
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDA) has a poor prognosis, with 5-year overall survival of approximately 6% [1-4]. PDA is often diagnosed at a late stage, at which point a curative resection is no longer possible and the approved systemic therapies have a relatively modest impact on survival. Despite recent introduction of more effective chemotherapy regimens including FOLFIRINOX and combination of gemcitabine with Nab-paclitaxel, veritably all patients with advanced disease ultimately progress [5, 6]. The only approved targeted therapy, erlotinib in combination with gemcitabine, has added approximately 4 weeks to patient’s overall survival, while other targeted agents including MEK, PI3K and mTOR inhibitors, have failed to demonstrate any efficacy in clinical studies [6-8]. Thus, there is an urgent and unmet need to develop rational, targeted approaches for the treatment of pancreatic cancer.
Genetic analysis of PDA has defined multiple oncogenic events that drive the initiation and progression of disease [9, 10]. Approximately 90% of pancreatic cancers are driven by KRAS, while the loss of the TP53 and SMAD4 tumor suppressors occurs in a large fraction of tumors. Unfortunately, these genetic events remain largely non-actionable due to the lack of drugs that selectively target these oncogenic events. An additional signature genetic/epigenetic event in PDA is the loss or silencing of the tumor suppressor CDKN2A [11]. Based on genetic analysis, mutation or homozygous deletion of CDKN2A occurs in ~50% of tumors, and epigenetic silencing is also frequently observed. Thus, the vast majority of PDA cases have functional loss of this tumor suppressor [8, 12].
The CDKN2A gene encodes the p16ink4a protein, which is a potent physiological inhibitor of CDK4/6 [13]. During cell cycle progression, CDK4/6 is activated downstream of multiple signaling cascades, and is required to initiate the phosphorylation of RB and subsequent entry into S-phase [14, 15]. The levels of p16ink4a are stimulated during specific stresses, leading to a blockade of proliferation and inducing features of senescence [13]. In particular, p16ink4a serves as a critical mediator of oncogene-induced senescence driven by KRAS mutation; therefore, there is strong selective pressure for its loss in PDA [16, 17]. Consequently, it is believed that the loss of p16ink4a is a key hallmark of RAS driven tumors and is frequently observed in a host of tumors with deregulation of the KRAS/RAF oncogenic singnaling pathway including non-small cell lung cancer, melanoma and colorectal cancer, in addition to PDA [18-20].
Since p16ink4a functions as an inhibitor of CDK4/6, it would be anticipated that this cell cycle regulated kinase family would be an important and actionable target in PDA. Over the last several years, drugs specifically targeting CDK4/6 have been developed [21, 22]. Presently, three highly-specific agents are now amenable to clinical use (PD-0332991, LEE-011, and LY2835219). These agents induce cell cycle arrest and delay progression/proliferation in tumor models [23-25]. This has been observed across a host of models and disease indications, including leukemias, breast cancer, and pediatric tumor types [21]. Surprisingly, in work published by several groups, single agent CDK4/6 inhibition had a particularly weak effect in models of pancreatic cancer [26-28]. Given the genetics of the disease, we sought to use patient-derived models to better delineate the significance of CDK4/6 inhibition in the treatment of PDA.
RESULTS
Established models of PDA have a relatively weak response to CDK4/6 inhibition
In order to evaluate the response to CDK4/6 inhibition, several well-established PDA cell lines were employed. Treatment with 1 µM PD-0332991 resulted in a modest suppression of proliferation, as measured by 5-bromo-2deoxyuridine (BrdU) incorporation, in established pancreatic cancer cell lines at 24 hours of treatment (Figure 1A). In these same cell lines, crystal violet staining after 7 days of treatment demonstrated variable inhibition of tumor cell growth amongst the PDA established lines, with the PANC1 line appearing relatively sensitive to CDK4/6 inhibition and the MIA-PACA2 line appearing relatively resistant (Figure 1B). Interestingly, BrdU incorporation in cell lines treated with PD-0332991 over a span of 2 weeks demonstrated proliferation in the presence of the agent, suggesting a durable response is not achieved in these models and there is rapid progression to a resistance (Figure 1C). To determine if this phenotype was selective to cells in culture, PL-45 and PL-5 xenografts were developed. The mice were treated with PD-0332991 via oral gavage. The treatment with PD-0332991 did elicit suppression of tumor growth in PL-5 xenografts, with tumor volume in the treatment group significantly lower than that of controls (p < 0.0001). However, the suppression of residual tumor proliferation was relatively limited and even in the presence of daily dosing, a proliferative index of 79.6% in PL-5 xenografts and 40.6% in PL-45 xenografts was observed (Figure 1D). Together, these data in established models is consistent with recently published studies that suggests that monotherapy with CDK4/6 inhibitors may not be particularly effective in PDA [26-28].
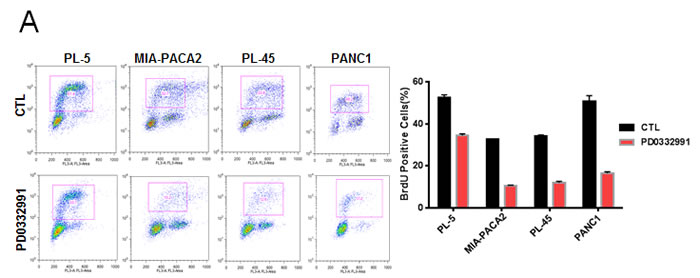
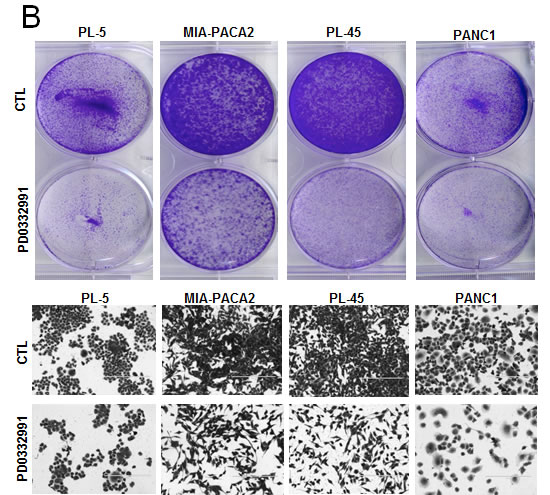
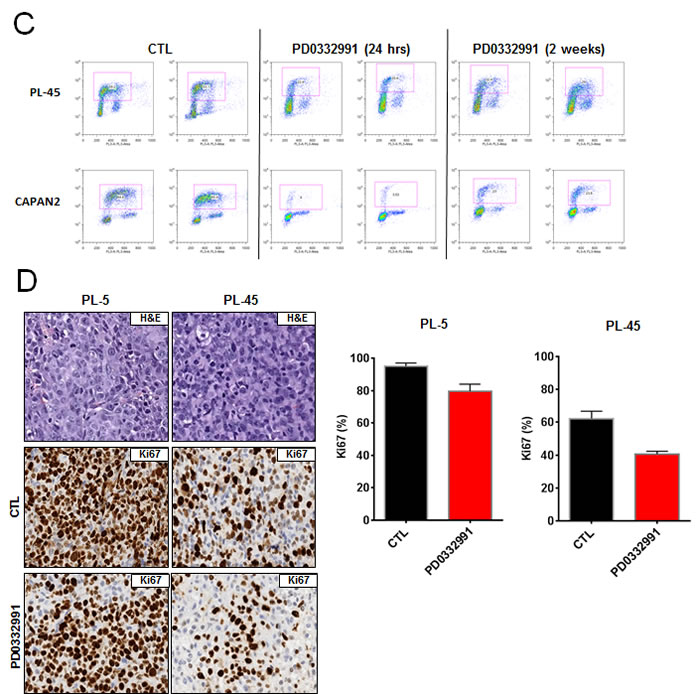
Figure 1: CDK4/6 inhibition yields modest efficacy in established models of PDA: A. Representative plots of BrdU incorporation in established PDA cell lines treated with PD-0332991 (1 μM) versus control (left panel). Quantification by flow cytometry of BrdU incorporation between control and PD-0332991 treated established cell lines (right panel). B. Crystal violet staining of established PDA cell lines after 7 days of treatment with control vs. PD-0332991 (upper panel). Photomicrographs of PDA cells in culture after 7 days of treatment with control vs. PD-0332991 (bottom panel). C. Representative BrdU incorporation after 24 hours and after 2 weeks of treatment with PD-0332991. D. H&E and Ki-67 staining of established PDA cell line xenografts treated with control and PD-0332991. Quantification of Ki-67 staining in established PDA cell line xenografts treated with PD-0332991 and control.
Explants cultures recapitulate the diversity of PDA
Many established cell lines have experienced selection for growth in culture, which is known to selectively perturb cell cycle regulatory mechanisms [29]. Furthermore, the desmoplastic architecture of PDA contributed by neighboring stromal cells is known to be particularly important for its biology [30, 31]. Therefore, we sought to develop a primary tumor explant model as a means to accurately recapitulate key features of PDA. The protocol used here was based on prior work with breast cancer specimens [32]. Fresh surgical tissue, not required for diagnosis, was dissected into approximately ~1 mm3 sections. These were cultured on a semi-solid support from 0-192 hours, during which time drugs could be applied. Explant cultures where then removed, fixed, and stained for relevant markers. After the establishment of this methodology, 15 unselected, consecutive PDA specimens were studied as explants (Figure 2A). The clinical and histopathological data of the subjects are summarized in Supplemental Figure 1. All explants retained tissue architecture and degrees of cellularity comparable to that observed in the tumor section obtained for clinical pathologic evaluation (Figure 2B). Morphological and architectural integrity of tissues was maintained in tumor samples for up to 192 hours of culture (Supplemental Figure 2A). The proliferative index was assessed by Ki67 immunostaining on day 1, 4, 6 and 8. The percentage of proliferating cells was stable across all time points, without a significant decrease up to 192 h (Supplemental Figure 2A). The number of apoptotic cells, visualized using cleaved caspase 3 staining also did not change significantly over time (Supplemental Figure 2A). The level of key molecular markers (p16ink4a, p53, EGFR) in the pathogenesis of PDA were evaluated in both primary tumor and explant, with representative staining between the surgical specimen and the cultured explants as shown (Figure 2B). P16ink4a loss or weak expression was seen in 14 of 15 cases (93%); strong p16 expression was seen in only one case (Case #5; Figure 2B). The TP53 tumor suppressor gene is frequently inactivated in PDA, with approximately 50% of these carcinomas showing intragenic mutation. Strong nuclear labeling for the p53 protein, which correlates with mutation stabilizing p53 protein, was present in 67% of primary tumors and corresponding explants cultures (Figure 2C). Strong membranous EGFR expression (score 3+), which stimulates downstream MAPK and PI3K/AKT signaling cascades to influence cell proliferation and metastasis [33], was seen in 5 of 15 (33.3%) of primary tumors and explants cultures. These findings demonstrate that ex vivo explants maintain the overall biomarker phenotype of the tumor. Given the theoretical possibility of high intra-explant variability, we evaluated six explant cases from duplicate and triplicate culture wells at 48 hours. As shown in Supplemental Figure 2B, there was significant concordance between multiple explants from a given case. To determine if the explant cultures retained the same proliferative nature of the primary tumor, Ki67 was evaluated in parallel. Among primary tumors, a range from 21 to 89.5% of Ki67 positive tumor cells was observed (Figure 2C). There was a strong positive correlation in Ki67 index between the original tumor and the corresponding explanted tissue in all cases (R2 = 0.97, p < 0.001). These data indicate that the ex vivo explants retained the proliferative potential similar to that of the primary tumor.
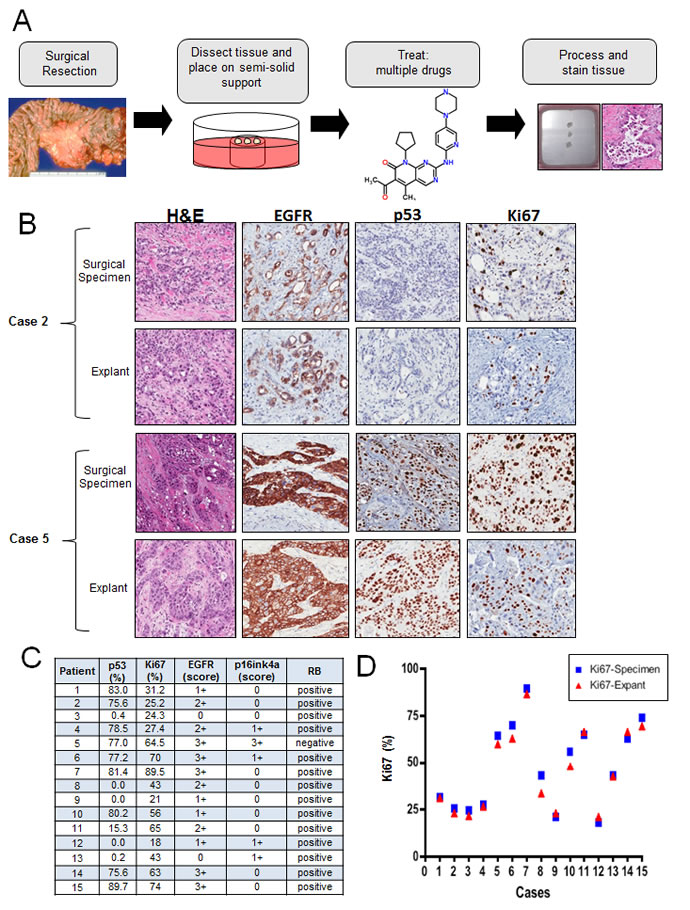
Figure 2: Primary tumor explants recapitulate multiple biological features of the disease: A. Schematic representation of the explant approach. B. Representative hematoxylin/eosin, EGFR, p53 and Ki67 staining between surgical specimens and tumor explants. C. Table summarizing the status of EGFR, p16, p53, RB and Ki67 index for the cases analyzed. D. Quantification of Ki67 staining from matched explant and surgical specimens demonstrating concordance.
CDK4/6 inhibition has potent activity in patient explants
Preservation of tissue architecture, viability and molecular phenotype suggested that treatment of explant cultures with therapeutic agents could reveal sensitivities that would be concordant with the primary tumor. In PDA explants, treatment with PD-0332991 showed profound suppression of Ki67 staining (Figure 3A and 3B). This effect was specific to the drug and was not observed in tissues treated with DMSO (vehicle) for the same period or with gemcitabine (Supplemental Figure 3A). Interestingly, there was some heterogeneity in the response to PD-0332991. Specifically, 13 of the 15 cases exhibited a greater than 5-fold suppression of Ki67 upon exposure to PD-0332991, and post-treatment Ki67 index below 7% (Figure 3B). In contrast, one case maintained a Ki67 index of 16.5% post-treatment, in spite of a greater than 5-fold reduction (case #7), and one case showed no reduction in Ki67 staining (case #5). Of note, the response to PD-0332991 did not depend on p53 status but was specific to the tumor cells, since lymphocytes present within the explants effectively responded as evidenced by suppression of Ki67 (not shown). Thus, the failure to respond to PD-0332991 appears to be a consequence of tumor-specific genetic events, and not due to distinctions in intrinsic drug sensitivity of a given individual. Since preclinical data suggest that an intact RB pathway is required for the cytostatic response to PD-0332991 [34-37], we further investigated the activation status of the RB pathway by immunohistochemical stains. Tumors that are RB deficient expressed exceedingly high levels of p16ink4a with high Ki67 proliferation index [13, 32]. Both the primary tumors and explants were stained for p16ink4a and RB. Within the cohort of evaluated cases, only one case exhibited high levels of p16ink4a and low RB (Case #5; Figure 3C). RB and p16ink4a expression was conserved between the primary tumors and the explants. These data indicate that the vast majority of PDA is responsive to PD-0332991 and only the infrequent loss of RB is associated with lack of response.
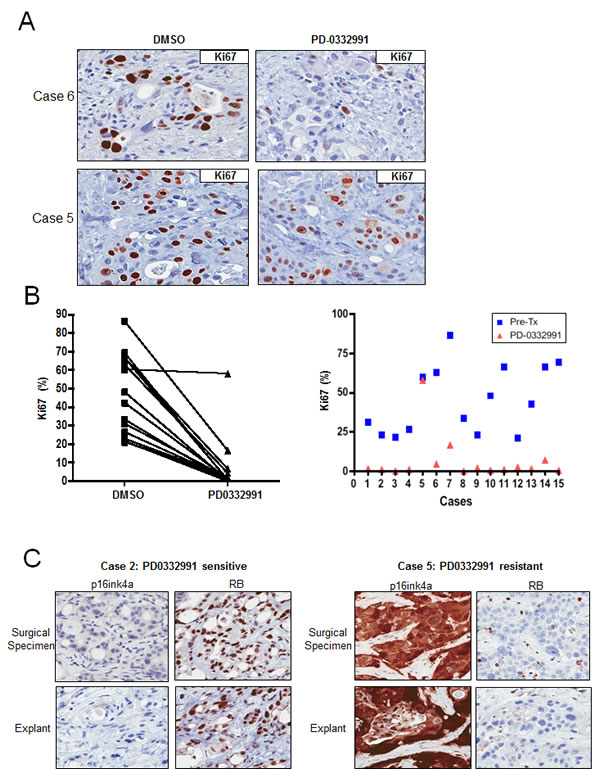
Figure 3: Tumor explants exhibit sensitivity to PD-0332991: A. Representative staining of Ki67 in drug-treated explants that exhibited significant response or lack of response (bottom panel) to PD-0332991 treatment (1 uM). B. Response to PD-0332991 across all explant cases evaluated. C. Representative staining for RB and p16ink4a in cases that responded (left panel) or did not respond (right panel) to PD-0332991.
Suppression of tumor growth in patient derived xenografts
Though tumor explants provide a striking recapitulation of the primary tumor, we sought to complement this approach with patient-derived xenografts to obtain in vivo data over a longer treatment period. In this model, fragments of primary tumor are subcutaneously implanted in NSG mice, allowing the tumors to grow and subsequently passaged into other NSG mice for expansion into treatment cohorts (Figure 4A). Unlike the PL-45 or PL-5 xenografts that harbor little tissue architecture reminiscent of PDA, the three patient-derived xenograft exhibited histologic architecture remarkably similar to their tumor of origin (Figure 4B). Treatment of the cohort with PD-0332991 resulted in marked suppression of tumor growth and in some cases a reduction in tumor volume (Figure 4C). Analysis of the tissue treated with PD-0332991 revealed complete suppression of Ki67 across all three PDX cohorts (Figure 4D). This profound anti-proliferative effect on Ki67 was not seen in the PDX cohort treated with gemcitabine similar to the observations in explant culture (Supplemental Figure 3B). Each tumor had a low baseline of apoptosis, as observed with cleaved caspase 3, and PD-0332991 either had no effect or elicited a slight increase in tumor cell turnover (Figure 4E). Together, these data indicate that CDK4/6 suppression is particularly effective in limiting the proliferation of patient-derived models of resectable disease.
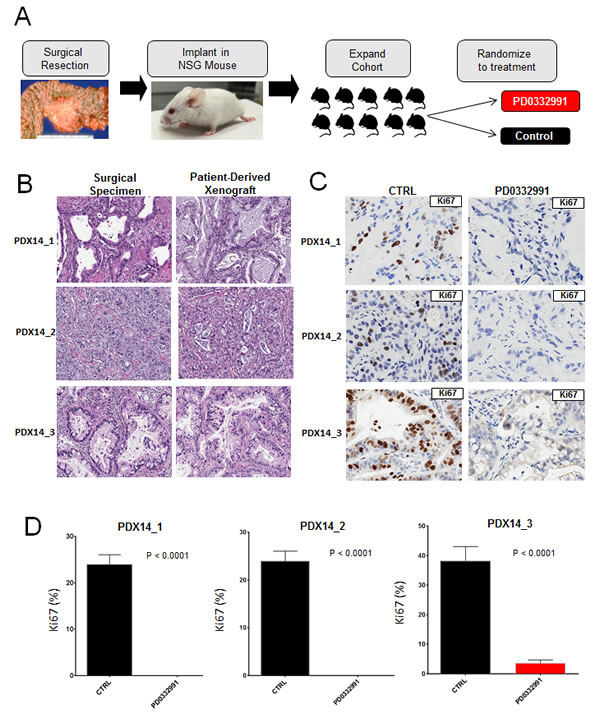
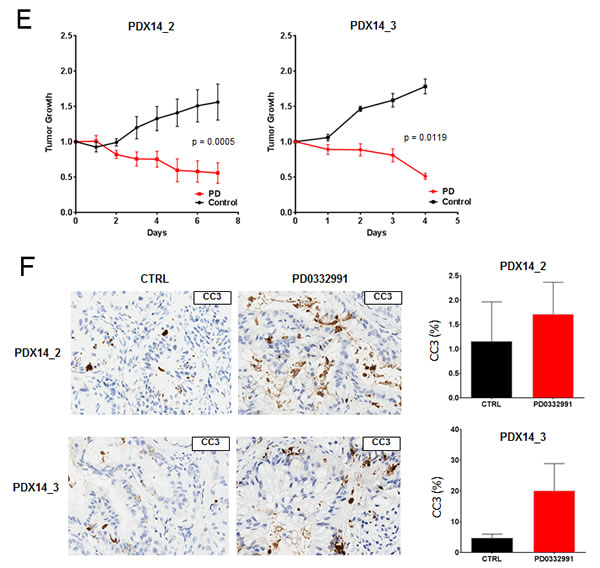
Figure 4: Patient-derived xenografts recapitulate primary tumor and demonstrate significant vulnerability to CDK4/6 inhibition: A. Schematic representation of the patient-derived xenograft approach. B. Representative hematoxylin/eosin staining between surgical specimens and patient-derived xenografts. C. Representative staining of Ki67 in control and PD-0332991 treated PDXs. D. Quantitation of Ki67 in the indicated PDX. E. Tumor growth of PDXs, normalized to initial tumor volume, while treated with PD-0332991 or control. F. Representative staining of cleaved caspase 3 in both control and PD-0332991 treated PDXs.
DISCUSSION
Treatment of PDA continues to present a significant challenge and in spite of multiple preclinical studies there has been only modest improvement in survival, with the principle gains coming from cytotoxic chemotherapies [1]. The vast majority of preclinical studies supporting drug development are carried out in established cell line models grown in culture or as xenografts. While facilitating rapid analysis, these models are not entirely representative of PDA. Many of these models, as shown here, grow very rapidly in cell culture and correspondingly produce xenografts with higher Ki67 indices than those reported in clinical PDA samples. The average Ki67 index in primary PDA is ~30% and the tumor exhibits an organized glandular architecture [38]. Additionally, the tumor is dominated by desmoplastic stroma that has recently emerged as a tumor suppressive feature of the disease though it also inhibits drug uptake [30, 39-41]. Despite the established models harboring genetic alterations that would be expected to confer substantial sensitivity to targeting CDK4/6, xenografts derived from established cell lines rapidly progress. This may reflect decades of selection for cell cycle deregulation in culture conditions. As result, high proliferation index and possibly cell cycle deregulation in these models can simply overwhelm cytostatic/targeted therapies through mechanisms that may not reflect primary tumor biology. While our study focused on CDK4/6 inhibition, cell cycle responses influence the activity of a range of targeted agents [42, 43], and this observation could be relevant to many targeted drug therapies.
It has become well accepted that tumors regulate the development of surrounding stroma through the induction of growth factor receptors and the production of their ligands [41, 44, 45], and that there is a degree of reciprocity involved in this communication, i.e., tumor stroma can influence tumor epithelial cells. The mutual interaction between tumor cells and surrounding environment appears particularly significant in PDA, where stromal cell promote tumor development and invasiveness but, in some cases, also limit rampant tumor cell growth [30, 46-48]. Thus, it is of importance to develop models that recapitulate these complex interactions. We employed two models that allowed us to study therapeutic efficacy from patient specimens: ex vivo tissue explants cultures and patient-derived xenografts. The histological features of both models are highly distinct from the fairly uniform collection of cells seen in xenografts established through injections of tumor lines such as PL-45 and PL-5. However, each of these models has intrinsic advantages/disadvantages. The explant model provides a window into the intact tumor of the patient. It fully retains human tumor-stroma interactions, which allows for studying the activity of anticancer drugs and small molecule inhibitors in the context of an individual patient’s tumor biology-in some ways reproducing the effect of performing serial biopsies to assess tissue response to therapy. In spite of the desirable features of the explant model, it does not recapitulate mechanisms of drug delivery, metabolism, and in vivo physiology. The PDX models employed accurately recapitulate the histological features of the pancreatic cancer from which they were derived and allow the study of tumor growth and inhibition over time. In the era of personalized medicine, these models could allow genetic alterations in tumor cells to be targeted within the context of their complex interactions with the stromal compartment.
Targeting cell cycle control is an attractive anti-tumor strategy, with the goal of preventing disease progression. The vast majority of human tumors exhibit some form of cell cycle deregulation, allowing tumors to proliferate unabated. Multiple aberrations affect the G1/S transition, including the amplification of D1 cyclin, deletions and mutations of RB and/or downregulation of CDK4/6 inhibitor p16ink4a [49, 50]. All these events lead to loss of proliferative control, with p16ink4a inactivation being exceedingly common in PDA [3, 13, 51]. Thus, it was unexpected that multiple established pancreatic cancer cell models would exhibit a relatively weak response to pharmacological CDK4/6 inhibit. In contrast, all explants that retained RB protein and showed low p16ink4A staining exhibited decrease in Ki67 proliferation index upon treatment with the CDK4/6 inhibitor PD-0332991. While RB loss has not been previously reported in pancreatic cancer cell models, the data here demonstrates that in some PDA cases functional expression of RB is lost. These data also indicate that the analyses of p16ink4a and RB status can be used prospectively to evaluate the response to CDK4/6 inhibitors in clinical specimens, and suggests an approach for developing markers for targeted therapies prior to clinical trials. Moreover, cytostatic response to PD-0332991 was confirmed in PDX models, lending further support to the possibility that CDK4/6 inhibitors may have potent activity in PDA.
A key question is whether the distinct response to PD-0332991 between established and patient-derived models reflects intrinsic PDA biology. While established models were initially developed from PDA lesions, some originated from metastatic disease, and most have been serially cultured for long periods of time, wherein mutations that enhance proliferation are progressively selected. It remains unclear if changes acquired during passaging in vitro mirror mutations occurring in primary tumor post chemotherapy and/or during progression to metastatic disease. The work herein demonstrates that our patient-derived models of PDA share remarkably similar biology to their pancreatic tumor of origin, and that in these models, CDK4/6 inhibition leads to potent suppression of tumor growth. Despite limited efficacy in established cell line models, this data suggests that CDK4/6 inhibitors should be considered for inclusion into emerging pancreatic cancer clinical trials.
MATERIALS AND METHODS
Cell culture, derivation and treatment
The established cell lines PL-45, MIA-PACA2, PL-5, PANC1, and CAPAN2 were cultured in DMEM supplemented with 10% FBS, as previously published [28]. Cells were treated with 1 µM CDK4/6 inhibitor PD-0332991, with drug replenished every 72 hours. Acute and durable responses to PD-0332991, measured at 24 hours and 2 weeks respectively, were assessed by 5-bromo-2-deoxyuridine (BrdU) incorporation and measured by flow cytometry. Following conclusion of treatment, PDA cell models were fixed in ice-cold 70% ethanol (in PBS) for 2 hours at 4ºC. Fixed cells were incubated with 2 M HCl and 0.3 mg/ml pepsin for 30 minutes at room temperature and neutralized with 0.1 M sodium borate (pH 8.5) for 2 minutes. Following neutralization, cells were washed with IFA buffer (10 mM HEPES [pH 7.4], 25 mM NaCl, 4% fetal calf serum) and then incubated in IFA containing 0.5% Tween for 10 min at room temperature. Cells were pelleted and re-suspended in FITC-anti-BrdU diluted in IFA buffer (1:10) (BD Biosciences, San Jose, CA) for 1 hour at room temperature. After antibody incubation, cells were washed once in IFA and incubated for 10 min on ice in 0.5 ml of PBS containing 20 μg/ml PI and 40 μg/ml RNase. Cells were then analyzed by FACSCalibur (BD Biosciences) instrument to determine PI (red) and FITC (green) staining for nuclear DNA and BrdU content, respectively.
Tissue preparation and explant cultures
PDA tumor tissue from resected surgical specimens, which was not needed for diagnostic purposes, was used. The collection of tissue was approved by the institutional review board (IRB). Tissues collected for explants culture were placed into 10 mL of complete explant culture medium for transport to the laboratory. Explant culture media consisted of RPMI 1640 base (Sigma) supplemented with 10% fetal calf serum (FCS). Antibiotic/antimycotic solution was added to a final concentration of 1 X (Sigma). Hydrocortisone (Sigma) was re-suspended in 95% EtOH and supplemented at a concentration of 1mg/100ml media. Lastly, recombinant human insulin was added at a final concentration of 1mg/100ml media (Gibco). Tissues were maintained in this medium at 4ºC until processing. All source tissues were plated as explants within 12 hours. Under sterile conditions and one hour prior to tissue dissection, 1 cm3 hemostatic gelatin dental sponges (Vetspon, Novartis) were hydrated in explant culture media at 37ºC. While the sponges were hydrating, tissues were transferred to a 10 cm cell culture dish with 10 ml complete explant media for dissection. Using a sterile scalpel and microdissection scissors, any apparent fat tissue was removed from the surface of the specimen and discarded. Samples were then cut into 1 mm3 pieces. Upon complete hydration of gelatin sponges, each was transferred to individual wells of a 12-well cell culture dish, and 2.5 ml of complete explant media was added to each well. One mm3 dissected specimen sections were chosen at random and placed on top of each sponge using sterile forceps. Tissues were placed near the edges of the sponge to allow for media perfusion in between tissue sections. Explant cultures were incubated at 37ºC and 5% CO2.
Each treatment condition was examined with three pieces of tissue per sponge. Fifteen independent PDA cases were evaluated. Only 12 explant cultures were evaluated for response to gemcitabine due to limited tissue. Following plating, explants were allowed to recover in explant media for 24 hours. Explants were then treated for 48 hours with vehicle control (DMSO), a selective CDK4/6 inhibitor PD-0332991 (1 μM), or gemcitabine (20 nM). Explants were removed from culture, fixed and processed for immunohistochemical analyses.
Histology and immunohistochemical stains
The original tumor tissue and the explant of the tumor tissue were fixed in 10% neutral-buffered formalin and embedded in paraffin. Four microm sections were stained with H&E for the histological evaluation. For immunohistochemical analysis four micron paraffin sections were prepared, deparaffinized in xylene, and rehydrated with graded ethanol. Antigen retrieval was performed by microwave pretreatment. The following antibodies were employed for the analysis: Ki67 (clone 30-9, dilution, Ventana Medical Systems), p53 (clone DO-7, dilution 1:100, DAKO), p16ink4a (clone E6H4, dilution 1:50, MTM Laboratories), RB (clone 1F8, dilution 1:50, Thermoscientific), EGFR (clone 5B7 prediluted, Ventana Medical Systems) and cleaved caspase 3 (clone 5A1E, dilution 1:300, Cell Signaling). All stains were performed using the BenchMark XT Slide Preparation System (Ventana Medical Systems).
Automated image analyses and scoring
Immunohistochemically stained primary tumor and explant slides were scanned using an Aperio ScanScope®CS instrument at 20X, (Aperio Technologies, Vista, CA, USA). Stains were quantified using analysis tools developed by Aperio. Nuclear algorithms were used to evaluate Ki67, p53 and RB stain; membranous and cytoplasmic algorithms were applied for EGFR and p16ink4a quantification, respectively. The mean staining intensity and percentage of staining cells were recorded for Ki67, p53, RB and p16ink4a stains. Immunolabeling for p53 was interpreted as positive and predictive of TP53 mutations if >75% tumors cell showed strong positive staining. RB expression was scored as negative (cancer cells showed no staining while normal cells were positive), weak (mean staining intensity was less than adjacent normal cells), or strong (staining intensity was equal to adjacent normal cells). RB loss was defined as a score of either negative, or weak. For p16ink4a the staining was graded as 0 (no cells staining), 1+ (diffuse weak cytoplasmic expression or < 25% cell showing strong staining), 2+ (25% to 75% of cells with strong staining), and 3+ (> 75% cells showing strong staining). A 3+ score was considered high expression. For EGFR the score (range 0-3) was reported.
Patient-derived xenografts
NOD-SCID IL2Rgammanull (NSG) mice were maintained in a barrier facility under HEPA-filtered air with food and water available as desired. Food, water and cage bedding were sterilized prior to use. Mice were manipulated under sterile conditions during surgery. Animal experiments fulfilled National Institutes of Health and UT Southwestern requirements for humane animal care and were approved by the UT Southwestern Institutional Animal Care and Use Committee.
Tumor tissue acquisition was performed under an Institutional Review Board (IRB) protocol approved at UT Southwestern Medical Center. Fresh tumor tissue was stored in sterile specimen cups and transported in RPMI-1640 (Sigma-Aldrich, St. Louis, MO) on ice. The tumor specimens were dissected in 3 mm x 3 mm fragments and coated in Matrigel® basement membrane matrix (Corning Life Sciences, Bedford, MA). NSG mice were anesthetized with isoflurane and, once sedated, their flank was shaved and cleansed with 70% alcohol and betadine. A 1 cm incision was made through the skin and subcutaneous tissue. A tumor fragment was then implanted into the flank and the incision was closed with 4-0 Vicryl suture. Mice were monitored daily for 2 consecutive days after surgery with particular attention paid to animal distress, wound dehiscence and signs of infections. Thereafter, they were examined 2-3 times per week. Tumor size was assessed by measurement with calipers once a week. Tumor volume was approximated using measurements of tumor length and width with the formula: (length x (width2))/2. The mice were sacrificed and tumors were harvested once they reached a diameter greater than 1.5 cm. The tissue was frozen in liquid N2 as well as kept fresh in RPMI for passaging into a larger cohort of mice. All three patient-derived xenografts were successfully passaged twice in order to develop a cohort large enough for treatment.
Cell line xenografts
Approximately 3.0x106 PL-45 and PL-5 cell lines were mixed with Matrigel® basement membrane matrix (Corning Life Sciences, Bedford, MA). Cells were injected subcutaneously into the flanks of NOD-SCID IL2Rgammanull (NSG) mice. Tumor progression was measured by calipers twice weekly and tumor volume was approximated using the formula: (length x (width2))/2.
Xenograft treatment
Treatment with PD-0332991 was performed on cohorts of at least 8 mice with tumors measuring between 150 and 500 mm3. The mice were randomized to treatment or control arms. The treated mice received palbociclib via oral gavage at a dose of 125 mg/kg in pH 4 lactate buffer (50 mM) daily. Gemcitabine was administered by intraperitoneal injection at a dose of 50 µg/kg in phosphate buffered saline. The control mice received lactate buffer via oral gavage once daily. Tumor sizes were assessed daily using calipers to determine tumor volume as described above. Treatment lasted for 5-8 days, after which the mice were sacrificed and the tumors were fixed in 10% formalin for histologic and immunohistological analysis.
Statistical analysis
All statistical analyses were performed using GraphPad Prism (GraphPad Software, Inc). Student’s t-test and Pearson correlation analyses were used and results with p < 0.05 were considered significant.
ACKNOWLEDGMENTS
We would like to thank all of the members of the laboratories for thought-provoking discussion and collegial support in this study. Derek Cox assisted with the animal studies. Matias Valsecchi, MD assisted with the explant studies. The University of Texas Southwestern Tissue Resource assisted with the collection of tissue specimens and is supported by the National Cancer Institute of the National Institutes of Health under award number 5P30CA142543.
FUNDING
The authors received no funding related to this work.
CONFLICTS OF INTEREST
There is no direct conflict related to this study. Over the last 5 years, ESK has received honoraria or research support from Permeon, Pfizer, and Eli Lilly.
REFERENCES
1. Paulson AS, Tran Cao HS, Tempero MA and Lowy AM. Therapeutic advances in pancreatic cancer. Gastroenterology. 2013; 144:1316-1326.
2. Tempero MA, Klimstra D, Berlin J, Hollingsworth T, Kim P, Merchant N, Moore M, Pleskow D, Wang-Gillam A and Lowy AM. Changing the way we do business: recommendations to accelerate biomarker development in pancreatic cancer. Clin Cancer Res. 2013; 19:538-540.
3. Bardeesy N and DePinho RA. Pancreatic cancer biology and genetics. Nat Rev Cancer. 2002; 2:897-909.
4. Yeo TP, Hruban RH, Leach SD, Wilentz RE, Sohn TA, Kern SE, Iacobuzio-Donahue CA, Maitra A, Goggins M, Canto MI, Abrams RA, Laheru D, Jaffee EM, Hidalgo M and Yeo CJ. Pancreatic cancer. Curr Probl Cancer. 2002; 26:176-275.
5. Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardiere C, Bennouna J, Bachet JB, Khemissa-Akouz F, Pere-Verge D, Delbaldo C, Assenat E, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. The New England journal of medicine. 2011; 364:1817-1825.
6. Kleger A, Perkhofer L and Seufferlein T. Smarter drugs emerging in pancreatic cancer therapy. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2014; 25: 1260-70.
7. Javle MM, Shroff RT, Xiong H, Varadhachary GA, Fogelman D, Reddy SA, Davis D, Zhang Y, Wolff RA and Abbruzzese JL. Inhibition of the mammalian target of rapamycin (mTOR) in advanced pancreatic cancer: results of two phase II studies. BMC Cancer. 2010; 10:368.
8. Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, Campos D, Lim R, Ding K, Clark G, Voskoglou-Nomikos T, Ptasynski M, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007; 25:1960-1966.
9. Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, Miller DK, Wilson PJ, Patch AM, Wu J, Chang DK, Cowley MJ, Gardiner BB, Song S, Harliwong I, Idrisoglu S, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012; 491:399-405.
10. Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, Hong SM, Fu B, Lin MT, Calhoun ES, Kamiyama M, Walter K, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008; 321:1801-1806.
11. Cowan RW and Maitra A. Genetic progression of pancreatic cancer. Cancer journal. 2014; 20:80-84.
12. Delpu Y, Hanoun N, Lulka H, Sicard F, Selves J, Buscail L, Torrisani J and Cordelier P. Genetic and epigenetic alterations in pancreatic carcinogenesis. Current genomics. 2011; 12:15-24.
13. Witkiewicz AK, Knudsen KE, Dicker AP and Knudsen ES. The meaning of p16(ink4a) expression in tumors: functional significance, clinical associations and future developments. Cell Cycle. 2011; 10:2497-2503.
14. Burkhart DL and Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer. 2008; 8:671-682.
15. Wang JY, Knudsen ES and Welch PJ. The retinoblastoma tumor suppressor protein. Advances in cancer research. 1994; 64:25-85.
16. Serrano M and Blasco MA. Putting the stress on senescence. Current opinion in cell biology. 2001; 13:748-753.
17. Serrano M, Lin AW, McCurrach ME, Beach D and Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997; 88:593-602.
18. Maitra A, Kern SE and Hruban RH. Molecular pathogenesis of pancreatic cancer. Best Pract Res Clin Gastroenterol. 2006; 20:211-226.
19. Witkiewicz AK, Cox D and Knudsen ES. CDK4/6 inhibition provides a potent adjunct to Her2-targeted therapies in preclinical breast cancer models. Genes & cancer. 2014; 5:261-272.
20. Young RJ, Waldeck K, Martin C, Foo JH, Cameron DP, Kirby L, Do H, Mitchell C, Cullinane C, Liu W, Fox SB, Dutton-Regester K, Hayward NK, Jene N, Dobrovic A, Pearson RB, et al. Loss of CDKN2A expression is a frequent event in primary invasive melanoma and correlates with sensitivity to the CDK4/6 inhibitor PD0332991 in melanoma cell lines. Pigment Cell Melanoma Res. 2014; 27:590-600.
21. Dickson MA. Molecular Pathways: CDK4 Inhibitors for Cancer Therapy. Clin Cancer Res. 2014; 20:3379-3383.
22. Witkiewicz AK and Knudsen ES. Retinoblastoma tumor suppressor pathway in breast cancer: prognosis, precision medicine, and therapeutic interventions. Breast Cancer Res. 2014; 16:207.
23. Gelbert LM, Cai S, Lin X, Sanchez-Martinez C, Del Prado M, Lallena MJ, Torres R, Ajamie RT, Wishart GN, Flack RS, Neubauer BL, Young J, Chan EM, Iversen P, Cronier D, Kreklau E, et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: in-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Investigational new drugs. 2014; 32:825-37.
24. Rader J, Russell MR, Hart LS, Nakazawa MS, Belcastro LT, Martinez D, Li Y, Carpenter EL, Attiyeh EF, Diskin SJ, Kim S, Parasuraman S, Caponigro G, Schnepp RW, Wood AC, Pawel B, et al. Dual CDK4/CDK6 inhibition induces cell-cycle arrest and senescence in neuroblastoma. Clin Cancer Res. 2013; 19:6173-6182.
25. Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, Albassam M, Zheng X, Leopold WR, Pryer NK and Toogood PL. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004; 3:1427-1438.
26. Heilmann AM, Perera RM, Ecker V, Nicolay BN, Bardeesy N, Benes CH and Dyson NJ. CDK4/6 and IGF1 Receptor Inhibitors Synergize to Suppress the Growth of p16INK4A-Deficient Pancreatic Cancers. Cancer Res. 2014; 74:3947-3958.
27. Liu F and Korc M. Cdk4/6 inhibition induces epithelial-mesenchymal transition and enhances invasiveness in pancreatic cancer cells. Mol Cancer Ther. 2012; 11:2138-2148.
28. Franco J, Witkiewicz AK and Knudsen ES. CDK4/6 inhibitors have potent activity in combination with pathway selective therapeutic agents in models of pancreatic cancer. Oncotarget. 2014; 5:6512-6525.
29. Dickson MA, Hahn WC, Ino Y, Ronfard V, Wu JY, Weinberg RA, Louis DN, Li FP and Rheinwald JG. Human keratinocytes that express hTERT and also bypass a p16(INK4a)-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Molecular and cellular biology. 2000; 20:1436-1447.
30. Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, Dekleva EN, Saunders T, Becerra CP, Tattersall IW, Westphalen CB, Kitajewski J, Fernandez-Barrena MG, Fernandez-Zapico ME, Iacobuzio-Donahue C, Olive KP, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014; 25:735-747.
31. Erkan M, Reiser-Erkan C, Michalski CW and Kleeff J. Tumor microenvironment and progression of pancreatic cancer. Experimental oncology. 2011; 32:128-131.
32. Dean JL, McClendon AK, Hickey TE, Butler LM, Tilley WD, Witkiewicz AK and Knudsen ES. Therapeutic response to CDK4/6 inhibition in breast cancer defined by ex vivo analyses of human tumors. Cell Cycle 2012. 11:2756-2761.
33. Lee J, Jang KT, Ki CS, Lim T, Park YS, Lim HY, Choi DW, Kang WK, Park K and Park JO. Impact of epidermal growth factor receptor (EGFR) kinase mutations, EGFR gene amplifications, and KRAS mutations on survival of pancreatic adenocarcinoma. Cancer. 2007; 109:1561-1569.
34. Dean JL, Thangavel C, McClendon AK, Reed CA and Knudsen ES. Therapeutic CDK4/6 inhibition in breast cancer: key mechanisms of response and failure. Oncogene 2010. 29:4018-4032.
35. Konecny GE, Winterhoff B, Kolarova T, Qi J, Manivong K, Dering J, Yang G, Chalukya M, Wang HJ, Anderson L, Kalli KR, Finn RS, Ginther C, Jones S, Velculescu VE, Riehle D, et al. Expression of p16 and retinoblastoma determines response to CDK4/6 inhibition in ovarian cancer. Clin Cancer Res. 2011; 17:1591-1602.
36. Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, Ginther C, Atefi M, Chen I, Fowst C, Los G and Slamon DJ. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009; 11:R77.
37. Michaud K, Solomon DA, Oermann E, Kim JS, Zhong WZ, Prados MD, Ozawa T, James CD and Waldman T. Pharmacologic inhibition of cyclin-dependent kinases 4 and 6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer Res. 2010; 70:3228-3238.
38. Ferrara C, Tessari G, Poletti A, Giacon C, Meggiato T, Martines D, Del Favero G and Naccarato R. Ki-67 and c-jun expression in pancreatic cancer: a prognostic marker? Oncol Rep. 1999; 6:1117-1122.
39. Chu GC, Kimmelman AC, Hezel AF and DePinho RA. Stromal biology of pancreatic cancer. Journal of cellular biochemistry. 2007; 101:887-907.
40. Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, Frese KK, Denicola G, Feig C, Combs C, Winter SP, Ireland-Zecchini H, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009; 324:1457-1461.
41. Neesse A, Michl P, Frese KK, Feig C, Cook N, Jacobetz MA, Lolkema MP, Buchholz M, Olive KP, Gress TM and Tuveson DA. Stromal biology and therapy in pancreatic cancer. Gut. 2010; 60:861-868.
42. Knudsen ES and Wang JY. Targeting the RB-pathway in cancer therapy. Clin Cancer Res. 16(4):1094-1099.
43. Knudsen ES and Knudsen KE. Tailoring to RB: tumour suppressor status and therapeutic response. Nat Rev Cancer. 2008; 8:714-24.
44. Hwang RF, Moore T, Arumugam T, Ramachandran V, Amos KD, Rivera A, Ji B, Evans DB and Logsdon CD. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008; 68:918-926.
45. Erkan M, Reiser-Erkan C, Michalski CW, Kong B, Esposito I, Friess H and Kleeff J. The impact of the activated stroma on pancreatic ductal adenocarcinoma biology and therapy resistance. Curr Mol Med. 2012; 12:288-303.
46. Bhowmick NA, Neilson EG and Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature. 2004; 432:332-337.
47. Shimoda M, Mellody KT and Orimo A. Carcinoma-associated fibroblasts are a rate-limiting determinant for tumour progression. Seminars in cell & developmental biology. 2010; 21:19-25.
48. Lee JJ, Perera RM, Wang H, Wu DC, Liu XS, Han S, Fitamant J, Jones PD, Ghanta KS, Kawano S, Nagle JM, Deshpande V, Boucher Y, Kato T, Chen JK, Willmann JK, et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc Natl Acad Sci U S A. 2014; 111:E3091-3100.
49. Sherr CJ. Cancer cell cycles. Science. 1996; 274:1672-1677.
50. Lim S and Kaldis P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development. 2013; 140:3079-3093.
51. Kanda M, Matthaei H, Wu J, Hong SM, Yu J, Borges M, Hruban RH, Maitra A, Kinzler K, Vogelstein B and Goggins M. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology. 2012; 142:730-733 e739.