
INTRODUCTION
Apoptosis is a cell death program intended for regulating cell number during development and tissue homeostasis as well as eliminating damaged/dangerous cells [1]. Evasion of apoptosis is typical of many cancers and a frequent cause of therapeutic resistance [2, 3]. This evasion is mostly due to impairment in the mitochondrial pathway of apoptosis. Proteins of the BCL-2 family strictly control this pathway by inducing the mitochondrial outer membrane permeabilization (MOMP). The latter indeed allows the release of apoptogenic factors responsible for the activation of caspases that are the executioners of the cellular demolition program [1, 2].
The BCL-2 family proteins are characterized by their domains of homology to BCL-2 (BH1 to 4) and their functional activities. They comprise the prosurvival members (BCL-2, BCL-XL, BCL-W, MCL-1 and A1) and the proapoptotic members. The latter include the MOMP effectors (mainly BAX and BAK) and the BH3-only proteins, so-called because they have only the BH3 domain (e.g., BIM, BID, PUMA, BAD, NOXA). The activity of the prosurvival proteins is to bind to proapoptotic molecules, notably the MOMP effectors BAX and BAK which are thus sequestered in an inactive form. All prosurvivals can bind BAX whereas only BCL-XL and MCL-1 bind to BAK. This antiapoptotic activity is antagonized by the BH3-only members: by inserting their helical BH3 region into the hydrophobic groove of the prosurvival proteins, they provoke the release of the sequestered BAX and BAK that are thus activated. These interactions are selective: for example NOXA binds only to MCL-1 and A1, BAD binds only to BCL-2, BCL-XL and BCL-W, whereas BIM, BID and PUMA can bind to all prosurvival proteins [1, 2]. Certain BH3-only proteins (such as BIM, BID and probably PUMA) can directly activate the proapoptotic proteins BAX and BAK.
Hence, the BCL-2 family proteins govern the MOMP by their specific interactions, and thus the fate of cells to either live or die. Targeting these proteins therefore appeared as an attractive apoptosis-based strategy to assist anticancer therapy [1, 2]. A particular attention has been focused on the development of agents capable of inhibiting the activity of prosurvival BCL-2 family members that are overexpressed in various malignancies [1, 2, 4]. This has led to the characterization of small molecules mimicking the BH3 domain of BH3-only proteins, likely to selectively bind to and antagonize prosurvival family members and thus induce apoptosis. These “BH3 mimetic” compounds are short peptides modeled on BH3 domains and small organic molecules. The latter are identified by screening libraries of either natural products or computer-designed molecules for binding to BCL-2 proteins [5-16]. Two major criteria were proposed by Lessene et al to define an authentic BH3 mimetic: high affinity binding to the targets (in the nM range) and induction of BAX/BAK-dependent apoptosis [5]. Some compounds have shown significant therapeutic effects in cancer patients.
The preclinical and clinical properties of the small molecules inhibiting prosurvival BCL-2 family proteins have been extensively reviewed [5-16]. Two of the most recent reviews have described the biological context to targeting these proteins and advances in therapeutic approaches with BH3 mimetics. In the one, Anderson et al have focused on the four agents that are in clinical evaluation, discussed the data in detail and pinpointed questions yet to be resolved for using these agents as part of combination therapy [15]. In the other, Roy et al have presented a comprehensive review of compounds that target the BCL-2 family-driven pathway [16]. The present article updates the small molecules targeting proteins of the BCL-2 family with the discovery of not only highly potent antagonists of prosurvival members but also direct activators of the MOMP effectors BAX and BAK and a dual prosurvival inhibitor/proapoptotic activator. These data bring a new dimension to the therapeutic targeting of BCL-2 family proteins.
INHIBITORS OF PROSURVIVAL BCL-2 PROTEINS
Small organic molecules
Obatoclax
This synthetic indol bipyrrole molecule derived from the natural product prodigiosin is capable of binding to all prosurvival BCL-2 family proteins with low affinity (in the µM range) and inducing apoptosis in tumor cells in vitro [17]. This putative pan-BH3 mimetic (or BIM-like BH3 mimetic) was the first to enter clinical trials but has shown only modest therapeutic effects [15, 18]. It is now known that obatoclax does not meet the two main criteria defining an authentic BH3 mimetic and that its proapoptotic activities result from off-target mechanisms [19, 20].
Gossypol family
Gossypol, a natural polyphenol, and its synthetic isomer AT-101 [21, 22] are also putative pan-BH3 mimetics: they do not fully meet the criteria for a bona fide BH3 mimetic and induce apoptosis via multiple mechanisms [19, 20, 23]. Like obatoclax, they showed limited anticancer activity in clinical trials [15]. Several gossypol and AT-101 derivatives such as sabutoclax (BI-97C1) and BI-97D6 were characterized in preclinical studies as exhibiting higher binding affinities (in the sub-µM range) and triggering predominantly BAX/BAK-dependent apoptosis; both sabutoclax and BI-97D6 show in vivo antitumor effects in animal models [24, 25]. Interestingly, sabutoclax has turned out to be a pan-BCL-2 inhibitor in some but not all cellular systems, displaying its best activity in inhibiting MCL-1 [26].
TW-37, a rationally designed benzoylsulphonyl analog of gossypol [22, 27], was also known to operate only in part as a pan-BH3 mimetic: it binds to BCL-2, BCL-XL and MCL-1 with moderate affinity (sub-µM), induces apoptosis depending partially on BAX/BAK activation and shows several off-target effects. However, a recent careful analysis has demonstrated that TW-37 (i) induces several typical features of mitochondrial apoptosis in MCL-1-dependent cells [but not BCL-2 or BCL-XL-dependent cells] and (ii) exhibits all the hallmarks of a NOXA-like BH3 mimetic antagonizing selectively MCL-1, although only at high concentrations [26]. This study suggested that derivatives of TW-37 with higher affinity for MCL-1 might be developed [26].
ABT-737 and navitoclax
The fragment-screening approach based on structure/activity relationship (SAR) by nuclear magnetic resonance (NMR) - initially described by Fesik and colleagues [28] - led to the discovery of ABT-737, a molecule with an acylsulfonamide moiety [29]. Its orally-bioavailable derivative ABT-263 (now navitoclax) was designed for clinical use [30]. Both molecules are authentic BH3 mimetics targeting BCL-2, BCL-XL and BCL-W but not MCL-1 or A1 (as the BH3-only protein BAD, so they are referred to as BAD-like BH3 mimetics). They were extensively characterized in preclinical studies [5, 16, 23]. The therapeutic activity of navitoclax in patients with hematologic malignancies (particularly chronic lymphocytic leukemia) and some solid cancers is now well established [15, 16].
ABT-199
Thrombocytopenia (i.e., an abnormal decrease in number of platelets in the blood) is a dose-limiting effect of navitoclax due to the inhibition of BCL-XL (a survival factor for platelets). To circumvent this side effect, navitoclax derivatives that bind specifically to BCL-2 (without significant binding to BCL-XL or BCL-W) were designed, leading to ABT-199. This true BH3 mimetic is the first highly selective inhibitor of BCL-2 [31]. Initial clinical studies of ABT-199 in chronic lymphocytic leukemia (CLL) and non-Hodgkin’s lymphoma have already shown impressive antitumor efficacy, with higher response rates than navitoclax and without thrombocytopenia [15, 16, 31]. The latest results from the trial in refractory or relapsed CLL patients have confirmed the effectiveness of ABT-199: 23% of the 78 patients recruited for 2 years had complete response (with no evidence of cancer in some patients), 54 % achieved a partial response and overall, 59% of the patients have survived for 2 years without leukemia progression; in addition, the complete response rates were similar in high-risk patients including those having p53 deficiency due to the deletion in chromosome 17 [32]. Moreover, several promising clinical data have been lastly reported: notably in phase I/II trials of ABT-199 in acute myeloid leukemia, and of its combination with bendamustine, rituximab or obinituzumab in non-Hodgkin’s lymphoma and CLL [33-36]. Phase III studies of ABT-199 (now venetoclax) are ongoing.
Other ABT-737 derivatives
Isosteric analogs of ABT-737 in which the acylsulfonamide moiety is replaced by a quinazoline group were synthesized: these quinazoline derivatives have reduced capacities to bind BCL-W while keeping their high-affinity binding to BCL-2 and BCL-XL [37]. The same characteristics were observed with other acylsulfonamide derivatives, as previously reviewed [16].
BM-957 and BM-1197
BM-957 is another ABT-737 derivative (a diphenyl-pyrrole-carboxamide scaffold linked to the nitrobenzene sulfonamide half of ABT-737): it also binds to BCL-2 and BCL-XL with sub-nM affinity, induces caspase 3-dependent apoptosis in tumor cell lines and causes tumor regression in xenograft models [38]. In order to optimize their compound BM-957, Wang and colleagues performed modifications of the pyrrole-carboxylic acid core structure leading to the new small molecule BM-1197 [39]. This compound binds to BCL-2 and BCL-XL with sub-nM affinity, inhibits their antiapoptotic activities and induces BAX/BAK-dependent apoptosis in small cell lung cancer cell lines. In addition, BM-1197 shows improved solubility and pharmacokinetic properties and elicits complete and long-term tumor regression in two different small cell lung cancer xenograft models [39]. Consequently, BM-1197 proves to be a new BAD-like BH3 mimetic.
WEHI-539 and A-1155463
High-throughput screening of small molecules targeting BCL-XL and structure-guided design led to the discovery of WEHI-539, a molecule with a benzothiazole hydrazone scaffold. This compound was the first highly selective antagonist of BCL-XL (through high affinity binding) over BCL-2 and BCL-W. Careful analysis of its mechanism of action has shown that WEHI-539 induces BAK-mediated apoptosis [40]. NMR fragment screening and structure-based design further led to the characterization of A-1155463, a more potent BCL-XL inhibitor than WEHI-539 while possessing none of its pharmaceutical liabilities. Importantly, this compound acts through an on-target mechanism and shows in vivo antitumor activity in mice [41]. The new small molecule A-155463 constitutes an actual BH3 mimetic specific for BCL-XL.
Arylsulfonamide derivatives
Hybridization of the arylsulfonamide portion of ABT-737 with an MCL-1 inhibitor resulted in a dual BCL-XL/MCL-1 ligand with moderate (sub-µM) affinity [42]. A substituted arylsulfonamide has shown selectivity for MCL-1 (albeit with sub-µM binding affinity): this molecule induces caspase-9 activation and apoptosis [43]. Interestingly, an approach similar to that used for the ABT series has recently allowed the design of a biphenyl arylsulfonamide molecule also targeting MCL-1 with an apparent affinity of 30 nM; despite it is not known whether this compound can induce apoptosis, it might serve as a promising lead for optimized effects [44].
MCL-1 inhibitor molecule-1 (MIM-1)
High-throughput screening of small molecules displacing a stapled peptide derived from the BH3 domain of MCL-1 has yielded a substituted thiazol molecule called MIM-1 (for MCL-1 inhibitor molecule-1); this small molecule specifically engages the binding groove of MCL-1 and induces BAK-dependent apoptosis in MCL-1-dependent cells [45]. However, the binding affinity for MCL-1 is modest (4.8 µM), high concentrations of MIM-1 are required for apoptosis, and this activity depends on the cell type [26].
Small molecule MCL-1 inhibitor
Using their NMR-based fragment screen approach (having led to ABT-737), Fesik and colleagues recently identified two chemically distinct compounds that bind to different sites of MCL-1; they then merged these compounds together to produce molecules binding MCL-1 selectively over BCL-2 and BCL-XL [46]. After optimization, they obtained a lead compound [indole-2-carboxylic acid] which binds and inhibits MCL-1 (with an affinity of 55 nM) and induces apoptosis. Crystal structure of the complex validates the strategy and provides detailed information to design more potent inhibitors [46].
A-1210477
A series of specific MCL-1 inhibitors exhibiting sub-nM affinity were obtained by high throughput screening and structure-guided design [47]. The compounds are indole-2-carboxylic acids like the small molecule MCL-1 inhibitor described by Fesik and colleagues [46], but have a 100-fold higher binding affinity. Leverson et al have further characterized the most potent of the compounds, A-1210477: it binds selectively to MCL-1 with an affinity of 0.45 nM, disrupts BIM/MCL-1 complexes in living cells, induces the hallmarks of mitochondrial apoptosis in MCL-1-dependent cancer cells and synergizes with navitoclax to induce apoptosis in various malignant cell lines [48]. Data also demonstrate that the compound acts through an on-target mechanism. Therefore, A-1210477 appears as the first BH3 mimetic targeting MCL-1.
Mcl-1 inhibitor-9
A high-throughput screening approach culminated in the characterization of a piperazine-substituted hydroxyquinoline that inhibits selectively MCL-1. This molecule called Mcl-1 inhibitor-9 was proposed to function as a BH3 mimetic in inducing mitochondrial apoptosis in cell lines [49] although the binding affinity and mechanism of action have not been evaluated.
Sesquiterpenoid derivatives
Screening a library of natural sesquiterpenoids for disrupting the MCL-1/BIM BH3 complex led to the identification of hexahydronaphtalene and the synthesis of substituted derivatives. The most potent MCL-1 inhibitor can engage the binding groove of MCL-1 in computational docking experiments, and it shows antagonizing activity in cancer cells [50]. This possible new class of MCL-1 inhibitor yet needs further development and biological characterization.
S1 family
The small molecule S1 (an oxo-phenalene-dicarbonitrile) binds to both BCL-2 and MCL-1 albeit with moderate affinity (in the sub-µM range), triggers BAX/BAK-dependent apoptosis, and shows antitumor activity in an animal model [51]. Although initially proposed as a pan-BH3 mimetic, S1 was reportedly found to act via multiple and off-target mechanisms [20, 52, 53]. A substituted derivative of S1 was designed and characterized as showing a higher affinity for binding MCL-1 (10 nM) and BCL-2 (20 nM) and a better apoptotic activity in tumor cell lines than the parental S1 [54]. Unless the demonstration that this activity is BAX/BAK-dependent, S1-derivative should be considered as a putative pan-BH3 mimetic. Other S1-derivatives were described as novel MCL-1 inhibitors [55, 56].
Acylsulfonamide molecules
A fragment-based NMR method (other than the Fesik’s approach) has allowed the generation of a series of acylsulfonamides (different from the ABT series). The small molecules exhibit high affinity (nM) for both BCL-XL and MCL-1 [57]. However, their biological validation has not been provided.
Compound JY-1-106
SAR analysis of oligoamide foldamers led to the design of JY-1-106, a substituted trisarylamide having the ability to induce apoptosis in vitro and to repress tumor growth in an animal model. Computational analyses indicate that JY-1-106 engages the BH3-binding grooves of both BCL-XL and MCL-1, suggesting that it may be a pan-BH3 mimetic [58]. Whether JY-1-106 meets the two criteria that define an authentic BH3 mimetic yet remains to be stated.
Other new small molecules
A number of small organic molecules designed to mimic BH3 peptides have just been reported such as a series of molecules containing a benzoylurea scaffold which selectively bind to BCL-XL albeit with µM affinity [59]. Is is also the case for several MCL-1 inhibitors including marinopyrrole derivatives, a single diastereomer of a macrolactam core and compounds with a 3-phenylthiophene-2-sulfonamide core moiety or with a phenyl-piperazine-triazine scaffold [60-63]. Interestingly, a small molecule targeting the prosurvival BCL-2 family protein A1 has been identified for the first time [64]. Although these compounds require further characterization and improvements, they may represent starting points for developing new classes of inhibitors.
BH3 domain-derived peptides
Peptides hardly penetrate into cells and are prone to rapid proteolytic degradation. Efforts focused to modify peptides in order to increase the cell penetration and stability and also to reproduce the helicity of BH3 domains needed for high affinity binding.
α/β foldamers
BIM and PUMA BH3 peptides were modified by incorporating non-natural amino acids: this resulted in α/β foldamer-peptides capable of binding to BCL-XL [65] and to both BCL-XL and MCL-1 with high affinity [66], as previously discussed [16]. It is furthermore noteworthy that peptides with non-canonical BH3 domain sequences were found to bind selectively to MCL-1, such as reverse BH3 motifs with helical conformation [67] and a variant of the BIM BH3 region, BimS2A [68]. However, the proapoptotic properties of these peptides have not been characterized with the exception of BimS2A which can induce BAK-dependent apoptosis when BCL-XL is absent or neutralized [68].
Cross-linked NOXA-BH3 peptide
A short peptide derived from the BH3-only protein NOXA was biphenyl cross-linked in order to stabilize the helical conformation. The cross-linked BH3 peptide is capable of binding selectively to MCL-1 with an affinity comparable to that of NOXA (in the nM range) and it can induce death in a lymphoma cell line [69]. Conjugating this compound with ubiquitin further led to a MCL-1 antagonist with increased cell penetration and killing activity [70]. Whether the biphenyl-NOXA BH3 peptide actually induces BAX/BAK-dependent apoptosis is not known.
BIM SAHB and other stapled BH3 peptides
Another approach to stabilize the helical conformation has been to staple BH3 peptides. The first “stabilized α-helix of BCL-2 domain” (SAHB) was generated from the BH3 domain of BID [71]. A SAHB derived from the BH3 helix of BIM was recently described [72]. This BIM SAHB (BIM 21-mer, residues 146-166) binds with high affinity to BCL-XL, BCL-W, MCL-1 and A1, and it induces mitochondrial apoptosis in leukemia/lymphoma cell lines and tumor regression in an animal model of human leukemia [72]. Data indicate that the BIM SAHB displays the hallmarks of an authentic pan-BH3 mimetic. However, the cell permeability, binding affinity and biological activity of SAHB compounds are much debated issues [73, 74]. They seem to depend on the cell type inasmuch as BIM SAHB (residues 146-166) shows activity in hematologic but not fibroblastic cell lines [75].
Other relevant stapled peptides were described such as a SAHB modeled on the MCL-1 BH3 domain which was characterized as a selective MCL-1 inhibitor with proapoptotic properties in leukemia cell lines [76]. A SAHB derived from the BIM BH3 region was reported to be able to bind the MOMP effector BAX at a site that directly triggers its activation [77, 78]. These two compounds have further served as probes to identify small molecules with BH3 mimicry properties, respectively MIM-1 (see previous section Small organic molecules) and BAM-7 (described in next section Activators of proapoptotic BCL-2 proteins.).
BIM BH3-derived peptides of the XXA series
By combining computational design, combinatorial library screening and rational mutagenesis, a series of peptides derived from the BH3 region of BIM were engineered. The lead compound XXA1 binds to BCL-XL with sub-nM affinity (and selectivity up to 1000-fold over other prosurvival proteins); it induces MOMP-dependent apoptosis in human platelets (for which BCL-XL is a survival factor) and malignant cell lines [79]. Therefore, XXA1 may be considered as a new, putative BH3 mimetic highly specific for BCL-XL.
ACTIVATORS OF PROAPOPTOTIC BCL-2 PROTEINS
Recent studies have used the BH3 mimetic concept to design small molecules having the ability to directly bind and activate proapoptotic members of the BCL-2 family, as described below.
BAM-7
Walensky and colleagues first characterized a SAHB modeled on the BIM BH3 region and capable of binding the MOMP effector BAX at a site that directly triggers its activation [77, 78]. They then used a computational screen of small molecules for displacing their SAHB from its ligand BAX, to obtain a substituted pirazolone molecule called BAM-7; this compound can engage the BAX trigger site and induce BAX activation and apoptosis [80]. This was the first description of a small molecule capable of directly activating a proapoptotic BCL-2 family protein. The compound BAM-7 could be used to generate direct activators of BAX with high binding affinity.
PUMA-BH3 peptide
Investigating the ability of the BH3-only proteins NOXA and PUMA to directly activate the MOMP effector BAK has highlighted a PUMA BH3 peptide that binds with nM affinity to the canonical binding pocket of BAK and induces its activation, leading to BAK-dependent MOMP and cell death [81, 82]. This study supports and extends others having indicated that different BH3 peptides as well as a BID SAHB can directly activate BAK [83-85]. These data therefore provide proof-of-concept that direct activators of BAK may be designed.
PUMA SAHB
A SAHB derived from the PUMA-BH3 helix was found to display a dual activity: it can both engage all the prosurvival BCL-2 family proteins (and inhibit their activity) and directly bind and activate the proapoptotic BAX [86]. In addition, a cell permeable analog of this PUMA SAHB (capable of binding to BCL-2, MCL-1 and BAX) induces mitochondrial apoptosis in neuroblastoma cells [86]. These data demonstrate for the first time that PUMA BH3 is a dual prosurvival inhibitor and proapoptotic direct activator. It is therefore conceivable to design BH3 mimetics having such a dual capacity.
CONCLUSIONS AND PERSPECTIVES
Since the emergence of the BH3 mimetic concept [4], targeting BCL-2 family proteins is considered to have a promising therapeutic potential especially for treating cancers [1, 2, 5]. Much progress has been made in this field of research with the identification of numerous inhibitors of prosurvival BCL-2 family proteins [6-16]. This field yet remains very challenging. The nature of the interaction between these compounds and their targets (protein/protein interaction, large and hydrophobic pockets) makes any program of drug discovery still difficult. Moreover, many putative BH3 mimetics were reported with minimal evidence of on-target effects. The low or modest binding affinity of some compounds is notably responsible for off-target effects. The difficulty to assess their mechanism of action can also be misleading [87].
Nevertheless, four small organic molecules targeting prosurvival BCL-2 family proteins have already entered clinical trials in cancer patients [15]. The clinical results with the two putative pan-BH3 mimetics obatoclax and AT-101 (that are now known to induce apoptosis mainly via off-target mechanisms) were disappointing. In contrast, the two authentic BH3 mimetics navitoclax and ABT-199 (which are both derived from the pioneer molecule ABT-737) proved to display anticancer efficacy particularly in hematologic malignancies: significant therapeutic effects were first observed with the BAD-like mimetic navitoclax and impressive results were further recorded with the BCL-2-specific ABT-199 that does not induce the thrombocytopenia exhibited by navitoclax (due to BCL-XL inhibition). These data have unequivocally demonstrated that BH3 mimetic agents can expand the anticancer armamentarium in combination with conventional treatments.
None of the characterized BH3-derived peptides have so far progressed towards clinical evaluation. This does not mean that the approach to identifying BH3-derived peptides is less promising than the approach of screening small organic molecules. The former compounds indeed display liability and pharmacologic difficulties, whereas the latter are often prone to off-target effects. To achieve high affinity binding to the targets is a frequent issue for both types of compounds. A general conclusion favoring either approach cannot be drawn yet.
Updating the small molecules targeting BCL-2 family proteins (as recapitulated in Table 1) highlights not only new antagonists of prosurvival proteins but also the possibility to design novel types of BH3 mimetics capable of directly activating proapoptotic proteins.
As regards the newly identified antagonists of prosurvival proteins, the first BH3 mimetic specific for BCL-XL has been discovered with the highly potent small-molecule inhibitor A-1155463 displaying on-target, antitumor activity in vivo. The BH3 peptide derivative XXA1 also appears as a candidate of interest. The emergence of the compound A-1210477 - a potent MCL-1 inhibitor with a high binding affinity and an on-target mechanism of action - indicates that a NOXA-like BH3 mimetic specific for MCL-1 will rapidly be obtained. Other MCL-1 antagonists might likewise be probed such as the pioneer MIM-1, the remarkable Small Molecule Mcl-1 Inhibitor, and a biphenyl cross-linked NOXA peptide. The further characterization of TW-37 has revealed that modifications enhancing its affinity for MCL-1 could also lead to an authentic NOXA-like BH3 mimetic. The generation of pan-BH3 mimetics (which are needed for replacing obatoclax and AT-101) seems possible from the BIM SAHB or the gossypol derivative sabutoclax that are known to inhibit a broad range of prosurvival proteins. The small molecules S1-derivative and JY-1-106 were also described as starting points for designing pan-BH3 mimetics. Therefore, after the emergence of the BAD-like BH3 mimetic navitoclax and the BCL-2-specific ABT-199, the developments of NOXA-like, BCL-XL-specific and pan-BH3 mimetics will likely be achieved. Lastly, BM-1197 is added to navitoclax as another, potent BAD-like BH3 mimetic displaying a particular therapeutic potential for small cell lung cancer.
Moreover, a new dimension to targeting BCL-2 family proteins is provided by the discovery of two novel types of small molecules capable of directly activating proapoptotic members of the family. First, after the identification of the small molecule BAM-7 as a BAX activator, the finding that a PUMA BH3 peptide can activate BAK confirms the possibility to generate BH3 mimetics capable of directly activating the MOMP effectors. Second, the characterization of a PUMA SAHB analog which both activates BAX and antagonizes BCL-2 and MCL-1 indicates that it is possible to design BH3 mimetics having the dual activity of prosurvival inhibitor and proapoptotic activator.
Many efforts are obviously required to implement the clinical application of the novel types of BH3 mimetics. Nevertheless, the therapeutic targeting of BCL-2 family proteins already appears as a potent weapon for overcoming the apoptotic resistance in cancers and improving the treatments of patients.
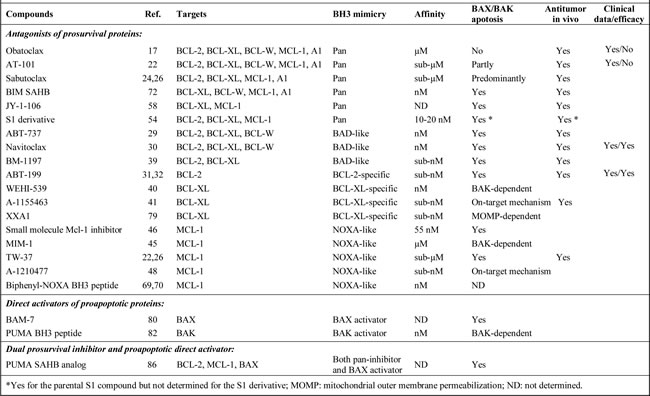
Table 1: Main small molecules targeting BCL-2 family proteins and their characteristics.
Conflicts of interest
The authors have no conflicts of interest to declare.
REFERENCES
1 Strasser A, Cory S, Adams JM. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. 2011; 30: 3667-3683.
2 Kelly GL, Strasser A. The essential role of evasion from cell death in cancer. Adv Cancer Res. 2011; 111: 39-96.
3 Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144: 646-674.
4. Baell JB, Huang DCS. Prospects for targeting the Bcl-2 family of proteins to develop novel cytotoxic drugs. Biochem Pharmacol. 2002; 64: 851-863.
5. Lessene G, Czabotar PE, Colman PM. Bcl-2 family antagonists for cancer therapy. Nature Rev. 2008; 7: 989-1000.
6. Azmi AA, Wang Z, Philip PA, Mohammad RM, Sarkar FH. Emerging Bcl-2 inhibitors for the treatment of cancer. Expert Opin Emerging Drugs. 2011; 16: 59-70.
7. Khaw SL, Huang DCS, Roberts AW. Overcoming blocks in apoptosis with BH3-mimetic therapy in hematological malignancies. Pathology. 2011; 43: 525-535.
8. Bajwa N, Chenzong L, Nikolovska-Coleska Z. Inhibitors of the anti-apoptotic Bcl-2 proteins: a patent review. Expert Opin Ther Patents. 2012; 22: 37-55.
9. Davids MS, Letai A. Targeting the B-cell lymphoma/leukemia 2 family in cancer. J Clin Oncol. 2012; 30: 3127-3135.
10. Balakrishnan K, Gandhi V. Bcl-2 antagonists: a proof of concept for CLL therapy. Invest New Drugs. 2013; 31: 1384-1393.
11. Thomas S, Quinn BA, Das SK, Dash R, Emdad L, Dasgupta S, et al. Targeting the Bcl-2 family for cancer therapy. Expert Opin Ther Targets. 2013; 17: 61-75.
12. Scarfo L, Ghia P. Reprogramming cell death: Bcl-2 family inhibition in hematological malignancies. Immun Lett. 2013; 155: 36-39.
13. Billard C. BH3 mimetics: status of the field and new developments. Mol Cancer Ther. 2013; 12: 1691-1700.
14. Belmar J, Fesik SW. Small molecule Mcl-1 inhibitors for the treatment of cancer. Pharmacol Ther. 2015; 145: 76-84.
15. Anderson MA, Huang D, Roberts A. Targeting BCL2 for the treatment of lymphoid malignancies. Sem Hematol. 2014; 51: 219-227.
16. Roy MJ, Vom A, Czabotar PE, Lessene G. Cell death and the mitochondria: therapeutic targeting of the BCL-2 family-driven pathway. Br J Pharmacol. 2014; 171: 1973-1987.
17. Nguyen M, Marcellus RC, Roulston A, Watson M, Serfass L, Madiraju SRM, et al. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Pro Natl Acad Sci USA. 2007; 104: 19512-19517.
18. Joudeh J, Claxton D. Obatoclax mesylate: pharmacology and potential for therapy of hematological neoplasms. Expert Opin Investig Drugs. 2012; 21: 363-373.
19. Vogler M, Weber K, Dinsdale D, Schmitz I, Schultze-Hosthoff K, Dyer MJS, et al. Different forms of cell death induced by putative BCL2 inhibitors. Cell Death Diff. 2009; 16: 1030-1039.
20. Albershardt TC, Salerni BL, Soderquist RS, Bates DJP, Pletnev AA, Kisselev AF, et al. Multiple BH3 mimetics antagonize antiapoptotic MCL-1 protein by inducing the endoplasmic reticulum stress response and up-regulating BH3-only protein NOXA. J Biol Chem. 2011; 286: 24882-24895.
21. Kitada S, Leone M, Sareth S, Zhai D, Reed JC, Pellecchia M. Discovery, characterization, and structure-activity relationships studies of proapoptotic polyphenols targeting B-cell lymphocyte/leukemia-2 proteins. J Med Chem. 2003; 46: 4259-4264.
22. Wang G, Nikolovska-Coleska Z, Yang CY, Wang R, Tang G, Guo J, et al. Structure-based design of potent small molecule inhibitors of anti-apoptotic Bcl-2 proteins. J Med Chem. 2006; 49: 6139-6142.
23. van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006; 10: 389-399.
24. Wei J, Stebbins JL, Kitada S, Dash R, Placzek W, Rega M, et al. BI-97C1, an optically pure apogossypol derivative as pan-active inhibitor of antiapoptotic B-cell lymphoma/leukemia-2 (Bcl-2) family proteins. J Med Chem. 2010; 53: 4166-4176.
25. Wei J, Stebbins JL, Kitada S, Sash R, Zhai D, Placzek WJ, et al. An optically pure apogossypolone derivative as potent pan-active inhibitor of anti-apoptotic Bcl-2 family proteins. Front Oncol. 2011; 1: 28.
26. Varadarajan S, Vogler M, Butterworth M, Dinsdale D, Walensky LD, Cohen GM. Evaluation and critical assessment of putative MCL-1 inhibitors. Cell Death Diff. 2013; 11: 1475-1484.
27. Mohammad RM, Goustin AS, Aboukameel A, Chen B, Banerjee S, Wang G, et al. Preclinical studies of TW-37, a new nonpeptidic small-molecule inhibitor of Bcl-2, in diffuse large cell lymphoma xenograft model reveal drug action on both Bcl-2 and Mcl-1. Clin Cancer Res. 2007; 13: 2226-2235.
28. Shuker SB, Hajduk PJ, Meadows PP, Fesik SW. Discovery high-affinity ligands for proteins: SAR by NMR. Science. 1996; 274: 1531-1534.
29. Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumors. Nature. 2005; 435; 677-681.
30. Tse C, Shoemaker AR, Adickers J, Chen J, Jin S, Johnson EF, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008; 68: 3421-3428.
31. Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013; 19: 202-208.
32. Seymour JF, Davids MS, Pagel JM, Kahl BS, Wierda WG, Puvvada S, et al. ABT-199 (GDC-0199) in relapsed/refractory chronic lymphocytic leukemia and small lymphocytic lymphoma: high response rates among patients with high risk disease features including unmutated IGVH. Presented at: 19th Congress of the European Hematology Association. Milan, Italy, June 12-16, 2014 (Abstract S702).
33. Konopleva M, Pollyea DA, Potluri J, Chyla BJ, Busman T, McKeegan E, et al. A phase 2 study of ABT-199 (GDC-0199) in patients with acute myelogenous leukemia (AML). Presented at: 56th ASH annual meeting and exposition. San Francisco, CA, December 6-9, 2014 (Abstract 118).
34. de Vos S, Flowers CR, Wang D, Swinnen LJ, Fowler N, Reid E, et al. The BCL-2 inhibitor ABT-199 (GDC-0199) in combination with bendamustine and rituximab in patients with relapsed or refractory non-Hodgin’s lymphoma. Presented at: 56th ASH annual meeting and exposition. San Francisco, CA, December 6-9, 2014 (Abstract 1722).
35. Roberts AW, Ma S, Brander DM, Kipps TJ, Barrientos JC, Davids MS, et al. Determination of recommended phase 2 dose of ABT-199 (GDC-0199) combined with rituximab (R) in patients with relapsed/refractory (R/R) chronic lymphocytic leukemia. Presented at: 56th ASH annual meeting and exposition. San Francisco, CA, December 6-9, 2014 (Abstract 325).
36. Flinn I, Brunvand M, Dyer MJS, Hillman P, Jones J, Lymp J, et al. Preliminary results of a phase 1b study (GP28331) combining GDC-0199 (ABT-199) and obinutuzumab in patients with relapsed/refractory or previously untreated chronic lymphocytic leukemia. Presented at: 56th ASH annual meeting and exposition. San Francisco, CA, December 6-9, 2014 (Abstract 4687).
37. Sleebs BE, Czabotar PE, Fairbrother WJ, Fairlie WD, Flygare JA, Huang DC, et al. Quinazoline sulfonamides ad dual binders of the B-cell lymphoma 2 and B-cell lymphoma extra long with potent proapoptotic cell-based activity. J Med Chem. 2011; 54: 1914-1926.
38. Chen J, Zhou H, Aguilar A, Liu L, Bai L, McEachern D, et al. Structure-based discovery of BM-957 as a potent small molecule inhibitor of Bcl-2 and Bcl-xL capable of achieving complete tumor regression. J Med Chem. 2012; 55: 8502-8514.
39. Bai L, Chen J, McEachern D, Liu L, Zhou H, Aguilar A, et al. BM-1197: a novel and specific Bcl-2/Bcl-xL inhibitor inducing complete and long-lasting tumor regression in vivo. PLoS One. 2014; 9: e99404.
40. Lessene G, Czabotar PE, Sleebs BE, Zobel K, Lowes KN, Adams JM, et al. Structure-guided design of a selective BCL-XL inhibitor. Nat Chem Biol. 2013; 9: 390-397.
41. Tao ZF, Hasvold L, Wang L, Wang X, Petros AM, Park CH, et al. Discovery of a potent and selective BCL-XL inhibitor with in vivo activity. ACS Med Chem Lett. 2014; 5: 1088-1093.
42. Tanaka Y, Aikawa K, Nishida G, Homma S, Sogabe S, Igaki S, et al. Discovery of potent Mcl-1/Bcl-xL dual inhibitors by using a hybridization strategy based on structural analysis of target proteins. J Med Chem. 2013; 56: 9635-9645.
43. Abulwerdi FA, Liao C, Mady AS, Gavin J, Shen C, Cierpicki T, et al. 3-substituted-N-(4-hydroxynaphtalen-1-yl)arylsulfonamides as a novel class of selective Mcl-1 inhibitors: structure-based design, synthesis, SAR, and biological evaluation. J Med Chem. 2014; 57: 4111-4133.
44. Petros AM, Swann SL, Song D, Swinger K, Park C, Zhang H, et al. Fragment-based discovery of potent inhibitors of the anti-apoptotic MCL-1 protein. Bioorg Med Chem Lett. 2014; 24: 1484-1488.
45. Cohen NA, Stewart ML, Gavathiotis E, Tepper JL, Bruekner SL, Koss B, et al. A competitive stapled peptide screen identifies a selective small molecule that overcomes Mcl-1-dependent leukemia cell survival. Chem Biol. 2012; 19: 1175-1186.
46. Friberg A, Vigil D, Zhao B, Daniels RN, Burke JP, Garcia-Barrantes PM, et al. Discovery of potent myeloid cell leukemia 1 (Mcl-1) inhibitors using fragment-based methods and structure-based design. J Med Chem. 2012; 56: 15-30.
47. Bruncko M, Wang L, Sheppard GS, Phillips DC, Tahir SK, Xue J, et al. Stucture-guided design of a series of MCL-1 inhibitors with high affinity and selectivity. J Med Chem. 2015; 58: 2180-2194.
48. Leverson JD, Zhang H, Chen J, Tahir SK, Phillips DC, Xue J, et al. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax). Cell Death Dis. 2015; 6: e1590.
49. Richards DJ, Lena R, Bannister T, Blake N, Perceall WE, Carlson NE, et al. Hydroxyquinoline-derived compounds and analoguing of selective Mcl-1 inhibitors using a functional biomarker. Bioorg Med Chem. 2013; 21: 6642-6649.
50. Kim YB, Balasis MA, Doi K, Berndt N, DuBoulay C, Hu CA, et al. Synthesis and evaluation of substituted hexahydronaphtalenes as novel inhibitors of the Mcl-1/BimBH3 interaction. Bioorg Med Chem Lett. 2012; 22: 5961-5965.
51. Zhang Z, Song T, Zhang T, Gao J, Wu G, An L, et al. A novel BH3 mimetic S1 potently induces Bax/Bak-dependent apoptosis by targeting both Bcl-2 and Mcl-1. Int J Cancer. 2011; 128: 1724-1735.
52. Soderquist R, Pletnev AA, Danilov AV, Eastman A. The putative BH3 mimetic S1 sensitizes leukemia to ABT-737 by increasing reactive oxygen species including endoplasmic reticulum stress, and upregulating the BH3-only protein NOXA. Apoptosis. 2014; 19: 201-209.
53. Zhong JT, Xu Y, Yi HW, Su J, Yu HM, Xiang XY, et al. The BH3 mimetic S1 induces autophagy through ER stress and disruption of Bcl-2/Beclin 1 interaction in human glioma U251 cells. Cancer Lett. 2012; 323: 180-187.
54. Song T, Li X, Chang X, Liang X, Zhao Y, Wu G, et al. 3-thiomorpholin-8-oxo-8H-acenaphto (1, 2-b) pyrrole-9-carbonitrile (S1) derivatives as pan-Bcl-2-inhibitors of Bcl-2, Bcl-xL and Mcl-1. Bioorg Med Chem. 2013; 21: 11-20.
55. Zhang Z, Song T, Li X, Wu Z, Feng Y, Xie F, et al. Novel soluble myeloid leukemia sequence 1 (Mcl-1) inhibitor (E, E)-2-(benzylaminocarbonyl)-3-styrylacrylonitrile (4g) developed using a fragment-based approach. Eur J Med Chem. 2013; 59: 141-149.
56. Zhang Z, Liu L; Li X, Song T, Wu Z, Liang X, et al. Fragment-based design, synthesis, and biological evaluation of N-substituted-5-(4-isopropylthiophenol)-2-hydroxynicotinamide derivatives as novel Mcl-1 inhibitors. Eur J Med Chem. 2013; 60: 410-420.
57. Rega MF, Wu B, Wei J, Zhang Z, Cellitti JF, Pellecchia M. SAR by interligand nuclear Overhauser effects (ILOEs) based discovery of acylsulfonamide compounds active against Bcl-xL and Mcl-1. J Med Chem. 2011; 54: 6000-6013.
58. Cao X, Yap JL, Newell-Rogers MK, Peddaboina C, Jiang W, Papaconstantinou HT, et al. The novel BH3 α-helix mimetic JY-1-106 induces apoptosis in a subset of cancer cells (lung cancer, colon cancer and mesothelioma) by disrupting Bcl-xL and Mcl-1 protein-protein interactions with Bak. Mol Cancer. 2013; 12: 42 (1-16).
59. Brady RM, Vom A, Roy MJ, Toovey N, Smith BJ, Moss RM, et al. De-novo designed library of benzoylureas as inhibitors of BCL-XL: synthesis, structural and biochemical characterization. J Med Chem. 2014; 57: 1323-1343.
60. Li R, Cheng C, Balasis ME, Liu Y, Garner TP, Daniel KG, et al. Design, synthesis and evaluation of marinopyrrole derivatives as selective inhibitors of Mcl-1 binding to pro-apoptotic Bim and dual Mcl-1/Bcl-xL inhibitors. Eur J Med Chem. 2015; 90: 315-331.
61. Fang C, D’Souza B, Thompson CF, Clifton MC, Fairman JW, Fulroth B, et al. Single diastereomer of a macrolactam core binds specifically to myeloid cell leukemia 1 (MCL1). ACS Med Chem Lett. 2014; 5: 1308-1312.
62. Yang C, Chen S, Zhou M, Li Y, Zhang Z, Liu Z, et al. Development of 3-phenyl-N-(2-(3-phenylureido)ethyl)-thiophene-2-sulfonamide compounds as inhibitors of antiapoptotic Bcl-2 family proteins. ChemMedChem. 2014; 9: 1436-1452.
63. Moon H, Lee WS, Oh M, Lee H, Im W, et al. Design, solid-phase synthesis, and evaluation of a phenyl-piperazine-triazine scaffold as α-helix mimetics. ACS Com Sci. 2014; 16: 695-701.
64. Mathieu AL, Sperandio O, Pottiez V, Balzarin S, Herledan A, Elkaïm JO, et al. Identification of small inhibitory molecules targeting the Bfl-1 anti-apoptotic protein that alleviates resistance to ABT-737. J Biomol Screen. 2014; 19: 1035-1046.
65. Boersma MD, Haase HS, Peterson-Kaufman KJ, Lee EF, Clarke OB, Colman PM, et al. Evaluation of diverse α-β-backbone patterns for functional α-helix mimicry: analogues of the Bim BH3 domain. J Am Chem Soc. 2012; 134: 315-323.
66. Smith BJ, Lee EF, Checco JW, Evangelista M, Gellman SH, Fairlie WD. Structure-guided rational design of α/β-peptide foldamers with high affinity for BCL-2 family prosurvival proteins. Chembiochem. 2013; 14: 1564-1572.
67. Placzek WJ, Sturlese M, Wu B, Cellitti JF, Wei J, Pellecchia M. Identification of a novel Mcl-1 Protein binding motif. J Biol Chem. 2011; 286: 39829-39835.
68. Lee EF, Czabotar PE, van Delft MF, Michalak EM, Boyle MJ, Willis SN, et al. A novel BH3 ligand that selectively targets Mcl-1 reveals that apoptosis can proceed without Mcl-1 degradation. J Cell Biol. 2008; 180: 341-355.
69. Muppidi A, Doi K, Edwardraja S, Drake EJ, Gulick AM, Wang HG, et al. Rational design of proteolytically stable, cell-permeable peptide-based selective Mcl-1 inhibitors. J Am Chem Soc. 2012; 134: 14734-14737.
70. Muppidi A, Doi K, Edwardraja S, Pulavarti SW, Szyperski T, Wang HG, et al. Targeted delivery of ubiquitin-conjugated BH3 peptide-based Mcl-1 inhibitors into cancer cells. Bioconjug Chem. 2014; 25: 424-432.
71. Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, et al. Activation of apoptosis in vivo by an hydrocarbon-stapled BH3 helix. Science. 2004; 305: 1466-1470.
72. LaBelle JL, Katz SG, Bird GH, Gavathiotis E, Stewart ML, Lawrence C, et al. A stapled Bim peptide overcomes apoptotic resistance in hematologic cancers. J Clin Invest. 2012; 122: 2018-2031.
73. Okamoto T, Zobel K, Federova A, Quan C, Yang H, Fairbrother WJ, et al. Stabilizing the pro-apoptotic BimBH3 helix (BimSAHB) does not necessarily enhance affinity or biological activity. ACS Chem Biol. 2013; 8: 297-302.
74. Bird GH, Gavathiotis E, LaBelle JL, Katz SG, Walensky LD. Distinct BimBH3 (BimSAHB) stapled peptides for structural and cellular studies. ACS Chem Biol. 2014; 9: 831-837.
75. Okamoto T, Segal D, Zobel K, Fedorova A, Yang H, Fairbrother WJ, et al. Further insights into the effects of pre-organizing the BimBH3 helix. ACS Chem Biol. 2014; 9: 838-839.
76. Stewart ML, Fire E, Keating AE, Walensky LD. The MCL-1 helix is an exclusive MCL-1 inhibitor and apoptosis sensitizer. Nat Chem Biol. 2010; 6: 595-601.
77. Gavathiotis E, Suzuki M, Davis ML, Pitter K, Bird GH, Katz SG, et al. BAX activation is initiated at a novel interaction site. Nature. 2008; 455: 1076-1082.
78. Gavathiotis E, Reyna DE, Davis ML, Bird GH, Walensky LD. BH3-triggered structural reorganization drives the interaction of proapoptotic BAX. Mol Cell. 2010; 40: 481-492.
79. Dutta S, Ryan J, Chen TS, Kougentakis C, Letai A, Keating AE. Potent and specific peptide inhibitors of human pro-survival protein Bcl-XL. J Mol Biol. 2015; 427: 1241-1253.
80. Gavathiotis E, Reyna DE, Bellairs JA, Leshchiner ES, Walensky LD. Direct and selective small-molecule activation of proapoptotic BAX. Nat Chem Biol. 2012; 8: 639-645.
81. Dai H, Smith A, Meng W, Schneider PA, Pang YP, Kaufmann SH. Transient binding of an activator BH3 domain to the Bak BH3-binding groove initiates Bak oligomerization. J Cell Biol. 2011; 194: 39-48.
82. Dai H, Pang YP, Ramirez-Alvarado M, Kaufmann SH. Evaluation of the BH3-only protein Puma as a direct Bak activator. J Biol Chem. 2014; 289: 89-99.
83. Du H, Wolf J, Schafer B, Moldoveanu T, Chipuk JE, Kuwana T. BH3 domains other than Bim and Bid can directly activate Bax/Bak. J Biol Chem. 2011; 286: 491-501.
84. Moldoveanu T, Grace CR, Llambi F, Nourse A, Fitzgerald P, Gehring K, et al. BID-induced structural changes in BAK promote apoptosis. Nat Struct Mol Biol. 2013; 20: 589-597.
85. Leshniner ES, Braun CR, Bird GH, Walensky LD. Direct activation of full-length proapoptotic BAK. Proc Nat Acad Sci USA. 2013; 110: E986-E995.
86. Edwards AL, Gavathiotis E, LaBelle JL, Braun CR, Opoku-Nsiah KA, Bird GH, et al. Multimodal interaction with BCL-2 family proteins underlies the proapoptotic activity of PUMA BH3. Chem Biol. 2013; 20: 888-902.
87. Erlanson DA. Learning from PAINful lessons. J Med Chem. 2015; 58: 2088-2090.