
INTRODUCTION
The control of human growth is a complex process involving multiple different biological pathways. Several conditions associated with human overgrowth are recognised and the underlying causes are extremely diverse [6]. More recently, GWAS studies have identified over 180 loci that contribute to human height, some of which overlap with Mendelian syndromes [1]. Weaver syndrome was first described in 1974 and is characterised by pre and postnatal overgrowth, variable learning disability and a distinctive facial appearance [2, 3]. The majority of cases are sporadic, though rare familial cases exhibiting an autosomal dominant pattern of inheritance have been reported [7-9]. Here we have undertaken exome sequencing and sanger sequencing to identify the cause of Weaver syndrome and to characterise the molecular and clinical associations of the causative gene.
RESULTS
To identify the cause of Weaver syndrome we first undertook exome capture and sequencing in four individuals with classic features of the condition, using the Agilent SureSelect Human All Exon kits and Illumina GAIIx platform (cases 3, 10, 14 and 16, Table 1). We used NextGENe software to detect sequence variants as previously described [10]. To prioritise variants for consideration we first evaluated only variants with coverage of at least 15 reads and we excluded variants that were intronic, synonymous, recorded in dbSNP and/or were present in 45 exomes we performed in other conditions. After these filters, 1,357 variants remained. We applied a script to ascertain genes with variants in all four individuals. This identified only one gene, EZH2, with each case carrying a different EZH2 mutation (Table 1). We confirmed the four mutations by Sanger sequencing and also showed that the mutations were not present in the seven parental samples available for study, establishing that the mutations had arisen de novo in at least three of the individuals with Weaver syndrome.
Case ID |
Mutation; protein alteration |
Inheritance |
Heighta |
OFCa |
Learning disability |
Malignancy |
1 |
c.401T>C;p.M134T |
de novo |
+2.2 |
+0.3 |
mild |
|
2 |
c.466A>G;p.K156E |
familial |
+3.3 |
+1.5 |
mild |
|
3 |
c.836A>G;p.H279R |
de novo |
+2.6 |
+2.3 |
mild |
|
4 |
c.1876G>A;p.V626M |
de novo |
+3.6 |
+3.2 |
no |
|
5 |
c.1915A>G;p.K639E |
de novo |
+6 |
+4.9 |
moderate |
|
6 |
c.1987T>A;p.Y663N |
de novo |
+5 |
+3 |
mild |
|
7 |
c.1991A>T;p.D664V |
nk |
+3.8 |
+3.2 |
moderate |
|
8 |
c.2044G>A;p.A682T |
de novo |
+4.2 |
+1.8 |
mild |
Neuroblastoma ALL |
9 |
c.2050C>T;p.R684C |
de novo |
+2.9 |
nk |
mild |
|
10 |
c.2050C>T;p.R684C |
nk |
+3.5 |
+1.5 |
nk |
|
11 |
c.2050C>T;p.R684C |
de novo |
+3 |
+2.6 |
mild |
|
12 |
c.2050C>T;p.R684C |
nk |
+5.6 |
+1.7 |
mild |
|
13 |
c.2084C>T;p.S695L |
de novo |
+5 |
nk |
mild |
|
14 |
c.2199C>G;p.Y733X |
de novo |
+7.6 |
+2.7 |
no |
|
15 |
c.2196-15_2196-2delTTCCTGTTGTTTCA |
nk |
+4.6 |
nk |
nk |
|
16 |
c.2204_2211dupAGGCTGAT |
de novo |
+4.6 |
+2.2 |
moderate |
|
17 |
c.2230_2232dupATC |
nk |
+3.4 |
+1.7 |
mild |
|
18 |
c.2222A>G;p.Y741C |
de novo |
+5.3 |
+2 |
no |
|
19 |
c.2233G>A;p.E745K |
nk |
+3.5 |
+1.4 |
mild |
Lymphoma |
aThe height and OFC measurements are the standard deviations relative to the mean |
||||||
To further evaluate the role of EZH2 in human overgrowth conditions we sequenced the full coding sequence and intron-exon boundaries of the gene by Sanger sequencing in an additional 300 individuals (Supplementary Table 1). These cases either had a clinical diagnosis of Weaver syndrome, or a non-specific overgrowth syndrome which we defined as height and/or head circumference at least two standard deviations above the mean, together with variable additional phenotypic features.
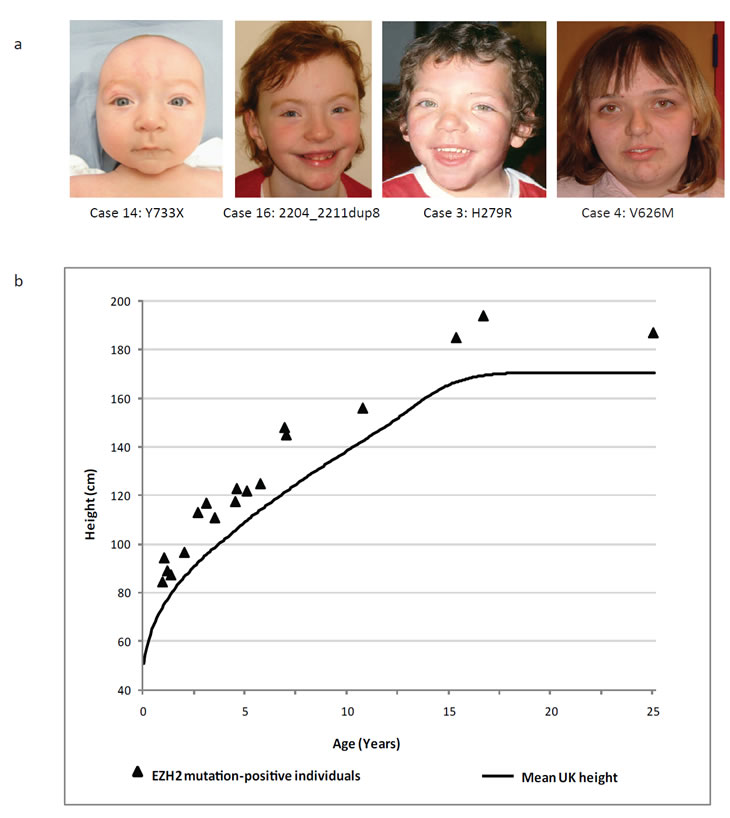
Figure 1: Facial features and height of EZH2 mutation-positive individuals. (a) Typical facial appearance of children with an EZH2 mutation and Weaver syndrome. (b) Height distribution of EZH2 mutation-positive individuals relative to the mean UK1990 height [22]
We identified mutations that we consider pathogenic in a further 15 individuals (Table 1, Figure 1, Figure 2, Supplementary Table 2). For nine individuals, analysis of parental samples demonstrated that the relevant mutation had arisen de novo, confirming pathogenicity. Case 2 had a family history of overgrowth and there was full segregation of the EZH2 mutation, K156E, with the overgrowth phenotype (Supplementary Figure 1). We identified one recurrent mutation, R684C, which was detected in four unrelated individuals. We were able to demonstrate that at least two had arisen de novo and therefore independently. This suggests that R684 is a mutational hotspot and likely reflects the increased mutability of the CpG site at this position. We believe the mutations 2230_2232dupATC, D664V, and E745K are pathogenic as they occur at highly conserved residues and were identified in individuals with a clinical diagnosis of Weaver syndrome. Parental samples were not available and thus it is possible that these are rare polymorphisms. However, analysis of 115 population controls through the full EZH2 sequence did not reveal either these, or any similar mutations, lending further support that they are pathogenic. The full list of EZH2 sequence variants we identified is given in Supplementary Table 2.
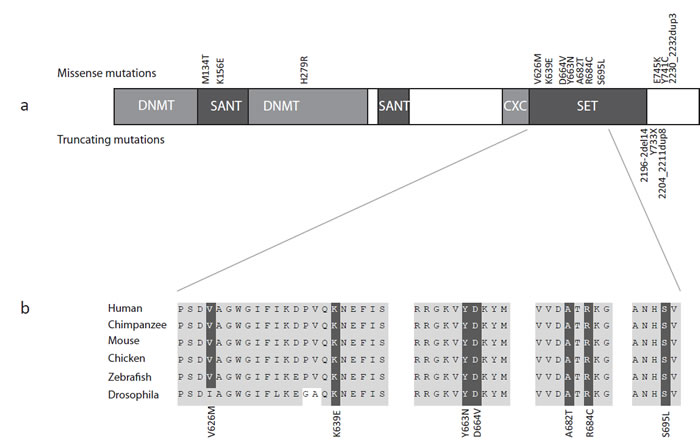
Figure 2: EZH2 structure, mutations and homology. (a) Schematic representation of the protein structure of EZH2 with domains and mutations. Missense mutations are represented above the protein and truncating mutations below the protein. (b) Across species homology of the EZH2 SET domain demonstrating missense mutations targeting conserved residues.
DISCUSSION
EZH2 (Enhancer of Zeste, Drosophila, homolog 2) encodes the catalytic component of the polycomb repressive complex 2 (PRC2), which epigenetically regulates chromatin structure and gene expression through trimethylation at H3K27 and recruitment of DNA methyltransferases, both of which act to repress transcription [11, 12]. EZH2 also has critical roles in stem cell maintenance and cell lineage determination, including osteogenesis, myogenesis, lymphopoiesis and hematopoiesis [13]. EZH2 contains a number of functional domains and it is noteworthy that the majority of the mutations we identified are missense mutations that target highly conserved residues within these domains (Figure 2).
Somatic gain-of-function and loss-of-function mutations in EZH2 have been reported in haematological malignancies [4, 5, 14, 15]. Activating somatic mutations at Y646, which increase H3K27 trimethylation, have been reported in 22% of diffuse large-cell B-cell lymphomas and in 7% of follicular lymphomas [14, 16]. Inactivating somatic EZH2 mutations have been reported in myeloid neoplasms such as poor prognosis myelodysplasia-myeloproliferative neoplasms (10-13%), myelofibrosis (13%) and various subtypes of myelodysplastic syndromes (6%) [4, 5, 15].
The mutations we identified in Weaver syndrome show considerable overlap with the mutations in myeloid malignancies. Indeed, three of the mutations, D664V, R684C and Y733X have also been detected as somatically acquired mutations in CMML and myelofibrosis [4]. Only two of the EZH2 mutation-positive individuals we report have developed malignancies; case 8 who carries a de novo missense mutation, A682T, in the SET domain, developed neuroblastoma and acute lymphoblastic lymphoma, both diagnosed at 13 months. Case 19 harbours a missense mutation, E745K, in the post-SET domain and developed lymphoma at 12 years. However, it should be noted that the oldest mutation-positive individual for whom we have follow up data is only 27 years old, and little long-term clinical data is currently available in Weaver syndrome. Given that the myeloid malignancies associated with somatic EZH2 mutations usually occur in later life it is possible that individuals with Weaver syndrome are at increased risk of myeloid and/or other malignancies.
Although there is overlap of the mutational spectra in Weaver syndrome and myeloid malignancies, there are also differences. The majority of mutations we identified are missense variants. We identified only three truncating mutations; all affect the last EZH2 exon and therefore may not initiate nonsense mediated RNA decay. By contrast, somatic truncating mutations have been reported throughout EZH2 [4, 5]. Additionally, germline mutations that result in full gene inactivation are predominantly truncating mutations that occur throughout the gene, for example NSD1 mutations in the overgrowth condition, Sotos syndrome [17]. These data suggest that the mutations in Weaver syndrome are not acting simply by generating haploinsufficiency. Larger, long-term studies of Weaver syndrome, together with functional analyses, should provide further insights into the nature of causative mutations and associated cancer risks.
There were consistent clinical features amongst the 19 individuals with EZH2 mutations. Most prominent was increased height. The height of all mutation-positive individuals was at least two standard deviations above the mean and in nine individuals it was greater than four standard deviations above the mean (Table 1, Figure 1). Increase in head circumference was less dramatic, in contrast to Sotos syndrome and the PTEN-associated syndromes in which macrocephaly is the more prominent and consistent manifestation of overgrowth (Table 1) [6, 17, 18]. Learning disability was frequent and usually in the mild-moderate spectrum, although some individuals had no reported learning difficulties. EZH2 mutation-positive individuals have a recognisable facial phenotype that includes a round face, high broad forehead, high hairline and hypertelorism (Figure 1). Although distinctive, indeed 16 of the 19 individuals we identified were referred with a clinical diagnosis of Weaver syndrome, overall the clinical characteristics can be relatively subtle and there may be considerable under-diagnosis of this condition.
EZH2 is the second histone methyltransferase associated with human overgrowth. Weaver syndrome shares many clinical similarities with the overgrowth condition, Sotos syndrome, which is due to mutations in the histone methyltransferase NSD1 [17]. Moreover, EZH2 is part of the PI3K/mTOR pathway which includes other genes that have been associated with dysregulated growth in humans [19]. For example, a somatic mutation in AKT1 causes Proteus syndrome [20] and germline PTEN mutations can cause macrocephaly and overgrowth [6, 18]. It is also of interest that these pathways were not strongly implicated in large-scale, genome-wide association studies of height [1]. This suggests that common variation in these genes may not contribute to human growth regulation and further emphasises the importance of studies of rare genetic syndromes in the explication of fundamental biological processes.
METHODS
Samples
Cases
Individuals with overgrowth were recruited through the Childhood Overgrowth Study. Informed consent was obtained from all participants and the research had approval from the London Multicentre Ethics Committee, (Reference: MREC/01/2/44). A full list of collaborators is given in the Supplementary Appendix. Cases were included in the EZH2 mutation screening if they had a clinical diagnosis of Weaver syndrome or if they had a non-specific overgrowth phenotype. Included amongst the individuals with Weaver syndrome were five who had previously been reported, cases 3, 5, 6, 7, and 17 [3, 21]. The group with non-specific overgrowth included individuals with global overgrowth, where both height and head circumference were at least two standard deviations above the mean, individuals with isolated macrocephaly where the head circumference was at least two standard deviations above the mean but height was below two standard deviations above the mean and individuals with height but not head circumference greater than two standard deviations above the mean. For figure 1, the height of EZH2 mutation-positive individuals was plotted with reference to the mean height which was calculated using sex-averaged data from male and female UK1990 growth charts, which was provided by the Child Growth Foundation [22].
Controls
DNA samples from UK controls were from the 1958 Birth Cohort, an ongoing follow-up of persons born in Great Britain in one week in 1958 which included a biomedical assessment during 2002-2004 at which blood samples and informed consent were obtained for creation of a genetic resource http://www.cls.ioe.ac.uk/studies.asp?section=000100020003.
Exome sequencing
The exome preparation and sequencing for cases 3, 10 and 16 was performed at Ambry Genetics Corp. (Aliso Viejo,CA, USA) using the Agilent SureSelect Human All Exon 38 Mb Kit. Each sample was run on two lanes of an Illumina Genome Analyzer IIx generating 2x76 bp reads. We performed the exome sequencing for case 14 in-house using the Illumina Genomic PE Sample Prep Kit (Ilumina, San Diego, CA, USA) and the Agilent SureSelect Human All Exon 50 Mb Kit. This sample was run on two lanes of an Illumina HiSeq Analyzer generating 2x100 bp reads.
Exome sequence analysis
We identified variants in the exomic sequence using NextGENe software version 2.10 (SoftGenetics, State College, PA, USA). We excluded variants that were intronic, synonymous, detected in a known pseudogene, recorded in dbSNP or detected in 45 in-house exomes from individuals with familial breast cancer that we have performed as part of a separate study. To prioritise evaluation of variants most likely to be real we first included only variants with coverage of at least 15 reads. In this final variant set we ran a script to identify genes with variants in all four individuals.
EZH2 mutation analysis
We performed Sanger sequencing of PCR products from genomic DNA to confirm the mutations identified by exome sequencing, and to mutationally analyse the full coding sequence in the overgrowth series. We designed PCR primers to amplify the 19 coding exons and intron-exon boundaries of EZH2 in 3 multiplex PCR reactions (Supplementary Table 1). The PCR was carried out using a Qiagen Multiplex PCR kit according to the manufacturer’s instructions. Products were sequenced with the original PCR primers or internal sequencing primers (exons 3 and 20) using the BigDye Terminator Cycle Sequencing Kit and an ABI 3730 Genetic Analyzer (Applied Biosystems, Foster City, CA,USA). Sequences were analyzed using Mutation Surveyor software v3.97 (SoftGenetics, State College, PA, USA). All mutations were confirmed by bidirectional sequencing of a second, independently amplified PCR product.
In silico analyses of identified variants
We computed the predicted effects of EZH2 nonsynonymous variants on protein function using PolyPhen [23] and SIFT [24]. All variants (intronic and coding) were analysed for their potential effect on splicing. Variants were analysed using two splice prediction algorithms NNsplice [25] and MaxEntScan, [26] via the Alamut software interface (Interactive Biosoftware). If both NNsplice and MaxEntScan scores were altered by >20% (i.e. a wildtype splice-site score decreases and/or a cryptic splice-site score increases) three further prediction algorithms were utilised; NetGene2 [27], HumanSplicingFinder [28] and Genscan [29]. A consensus decrease in a wildtype splice-site score and/or a consensus increase in a cryptic splice-site score across all algorithms was considered indicative of disruption of normal splicing. To evaluate the conservation of variants we used the HomoloGene system for automated detection of homologs among the annotated genes of completely sequenced eukaryotic genomes. http://www.ncbi.nlm.nih.gov/homologene/37926.
ACKNOWLEDGMENTS
We thank the families and the physicians and nurses that recruited them, for their participation in this study which was funded by the Institute of Cancer Research. We thank Deborah Hughes and Bernadette Ebbs for assistance in DNA extraction and running the ABI sequencers. We thank Ann Strydom for assistance with the manuscript. The case samples were collected through the Childhood Overgrowth Collaboration, the full list of contributors is given in the Supplementary Appendix. We acknowledge NHS funding to the ICR/RMH NIHR Specialist Biomedical Cancer Research Centre.
Accession codes
EZH2 mutation nomenclature corresponds to Ensembl Transcript ID ENST00000337432.
URLs
dbSNP: ncbi.nlm.nih.gov/projects/SNP/
HomoloGene: ncbi.nlm.nih.gov/homologene/37926.
Mutation Surveyor and NextGENe software: softgenetics.com
AUTHOR CONTRIBUTIONS
N.R. and K.T.B. designed the experiment. K.T.B. undertook phenotypic assessment and case stratification. S.H., A.M. and J.D. performed exome sequencing. E.R. K.S. and J.D. performed data management and analysis. J.D., S.H., S.DVD., E.R.P., E.Ra., S.S. and C.L. performed EZH2 mutation analysis. A.Z., K.T.B. and N.R. coordinated case recruitment to the Childhood Overgrowth Study; the full list of collaborators is given in the Supplementary Appendix. S.B., C.C., F.F., A.M., V.McC., M.P., W.R., J.R., M.S., C.T.,V.S., P.W., I.K.T., K.T.B. and T.C. provided samples and clinical information. K.T.B. and N.R. wrote the manuscript with input from J.D. and other authors. N.R. oversaw and managed all aspects of the study.
REFERENCES
1. Lango Allen H, Estrada K, Lettre G, Berndt SI, Weedon MN, Rivadeneira F, Willer CJ, Jackson AU, Vedantam S, Raychaudhuri S, Ferreira T, Wood AR, Weyant RJ, Segre AV, Speliotes EK, Wheeler E et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature. 2010; 467:832-838.
2. Weaver DD, Graham CB, Thomas IT, Smith DW. A new overgrowth syndrome with accelerated skeletal maturation, unusual facies, and camptodactyly. J Pediatr. 1974; 84:547-552.
3. Cole TR, Dennis NR, Hughes HE. Weaver syndrome. J Med Genet. 1992; 29:332-337.
4. Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, Waghorn K, Zoi K, Ross FM, Reiter A, Hochhaus A, Drexler HG, Duncombe A, Cervantes F, Oscier D, Boultwood J et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010; 42:722-726.
5. Chase A, Cross NC. Aberrations of EZH2 in cancer. Clin Cancer Res. 2011; 17:2613-2618.
6. Patton M: Genetic and Dysmorphic syndromes of Tall Stature. In: Growth Disorders - Pathophysiology and Treatment Edited by Kelnar, Stirling, Savage, Second edn: Chapman & Hall; 1998.
7. Dumic M, Vukovic J, Cvitkovic M, Medica I. Twins and their mildly affected mother with Weaver syndrome. Clin Genet. 1993; 44:338-340.
8. Fryer A, Smith C, Rosenbloom L, Cole T. Autosomal dominant inheritance of Weaver syndrome. J Med Genet. 1997; 34:418-419.
9. Proud VK, Braddock SR, Cook L, Weaver DD. Weaver syndrome: autosomal dominant inheritance of the disorder. Am J Med Genet. 1998; 79:305-310.
10. Snape K, Hanks S, Ruark E, Barros-Nunez P, Elliott A, Murray A, Lane AH, Shannon N, Callier P, Chitayat D, Clayton-Smith J, Fitzpatrick DR, Gisselsson D, Jacquemont S, Asakura-Hay K, Micale MA et al. Mutations in CEP57 cause mosaic variegated aneuploidy syndrome. Nat Genet. 2011; 43:527-529.
11. Cao R, Zhang Y. The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr Opin Genet Dev. 2004; 14:155-164.
12. Vire E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, Morey L, Van Eynde A, Bernard D, Vanderwinden JM, Bollen M, Esteller M, Di Croce L, de Launoit Y, Fuks F. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006; 439:871-874.
13. Chou RH, Yu YL, Hung MC. The roles of EZH2 in cell lineage commitment. Am J Transl Res. 2011; 3:243-250.
14. Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F, Yap D, Humphries RK, Griffith OL, Shah S, Zhu H, Kimbara M et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010; 42:181-185.
15. Nikoloski G, Langemeijer SM, Kuiper RP, Knops R, Massop M, Tonnissen ER, van der Heijden A, Scheele TN, Vandenberghe P, de Witte T, van der Reijden BA, Jansen JH. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet. 2010; 42:665-667.
16. Sneeringer CJ, Scott MP, Kuntz KW, Knutson SK, Pollock RM, Richon VM, Copeland RA. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc Natl Acad Sci U S A. 2010; 107:20980-20985.
17. Tatton-Brown K, Douglas J, Coleman K, Baujat G, Cole TR, Das S, Horn D, Hughes HE, Temple IK, Faravelli F, Waggoner D, Turkmen S, Cormier-Daire V, Irrthum A, Rahman N. Genotype-phenotype associations in Sotos syndrome: an analysis of 266 individuals with NSD1 aberrations. Am J Hum Genet. 2005; 77:193-204.
18. Mester JL, Tilot AK, Rybicki LA, Frazier TW, 2nd, Eng C. Analysis of prevalence and degree of macrocephaly in patients with germline PTEN mutations and of brain weight in Pten knock-in murine model. Eur J Hum Genet. 2011; 19:763-768.
19. Dazert E, Hall MN. mTOR signaling in disease. Curr Opin Cell Biol. 2011.
20. Lindhurst MJ, Sapp JC, Teer JK, Johnston JJ, Finn EM, Peters K, Turner J, Cannons JL, Bick D, Blakemore L, Blumhorst C, Brockmann K, Calder P, Cherman N, Deardorff MA, Everman DB et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med. 2011; 365:611-619.
21. Greenberg F, Wasiewski W, McCabe ER. Weaver syndrome: the changing phenotype in an adult. Am J Med Genet. 1989; 33:127-129.
22. Child Growth Foundation UK: The British 1990 growth reference. Revised September 1996. In.: Child Growth Foundation, 2 Mayfield Avenue, London W4 1PW, UK.
23. Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002; 30:3894-3900.
24. Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001; 11:863-874.
25. Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. J Comput Biol. 1997; 4:311-323.
26. Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol. 2004; 11:377-394.
27. Brunak S, Engelbrecht J, Knudsen S. Prediction of human mRNA donor and acceptor sites from the DNA sequence. J Mol Biol. 1991; 220:49-65.
28. Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009; 37:e67.
29. Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J Mol Biol. 1997; 268:78-94.