine coated coverslips were fixed according to pH-shift protocol as described [57] and stained for AR (AR441, Santa Cruz Biotechnology) and LMTK2 (HPA010657, Sigma-Aldrich). Coverslips were mounted onto a slide using Prolong Gold with DAPI (Invitrogen). The cellular signal was visualized using a PlanApo 60 ×, 1.42 NA oil immersion objective of an Olympus IX71 inverted microscope (Olympus, Center Valley, PA) coupled to a VT-Infinity 3 confocal system (VisiTech International, Sunderland, UK). For each sample, multiple coverslips were imaged (≥30 cells per coverslip) under identical settings.
Fractionation of membrane/cytoplasmic and nuclear proteins
LNCaP cells were starved of androgen for 3 days by replacing complete media containing FBS with Charcoal Stripped Fetal Bovine Serum (CSFBS). On day 3, cells were treated either with DMSO or R1881 (Sigma-Aldrich) as described in results. Nuclear and non-nuclear fractions were separated as per manufacture’s recommendation (GeneTex, Inc., Irvine, CA) and were probed for LMTK2 and AR using Immunoblot analyses. Anti-human GAPDH antibody (SC-25778) was used for analyzing non-nuclear fraction while Lamin-A (SC-20680) was used for nuclear fraction.
Co-immunoprecipitation and immunoblot analyses
LNCaP cells were lysed in 1% NP-40 lysis buffer and endogenous complexes of AR were immunoprecipitated using mouse anti-AR 441 antibody. Immunprecipitated complexes were then probed for LMTK2 (rabbit anti-LMTK2) using immunoblot assay. AR pull down was confirmed by blotting with rabbit anti-AR N20 antibody. For immunoblot analyses samples were prepared in 4 × LDS sample buffer, resolved on 4–12% gradient Tris-Glycine Gel and transferred on to polyvinyldiflouride (PVDF) membrane. Membranes were probed for several proteins using human anti-AR, anti-LMTK2, anti-β-ACTIN (LI-COR Biotechnology, Lincoln, NE) and anti-FKBP51 (ab-2901, ABCAM, Cambridge, MA) antibodies. Proteins were detected and analyzed using Odyssey Sa-Infrared imaging system™ (Li-COR Biotechnology).
Tissue immunohistochemistry
Tissue arrays (BC19021a) obtained from US-Biomax (Rockville, MD) containing normal, malignant and metastatic human prostate tissue was used. The arrays were probed for LMTK2 and AR using a Tyramide Signal Amplification Kit (TSA, Invitrogen) as per manufacturer’s recommendation.
Proximity ligation assay
A Proximity Ligation Assay kit (Olink Bioscience, Uppsala, Sweden) was used to study the interaction between AR and LMTK2 in LNCaP cells [58]. Staining procedure were carried out following the manufacturer’s instructions using rabbit anti-LMTK2 and mouse anti-AR antibodies to detect AR/LMTK2 interactions. BAX (Bcl2 associated protein-X) and CFTR (Cystic Fibrosis Trans membrane Conductance Regulator) are non-interacting proteins, and hence rabbit anti-BAX (sc-493) and mouse anti-CFTR (596, UNC at Chapel Hill, NC) were used as negative control. The cellular PLA signal was visualized using the Olympus IX71 inverted microscope as described above and quantified using Image-J software [59].
Real-Time RT-PCR
Total cellular RNA was extracted from cells using the Trizol RNA isolation reagent according to manufacturer’s instruction (Invitrogen). Complementary DNA synthesis reactions were performed with 1 μg of RNA using SuperScript® III First-Strand Synthesis System (Invitrogen) according to manufacturer instruction. cDNA samples were amplified using SYBR® Green PCR Master Mix on the Applied Biosystems 7500 Detection System. Gene-specific forward and reverse primers (Supplementary Table 1) used have been reported in earlier studies [60, 61]. Furthermore, specificity and efficiency for primers were analyzed by running qPCR with series of cDNA dilutions and specific amplification for every assay were confirmed by melt curve analysis. All assays were run in duplicates and were repeated 3 times. The amplified transcripts were quantified using the comparative ΔΔCt method.
PSA measurement
PSA (Prostate Specific Antigen) protein levels were detected using an enzyme-linked immunosorbent assay (ELISA) technique [62]. Cells were plated into 96-well plates at density of 1 × 105 per well in RPMI media supplemented with 10% CSFBS. After 3 days, cells were treated for 16 h as detailed in results. PSA in the cultural supernatant and cellular PSA was quantified with an ELISA kit (Abcam) following the manufacturer’s instruction. All assays were run in duplicates and repeated 3 times.
Dual luciferase assay
Cells (1 × 105 per well in 96-well plates) were co-transfected with Halo-AR and/or either AR reporter, negative control or positive control in 2:1 ratio, supplied with cignal androgen receptor reporter kit (Qiagen, Valencia, CA). Cells were grown in charcoal-stripped media for 2 days and were treated with 1 nm R1881 for 16 h. Luciferase activities were measured by using the dual-luciferase reporter gene assay system (Promega, Madison, WI) following the manufacturer’s instruction in plate reader (POLARstar Omega, BMG Labtech, Germany). Final results were normalized for transfection efficiencies using the Renillia Luciferase Assay Value. Luciferase assay were repeated for 3 times.
Cell viability assays
1 × 105 cells were plated per well onto flat clear bottom white polystyrene 96-well plate (Nunc) in RPMI media supplemented with 10% CSFBS. Cells were treated as discussed in the result section. Cell viability was measured using ATP-GloTM Bioluminometric cell viability assay kit (Biotium, CA.) according to manufacturer’s instruction [63]. Each assay was performed in triplicate and repeated 3 times (n = 3). Luminescence was measured using POLARstar Omega.
Tumorigenicity assay
Cells were harvested and resulting pellet was washed in PBS. 1 × 105 cells per well were plated onto 6-well Ultra-Low Attachment Plates (Corning, NY) in Tumorsphere medium (PrEGM media with supplied growth factors and supplements+1% N2 (Invitrogen) and 1% B27 (Invitrogen). After 7 days number of spheroids (solid, rounded structures) were counted under light microscope. Each experiment was carried out in triplicates.
ACKNOWLEDGMENTS
Authors are particularly indebted to the reviewers of our manuscript for many helpful suggestions.
FUNDING SOURCE
Work in the authors’ laboratory is supported by Cystic Fibrosis Foundation Grant CFF BRADBU05G0 and National Institute of Health National Heart, Lung and Blood Institute Grant HL-102208 awarded to Neil Bradbury.
CONFLICTS OF INTEREST
No conflicts of interest, financial or otherwise, are declared by the author(s).
REFERENCES
1. Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM. The nuclear receptor superfamily: the second decade. Cell. 1995; 83:835–839.
2. Tsai MJ, O'Malley BW. Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annual review of biochemistry. 1994; 63:451–486.
3. Chang CS, Kokontis J, Liao ST. Molecular cloning of human and rat complementary DNA encoding androgen receptors. Science. 1988; 240:324–326.
4. Yeh S, Tsai MY, Xu Q, Mu XM, Lardy H, Huang KE, Lin H, Yeh SD, Altuwaijri S, Zhou X, Xing L, Boyce BF, Hung MC, Zhang S, Gan L, Chang C. Generation and characterization of androgen receptor knockout (ARKO) mice: an in vivo model for the study of androgen functions in selective tissues. Proceedings of the National Academy of Sciences of the United States of America. 2002; 99:13498–13503.
5. De Marzo AM, Nelson WG, Meeker AK, Coffey DS. Stem cell features of benign and malignant prostate epithelial cells. The Journal of urology. 1998; 160:2381–2392.
6. Huang H, Tindall DJ. The role of the androgen receptor in prostate cancer. Critical reviews in eukaryotic gene expression. 2002; 12:193–207.
7. Institute NC. 2014; SEER Stat Fact Sheets: Prostate Cancer. http://seer.cancer.gov/statfacts/html/prost.html.
8. Scherr D, Swindle PW, Scardino PT, National Comprehensive Cancer N. National Comprehensive Cancer Network guidelines for the management of prostate cancer. Urology. 2003; 61:14–24.
9. Mark A, Perlmutter HL. Androgen Deprivation Therapy in the Treatment of Advanced Prostate Cancer. Reviews in Urology. 2007; 9:S3–S8.
10. Amler LC, Agus DB, LeDuc C, Sapinoso ML, Fox WD, Kern S, Lee D, Wang V, Leysens M, Higgins B, Martin J, Gerald W, Dracopoli N, Cordon-Cardo C, Scher HI, Hampton GM. Dysregulated expression of androgen-responsive and nonresponsive genes in the androgen-independent prostate cancer xenograft model CWR22-R1. Cancer research. 2000; 60:6134–6141.
11. Smaletz O, Scher HI. Outcome predictions for patients with metastatic prostate cancer. Seminars in urologic oncology. 2002; 20:155–163.
12. Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nature reviews Cancer. 2001; 1:34–45.
13. Seruga B, Ocana A, Tannock IF. Drug resistance in metastatic castration-resistant prostate cancer. Nature reviews Clinical oncology. 2011; 8:12–23.
14. Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nature medicine. 2004; 10:33–39.
15. Sirotnak FM, She Y, Khokhar NZ, Hayes P, Gerald W, Scher HI. Microarray analysis of prostate cancer progression to reduced androgen dependence: studies in unique models contrasts early and late molecular events. Molecular carcinogenesis. 2004; 41:150–163.
16. Whitworth H, Bhadel S, Ivey M, Conaway M, Spencer A, Hernan R, Holemon H, Gioeli D. Identification of kinases regulating prostate cancer cell growth using an RNAi phenotypic screen. PloS one. 2012; 7:e38950.
17. Gordon V, Bhadel S, Wunderlich W, Zhang J, Ficarro SB, Mollah SA, Shabanowitz J, Hunt DF, Xenarios I, Hahn WC, Conaway M, Carey MF, Gioeli D. CDK9 regulates AR promoter selectivity and cell growth through serine 81 phosphorylation. Molecular endocrinology. 2010; 24:2267–2280.
18. Atanaskova N, Keshamouni VG, Krueger JS, Schwartz JA, Miller F, Reddy KB. MAP kinase/estrogen receptor cross-talk enhances estrogen-mediated signaling and tumor growth but does not confer tamoxifen resistance. Oncogene. 2002; 21:4000–4008.
19. Grivas PD, Tzelepi V, Sotiropoulou-Bonikou G, Kefalopoulou Z, Papavassiliou AG, Kalofonos H. Estrogen receptor alpha/beta, AIB1, and TIF2 in colorectal carcinogenesis: do coregulators have prognostic significance? International journal of colorectal disease. 2009; 24:613–622.
20. Osborne CK, Bardou V, Hopp TA, Chamness GC, Hilsenbeck SG, Fuqua SA, Wong J, Allred DC, Clark GM, Schiff R. Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. Journal of the National Cancer Institute. 2003; 95:353–361.
21. Gioeli D, Black BE, Gordon V, Spencer A, Kesler CT, Eblen ST, Paschal BM, Weber MJ. Stress kinase signaling regulates androgen receptor phosphorylation, transcription, and localization. Mol Endocrinol. 2006; 20:503–515.
22. Chen S, Kesler CT, Paschal BM, Balk SP. Androgen receptor phosphorylation and activity are regulated by an association with protein phosphatase 1. J Biol Chem. 2009; 284:25576–25584.
23. Mellinghoff IK, Vivanco I, Kwon A, Tran C, Wongvipat J, Sawyers CL. HER2/neu kinase-dependent modulation of androgen receptor function through effects on DNA binding and stability. Cancer cell. 2004; 6:517–527.
24. Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocrine reviews. 2004; 25:276–308.
25. Wang H, Brautigan DL. A novel transmembrane Ser/Thr kinase complexes with protein phosphatase-1 and inhibitor-2. The Journal of biological chemistry. 2002; 277:49605–49612.
26. Nixon A, Jia Y, White C, Bradbury NA. Determination of the membrane topology of lemur tyrosine kinase2 (LMTK2) by fluorescence protease protection. American journal of physiology Cell physiology. 2013; 304:C164–169.
27. Kesavapany S, Lau KF, Ackerley S, Banner SJ, Shemilt SJ, Cooper JD, Leigh PN, Shaw CE, McLoughlin DM, Miller CC. Identification of a novel, membrane-associated neuronal kinase, cyclin-dependent kinase 5/p35-regulated kinase. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2003; 23:4975–4983.
28. Wang H, Brautigan DL. Peptide microarray analysis of substrate specificity of the transmembrane Ser/Thr kinase KPI-2 reveals reactivity with cystic fibrosis transmembrane conductance regulator and phosphorylase. Molecular & cellular proteomics: MCP. 2006; 5:2124–2130.
29. Harries LW, Perry JR, McCullagh P, Crundwell M. Alterations in LMTK2, MSMB and HNF1B gene expression are associated with the development of prostate cancer. BMC Cancer. 2010; 10:315.
30. Wang NN, Wang JY, Shi XH, Zhang YG, Liu M, Wang X, Hui J, Chen X, Liang SY, Wei D, Yang F, Zhao F, Zhang YH, Yang Z. [Association of TET2, LMTK2 and FAM84B gene expression with prostate cancer risk in Chinese patients] Zhonghua zhong liu za zhi. Chinese journal of oncology. 2013; 35:262–267.
31. Eeles RA, Kote-Jarai Z, Giles GG, Olama AA, Guy M, Jugurnauth SK, Mulholland S, Leongamornlert DA, Edwards SM, Morrison J, Field HI, Southey MC, Severi G, Donovan JL, Hamdy FC, Dearnaley DP, et al. Multiple newly identified loci associated with prostate cancer susceptibility. Nat Genet. 2008; 40:316–321.
32. Shui IM, Lindstrom S, Kibel AS, Berndt SI, Campa D, Gerke T, Penney KL, Albanes D, Berg C, Bueno-de-Mesquita HB, Chanock S, Crawford ED, Diver WR, Gapstur SM, Gaziano JM, Giles GG, et al. Prostate cancer (PCa) risk variants and risk of fatal PCa in the National Cancer Institute Breast and Prostate Cancer Cohort Consortium. European urology. 2014; 65:1069–1075.
33. Chibalina MV, Seaman MN, Miller CC, Kendrick-Jones J, Buss F. Myosin VI and its interacting protein LMTK2 regulate tubule formation and transport to the endocytic recycling compartment. J Cell Sci. 2007; 120:4278–4288.
34. Inoue T, Kon T, Ohkura R, Yamakawa H, Ohara O, Yokota J, Sutoh K. BREK/LMTK2 is a myosin VI-binding protein involved in endosomal membrane trafficking. Genes Cells. 2008; 13:483–495.
35. Manser C, Vagnoni A, Guillot F, Davies J, Miller CC. Cdk5/p35 phosphorylates lemur tyrosine kinase-2 to regulate protein phosphatase-1C phosphorylation and activity. J Neurochem. 2012; 121:343–348.
36. Rattray M. New insights on regulation of LMTK2, a membrane kinase integrating pathways central to neurodegeneration. J Neurochem. 2012; 121:327–328.
37. Jenster G, Trapman J, Brinkmann AO. Nuclear import of the human androgen receptor. The Biochemical journal. 1993; 293:761–768.
38. Andersson MGA-C. Visualization and quantification of protein-protein interactions in cells and tissues. Nature Methods. 2010; 7.
39. Kim J, Coetzee GA. Prostate specific antigen gene regulation by androgen receptor. Journal of cellular biochemistry. 2004; 93:233–241.
40. Holzbeierlein J, Lal P, LaTulippe E, Smith A, Satagopan J, Zhang L, Ryan C, Smith S, Scher H, Scardino P, Reuter V, Gerald WL. Gene expression analysis of human prostate carcinoma during hormonal therapy identifies androgen-responsive genes and mechanisms of therapy resistance. The American journal of pathology. 2004; 164:217–227.
41. Wang S, Huang S, Zhao X, Zhang Q, Wu M, Sun F, Han G, Wu D. Enrichment of prostate cancer stem cells from primary prostate cancer cultures of biopsy samples. International journal of clinical and experimental pathology. 2014; 7:184–193.
42. Rybak AP, He L, Kapoor A, Cutz JC, Tang D. Characterization of sphere-propagating cells with stem-like properties from DU145 prostate cancer cells. Biochimica et biophysica acta. 2011; 1813:683–694.
43. Sharifi N, Kawasaki BT, Hurt EM, Farrar WL. Stem cells in prostate cancer: resolving the castrate-resistant conundrum and implications for hormonal therapy. Cancer biology & therapy. 2006; 5:901–906.
44. Kosugi S, Hasebe M, Entani T, Takayama S, Tomita M, Yanagawa H. Design of peptide inhibitors for the importin alpha/beta nuclear import pathway by activity-based profiling. Chemistry & biology. 2008; 15:940–949.
45. Kosugi S, Hasebe M, Matsumura N, Takashima H, Miyamoto-Sato E, Tomita M, Yanagawa H. Six classes of nuclear localization signals specific to different binding grooves of importin alpha. The Journal of biological chemistry. 2009; 284:478–485.
46. Kosugi S, Hasebe M, Tomita M, Yanagawa H. Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proceedings of the National Academy of Sciences of the United States of America. 2009; 106:10171–10176.
47. la Cour T, Kiemer L, Molgaard A, Gupta R, Skriver K, Brunak S. Analysis and prediction of leucine-rich nuclear export signals. Protein engineering, design & selection: PEDS. 2004; 17:527–536.
48. Harbour ME, Breusegem SY, Seaman MN. Recruitment of the endosomal WASH complex is mediated by the extended 'tail' of Fam21 binding to the retromer protein Vps35. The Biochemical journal. 2012; 442:209–220.
49. Deng ZH, Gomez TS, Osborne DG, Phillips-Krawczak CA, Zhang JS, Billadeau DD. Nuclear FAM21 participates in NF-kappaB-dependent gene regulation in pancreatic cancer cells. Journal of cell science. 2015; 128:373–384.
50. Buell G, Schulz MF, Arkinstall SJ, Maury K, Missotten M, Adami N, Talabot F, Kawashima E. Molecular characterisation, expression and localisation of human neurokinin-3 receptor. FEBS letters. 1992; 299:90–95.
51. Jensen DD, Sundstrom K, Flynn FW. Expression of the nuclear transport protein importin ss-1 and its association with the neurokinin 3 receptor in the rat hypothalamus following acute hyperosmotic challenge. Neuroscience. 2010; 170:1020–1027.
52. Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinanen R, Palmberg C, Palotie A, Tammela T, Isola J, Kallioniemi OP. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nature genetics. 1995; 9:401–406.
53. Rubin MA, Varambally S, Beroukhim R, Tomlins SA, Rhodes DR, Paris PL, Hofer MD, Storz-Schweizer M, Kuefer R, Fletcher JA, Hsi BL, Byrne JA, Pienta KJ, Collins C, Sellers WR, Chinnaiyan AM. Overexpression, amplification, and androgen regulation of TPD52 in prostate cancer. Cancer research. 2004; 64:3814–3822.
54. Periyasamy S, Hinds T Jr, Shemshedini L, Shou W, Sanchez ER. FKBP51 and Cyp40 are positive regulators of androgen-dependent prostate cancer cell growth and the targets of FK506 and cyclosporin A. Oncogene. 2010; 29:1691–1701.
55. Lukacs RU, Goldstein AS, Lawson DA, Cheng D, Witte ON. Isolation, cultivation and characterization of adult murine prostate stem cells. Nature protocols. 2010; 5:702–713.
56. Peehl DM, Leung GK, Wong ST. Keratin expression: a measure of phenotypic modulation of human prostatic epithelial cells by growth inhibitory factors. Cell and tissue research. 1994; 277:11–18.
57. Apodaca G, Katz LA, Mostov KE. Receptor-mediated transcytosis of IgA in MDCK cells is via apical recycling endosomes. The Journal of cell biology. 1994; 125:67–86.
58. Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, Landegren U. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nature methods. 2006; 3:995–1000.
59. Huang H, Shah K, Bradbury NA, Li C, White C. Mcl-1 promotes lung cancer cell migration by directly interacting with VDAC to increase mitochondrial Ca2+ uptake and reactive oxygen species generation. Cell death & disease. 2014; 5:e1482.
60. Ni L, Yang CS, Gioeli D, Frierson H, Toft DO, Paschal BM. FKBP51 promotes assembly of the Hsp90 chaperone complex and regulates androgen receptor signaling in prostate cancer cells. Molecular and cellular biology. 2010; 30:1243–1253.
61. Shah K, Cheng Y, Hahn B, Bridges R, Bradbury NA, Mueller DM. Synonymous Codon Usage Affects the Expression of Wild Type and F508del CFTR. Journal of molecular biology. 2015; 427:1464–1479.
62. Squillace RM, Miller D, Wardwell SD, Wang F, Clackson T, Rivera VM. Synergistic activity of the mTOR inhibitor ridaforolimus and the antiandrogen bicalutamide in prostate cancer models. International journal of oncology. 2012; 41:425–432.
63. Riss TL, Moravec RA, Niles AL, Benink HA, Worzella TJ, Minor L. (2004). Cell Viability Assays. In:Sittampalam GS, Gal-Edd N, Arkin M, Auld D, Austin C, Bejcek B, Glicksman M, Inglese J, Lemmon V, Li Z, McGee J, McManus O, Minor L, Napper A, Riss T, Trask OJ, et al., eds. Assay Guidance Manual. (Bethesda (MD)).