
INTRODUCTION
Anti-Epithelial Growth Factor Receptor (EGFR) targeted therapy, such as panitumumab and cetuximab, is effectively reducing the risk of tumor progression and improving overall survival (OS), progression free survival (PFS) and quality of life in metastatic colorectal cancer (mCRC) patients whose tumor is RAS wild-type [1, 2]. Conversely, mCRC patients with mutated RAS tumors (KRAS and NRAS mutations) who received panitumumab in combination with oxaliplatin-based chemotherapy had a significantly worse outcome in OS and PFS [3]. This notion has led to a new incentive by the European Medicine Agency (EMA) for Vectibix (panitumumab) in June 2013 [4] and for Erbitux (cetuximab) in November 2013 [5], to indicate administration of EGFR targeted therapy only to patients with wild-type RAS mCRC. This has increased the need for reliable RAS mutation testing methods to assure the quality of RAS status determination.
Most molecular testing methods that are used nowadays accurately assess mutational status of RAS genes in samples with >30-50% tumor cells, or alternatively with 15-25% of mutated alleles in the test sample [6, 7]. However, with lower number of mutated alleles in the sample, the limit of accurate detection of a method declines depending on the test method used [8]. Even when using the same method, differences in protocols between laboratories can result in different outcomes. It has been suggested that the reproducibility amongst different testing methods is not as high as anticipated for based on previous EQA schemes for KRAS exon 2 testing [9]. In a recent study, in 29 out of 182 KRAS exon 2 wild-type tumors (15.9%), as assessed with Sanger sequencing, a KRAS exon 2 mutation was found with next generation sequencing (NGS) [10]. This suggests a higher variability in reproducibility between test methods and laboratories than initially measured [11].
Given the clinical impact of RAS-testing, it is of utmost importance to control for reliable performance of routine RAS-testing methods used in clinical practice. In this study, we evaluated the inter-laboratory agreement of RAS-testing amongst 171 mCRC patients of 17 Dutch laboratories.
RESULTS
Integration of full RAS-testing in the Netherlands
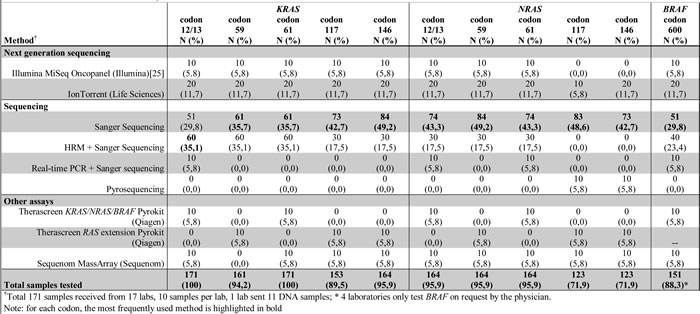
In January 2014, 11 of 17 participating Dutch laboratories had introduced full RAS testing (65%). All 17 participating centers performed KRAS exon 2 (codon 12 and 13) and KRAS exon 3 (codon 61) testing. The method most frequently used for KRAS exon 2 was Sanger sequencing of PCR products, either directly (5 laboratories; 51 samples (29.8%)) or to specify the mutation detected with high resolution melting (HRM) analysis or to confirm a real-time PCR result (7 laboratories; 70 samples (40.9%)) [Table 1].
Of the 6 laboratories that had not introduced full RAS-testing, one laboratory did not test for NRAS exon 2, 3, 4 and KRAS exon 4, three had not introduced NRAS exon 4 (codon 117 and 146), one laboratory did not have KRAS exon 4 codon 117 in their test panel and one laboratory did not test for KRAS exon 3 codon 59 [see Table 1]. One of these 6 laboratories introduced full RAS testing early 2014 and had only tested 3 out of 10 (30%) CRC cases with full RAS-testing.
For full RAS-testing direct Sanger sequencing of PCR products was used most often either with or without a prescreen (range 35.7% - 49.2%) [Table 1]. The use of Mass Spectrometry (Sequenom) or CE-IVD kits (Therascreen) was reported by 2 laboratories (11.6%). In 3 laboratories (17.5%) next generation sequencing (NGS) with MiSeq (Illumina) or Ion Torrent (Life technologies) was used for RAS testing. In conclusion, full RAS testing had been introduced in the majority of laboratories participating in this study and relied mostly on Sanger sequencing methods.
Table 1: Methods used to test for KRAS, NRAS and BRAF mutations.
Mutation frequencies of KRAS, NRAS and BRAF
The amount and quality of 167 out of 171 received DNA samples was sufficient for successful evaluation by the reference laboratory using NGS of at least one of the target sites (165 samples for KRAS, 163 for NRAS and 163 for BRAF; see materials and methods). Overall, in 102 samples (61.1% (95% CI 53.5 – 68.2)) a mutation in KRAS, NRAS or BRAF was found at the reference laboratory [Figure 1A].
A KRAS exon 2 mutation was detected in 59 mCRC cases (35.8%), other RAS mutations were found in KRAS exon 3 (n = 3 (3.6%)), KRAS exon 4 (n = 3 (3.6%)) NRAS exon 2 (n = 1 (1.2%)) and NRAS exon 3 (n = 3 (3.6%)). No mutations in NRAS exon 4 were detected. This resulted in a total 79 RAS mutations (47.6% (95% CI 40.1-55.2)) of which only 8 were NRAS mutations. The mean percentage of mutated alleles was 42.1% (SD 15.7%) for KRAS and 46.1% (SD 23.25%) for NRAS [Supplementary Table 1]. The majority of KRAS mutations affected codon 12 (70.5%), especially p.Gly12Asp (23.9%) and p.Gly12Val were common (29.6%) [Figure 1B]; 5 out of 8 NRAS mutations were found in codon 61 (62.5%) [Figure 1C]. None of the samples harbored both or more than one KRAS and/or NRAS mutation.
BRAF mutations occurred in 23 of 163 CRC cases (14.1% (95%CI 9.4-20.1)) [Figure 1A] and were all c.1799T>A (p.Val600Glu) mutations in exon 15. The mean percentage of mutated alleles for BRAF was 34.5% (SD 14.3%) [Supplementary Table 1]. Both KRAS and NRAS mutations in our sample were mutually exclusive with BRAF mutations (OR =2.15 (95%CI 1.80-2.57); Chi-square p<0.01).
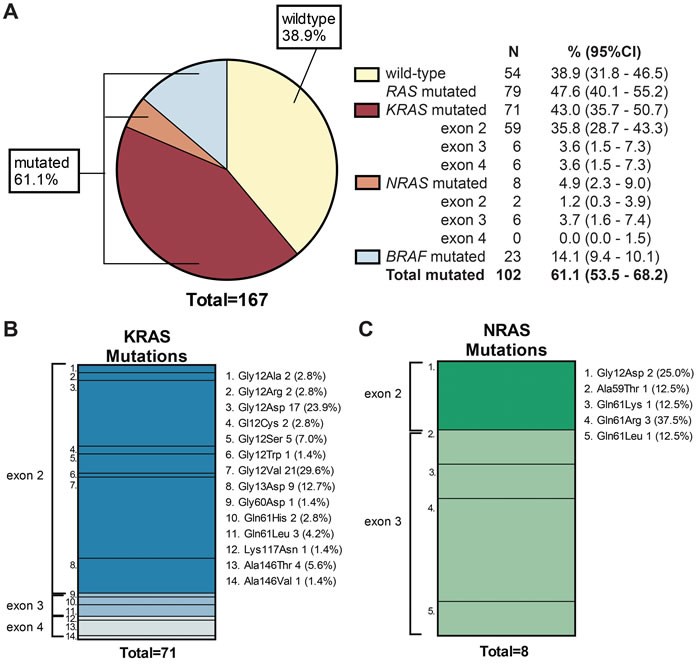
Figure 1: Mutation prevalence of KRAS, NRAS and BRAF. A) Pie chart showing all evaluated CRC cases (n = 167; 167 of 171 samples could be evaluated for at least one of the target sites). Of these CRC cases, 61.1% had a mutation. KRAS mutations alone were most frequently observed (43.0%). BRAF mutations were found in 23 CRC cases (All in exon 15; c.1799T>A (p.Val600Glu)). The 95% CI was calculated with Jeffry’s method. B) A total of 71 KRAS mutations was found; 59 in KRAS exon 2 (83.2%), 6 in KRAS exon 3 (8.4%), and 6 in KRAS exon 4 (8.4%). Gly12Val (29.6%) and Gly12Asp (23.9%) were the most common mutations found followed by Gly13Asp (12.7%). C) A total of 8 NRAS mutations was detected of which 2 were detected in exon 2 (Gly12Asp) and 6 in exon 3. The most common mutation in NRAS was Gln61Arg (37.5%).
High concordance of RAS and BRAF-testing
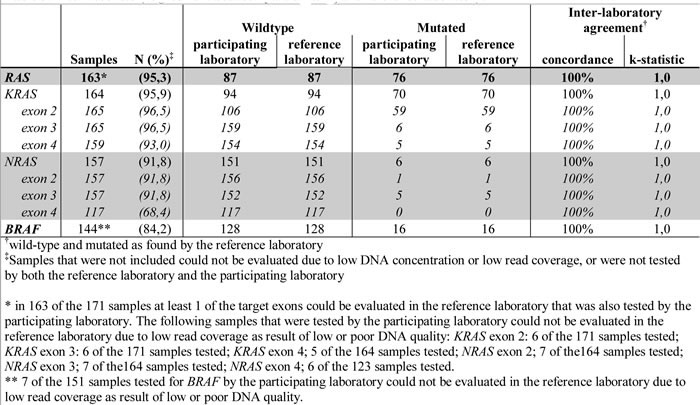
For 167 samples that could be evaluated by the reference laboratory, all mutations that had been found in the participating laboratories were verified. In addition, the reference lab detected three RAS and seven BRAF mutations that had not been detected in the participating laboratories. This concerned mutations in KRAS exon 4 (c.436G>A; p.Ala146Thr), NRAS exon 2 (c.35G>A; p.Gly12Asp), NRAS exon 3 (c.181C>A; p.Gln61Lys) and seven mutations in BRAF (c.1799T>A; p.Val600GLu) that the participating laboratory had not tested for [Table 2]. Altogether, the inter-laboratory agreement for tests performed by both the reference and participating laboratory was 100% (163/163) (κ statistic 1.0) for full RAS and 100% (144/144) (κ statistic 1.0) for BRAF-testing [Table 3]. All three additionally found RAS-mutations were reported back to the respective participating laboratory, and were confirmed with their newly installed Sanger sequencing or NGS approaches for the respective target sites.
Table 2: Additional mutations found by the reference laboratory.
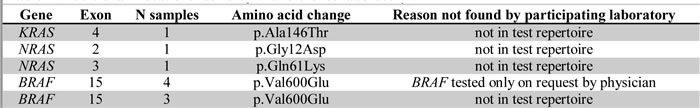
Table 3: Inter-laboratory agreement between participating and reference laboratory.
Association between percentage of neoplastic cells and mutation detection
Testing sensitivity of RAS-mutations is dependent on the percentage of neoplastic cells represented in the test sample; low neoplastic cell percentages may result in missing of RAS-mutations depending on the technique that is used [8]. For 158 samples the percentage of neoplastic cells as estimated by the pathologist of the participating laboratory was known. The median estimated neoplastic cell percentage represented in the DNA samples was 50% (inter quartile range (IQR)=30). Of the 158 samples, 47 (29.7%) had neoplastic cell percentages below 40%. When comparing the distribution of estimated neoplastic cell percentages amongst RAS-mutated (N = 73) and RAS-wild-type (N = 85) samples, the median neoplastic cell percentage was significantly lower in RAS-mutated samples (50.0% (IQR=28) compared to RAS-wild type (60.0% (IQR=20)) samples (Mann-Whitney U, p < 0.01) [Figure 2]. In RAS-mutated samples, the mean percentage of mutated alleles correlated with the percentage of neoplastic cells in the test sample (Pearson r = 0.433 (p < 0.01) for KRAS and Pearson r = 0.792 (p = 0.034) for NRAS; Supplementary Figure 1).
The frequency of RAS mutations amongst samples with neoplastic cell percentages ≤40% was significantly higher than in samples >40% neoplastic cells (OR 2.45 (95%CI 1.22 – 4.94), chi-square p = 0.011). A similar result was obtained when lowering the cut-off to 30 or 20% neoplastic cells, suggesting that the sensitivity of the RAS mutation analyses were adequate, even in samples with low neoplastic cells. However, care should be taken while reporting wild-type in samples with low neoplastic cells depending on the lower limit of detection of the technique applied to prevent reporting false-negatives.
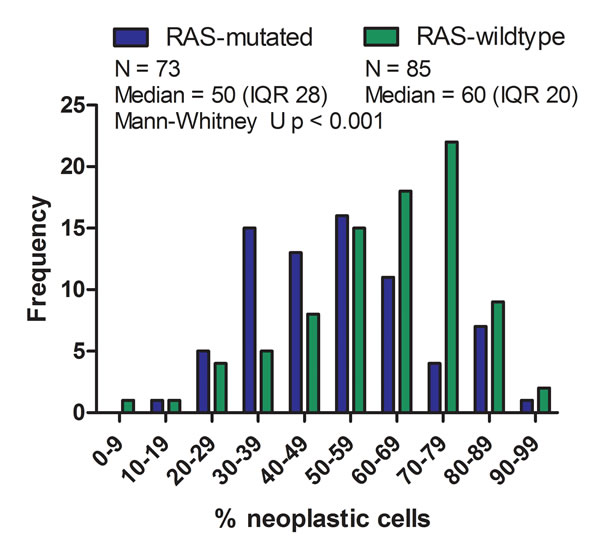
Figure 2: Neoplastic cell percentage in RAS-mutated and RAS-wild-type tumors. The distribution of the estimated percentage of tumor cells in the test sample was compared between RAS-mutated and RAS-wild type CRC cases. Frequency of CRC cases is shown on the Y-axis; the X-axis represents the percentage of neoplastic cells. Mann-Whitney U test of the comparison indicates that the median neoplastic cell percentage of RAS-mutated mCRC cases is significantly lower (Median 50 (IQR 28), Mean Rank = 66,65) than RAS-wild-type mCRC cases (Median 60 (IQR 20), Mean Rank = 90,54; p = 0.001) and thus that the populations have distinct neoplastic cell percentage distributions.
DISCUSSION
Our results show an excellent concordance (100%, kappa 1.0) of RAS-test results of the reference lab oratory and 17 other Dutch laboratories in routine clinical practice, despite differences in testing methods used. Overall, full RAS-testing of KRAS exon 2, 3 and 4 and NRAS exon 2, 3 and 4 has been introduced in the majority of the participating laboratories (65%). Three RAS mutations and seven BRAF mutation were missed due to incomplete testing.
The frequency of RAS mutations reported in our study matches those previously reported in the literature. Approximately 35-45% of all CRCs contain RAS-mutations in KRAS exon 2 [3, 12-16], whereas approximately 10% of RAS-mutations occurs in KRAS exons 3 and 4 or NRAS exons 2, 3 and 4 [3, 17-19]. In addition, KRAS, NRAS and BRAF mutations have a strong tendency towards mutual exclusivity [20]. BRAF mutations occurred in 14% of our samples, which seems higher than the reported 9% in the Cancer Genome Atlas [20]. While BRAF mutation analysis is incorporated in the test repertoire of most laboratories (71%), some laboratories only test for BRAF on special request by the physician when tumors were previously tested RAS wild-type. BRAF mutation frequency amongst KRAS exon 2 wild-type tumors is reported to be around 8-15% [10, 18].
To assure accurate determination of RAS wild-type and mutant status, the quality of mutation detection for each KRAS and NRAS exon needs to be determined. Reproducibility is one of the measures that signify the quality of diagnostic tests. Poor reproducibility can have several causes: 1) the testing methods used have a difference in sensitivity (ability to identify tumors with the mutation) or, 2) there is variation between different persons/laboratories performing the test (inter-observer variability). In a recently published Italian study, it was found that amongst KRAS exon 2 wild-type tumors, as assessed with Sanger sequencing, a KRAS exon 2 mutation was found with NGS in 28 out of 182 mCRC cases (15.9%). The difference in sensitivity between the testing methods could have partly accounted for the discrepancy found in this study [10]. In our study the discrepancy between the test results generated in 17 different laboratories using a variety of testing methods and the reference laboratory using a NGS approach was naught, even in samples with a neoplastic cell percentage between 20-40% and in laboratories using Sanger sequencing. In the 2012 KRAS external quality assessment program of the European Society of Pathology, less than 5% of all samples were wrongly genotyped for KRAS amongst 100 laboratories in 26 countries [11].
Testing sensitivity is reflected by the limit of detection of the method, but is also limited by the percentage of neoplastic cells represented in the test sample. In fact, low neoplastic cell percentage (~10%) led to 16 of the 29 false-negatives in the 2012 KRAS EQA scheme [11]. For PCR and Sanger sequencing a minimum amount of 20-30% of tumor cells is required [21]. Our study has high reproducibility even in samples with a low neoplastic cell percentage and the percentage of mutated alleles correlates with the neoplastic cell content in the sample [Supplementary Figure 1]. In fact, the percentage of RAS mutant samples was significantly higher in samples with neoplastic cell percentages below 40%, which demonstrates that mutations could also be detected in samples with a low neoplastic cell percentage. However, this unexpected observation could be merely coincidental due to small subgroups and difference in tumor sampling in combination with variation in estimating the amount of neoplastic cells, which is known to be high among pathologists [22, 23].
Potential sources of bias in our study could have affected the reported reproducibility. First, centers were asked to send DNA of the first 10 CRC cases tested in 2014. Because KRAS exon 2 mutations are the most frequent, there were some laboratories where only KRAS exon 2 mutations or wild-type RAS-status was reported. This could have led to an overestimation of the reproducibility because other RAS-mutations were not present. Secondly, the likelihood of finding false-negatives is higher when only wild-type samples are re-tested. In this series only 54 samples were wild-type for all the tested hotspot mutations and thus could have led to an underestimation of false-negative wild-type samples. The small number of NRAS-mutations found has likely overestimated the reproducibility of this group of mutations. Future studies need to verify whether newly installed methods reproducibly detect NRAS mutations, especially in laboratories that did not report any NRAS mutations in the tested CRC panel.
In conclusion, our study clearly shows that RAS-status can be reproducibly assessed between laboratories in routine clinical practice using similar or different testing methods. With constant improvement of testing methodologies and quality controls, this offers good expectations for the future of molecular testing in mCRC. Nevertheless, more in depth analyses with regard to the effect of testing sensitivity and the percentage of tumor cells in the test sample on RAS mutation detection are warranted.
PATIENTS AND METHODS
Patient selection and data collection
All 22 Dutch institutes that participated in the European Quality Assurance (EQA) scheme of the European society of pathology (ESP) in 2013 were invited to participate in the study. Seventeen laboratories responded and were subsequently requested to send 10 µl DNA (≥30 ng) of the first 10 mCRCs tested in routine diagnostics for anti-EGFR targeted therapy from 1st January 2014. In addition, each laboratory was asked to indicate the testing method used, the mutations tested for and the percentage of neoplastic cells in the tissue the DNA was extracted from. Moreover, the KRAS, NRAS and BRAF mutations found by the participating laboratories were reported and stored by a third party until study end. Retesting of the samples at the reference laboratory was performed blinded. Approval by a medical ethics committee was not required. All data were reported in de-identified form and are in agreement with the Dutch Data Protection Act.
Next generation sequencing
A total of 171 DNA samples received from the participating laboratories (one lab contributed 11 samples) were quantified on the Qubit platform (Life technologies). Samples with a DNA concentration below 0.5 ng/µl were excluded from further analysis. Bar-coded libraries were prepared from 10-100 ng DNA using a custom AmpliSeq panel targeting frequently mutated regions in the KRAS, NRAS, EGFR, PIK3CA, ERBB2, AKT1, BRAF genes and the AMELX/Y gene as a control gene to determine the sex of the patient. Libraries were equimolar pooled and clonal amplification was performed by emulsion PCR using the One Touch 2 system (Life Technologies) and subsequently run on the Ion Torrent Personal Genome Machine (Life Technologies). Torrent Suite Software v.3.4.2. was used to pre-process the raw data. Subsequent mapping and variant calling was performed using SeqNext software v.4.1.2. (JSI medical systems GmbH). The following genes and exons were included in the data analysis: KRAS exon 2, 3 and 4 (NM_004985.3), NRAS exon 2,3 and 4 (NM_002524.3) and BRAF exon 15 (NM_004333.4). The minimum read coverage allowed to call mutant alleles was set at 100 reads. In general, read coverage was more than 500 resulting in a sensitivity of 5% mutant alleles. For a read coverage between 100-500 reads, a limit of 10% mutant alleles was employed. When coverage was low or mutation calling could not be established due to other technical reasons, the run was repeated. When there was still insufficient coverage of the target sites after repeating the run, the samples were excluded from analysis. In total 6 KRAS, 8 NRAS and 8 BRAF target sites had insufficient coverage due to low amount or poor DNA quality. Therefore, we could successfully evaluate a total of 165 KRAS, 163 NRAS and 163 BRAF target sites; for 167 samples at least one of the target sites could be successfully evaluated.
Data analysis
Variants were filtered for known SNPs and systematic sequencing artifacts. Next, the somatic mutations found in the DNA-samples by the reference laboratory were compared with the mutations originally found by the participating laboratories to assess the percentage of agreement of the results. The mutations were compared at the genotype level. Each participating pathology center was informed about their individual results and performance.
Statistical analysis
All statistical analyses were performed in IBM SPSS statistics version 20. Mutation frequencies were calculated for KRAS, NRAS and BRAF. Binominal confidence intervals were calculated using the Jeffrey’s interval. The detected mutation(s) in the DNA-samples at the genotype level were compared between the reference laboratory and the participating laboratories and expressed as percentage agreement. Data were evaluated for hotspot mutations in KRAS exon 2, 3 and 4, NRAS exon 2, 3 and 4 and BRAF exon 15. Concordance (inter-laboratory agreement, kappa-statistic [24]) was only calculated when the mutation was targeted in the test panel of both the reference and participating laboratory. Odds ratios with 95% confidence intervals and chi-square statistics were calculated to evaluate associations between RAS-mutations and BRAF. The distribution of tumor cell percentage in the DNA-samples amongst RAS-mutated and RAS-wild-type samples was assessed with the Mann-Whitney U test.
ACKNOWLEDGMENTS
We thank all the laboratories that participated in this study for their cooperation and fruitful discussions on the topic. We thank Véronique Tack from the University of Leuven for communication with and recruitment of the laboratories.
FUNDING
This study was funded by MERCK Group KGaA. The source of funding did not have any influence on the design and the analysis of the results.
DISCLOSURE
E. Schuuring received honoraria for expertise from commercial partners, is part of scientific advisory boards from Amgen, Abbott, Roche and Pfizer, and received travel reimbursements (on invitation) from Abbott and Roche. T. van Wezel is part of the scientific advisory board of Amgen. N ‘t Hart is part of the scientific advisory boards of Amgen, Pfizer, Astra and Roche. Han van Krieken is part of the scientific advisory boards of Amgen and Merck KGaA. All remaining authors have declared no conflict of interest.
REFERENCES
1. Stintzing S, Kapaun C, Laubender RP, Jung A, Neumann J, Modest DP, Giessen C, Moosmann N, Wollenberg A, Kirchner T, Heinemann V. Prognostic value of cetuximab-related skin toxicity in metastatic colorectal cancer patients and its correlation with parameters of the epidermal growth factor receptor signal transduction pathway: results from a randomized trial of the GERMAN AIO CRC Stu. Int. J. Cancer 2013; 132: 236–245.
2. Schwartzberg LS, Rivera F, Karthaus M, Fasola G, Canon J-L, Hecht JR, Yu H, Oliner KS, Go WY. PEAK: a randomized, multicenter phase II study of panitumumab plus modified fluorouracil, leucovorin, and oxaliplatin (mFOLFOX6) or bevacizumab plus mFOLFOX6 in patients with previously untreated, unresectable, wild-type KRAS exon 2 metastatic colorectal . J. Clin. Oncol. 2014; 32: 2240–2247.
3. Douillard J-Y, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, Rivera F, Kocákova I, Ruff P, Błasińska-Morawiec M, Šmakal M, Canon JL, Rother M, Williams R, Rong A, Wiezorek J, Sidhu R, Patterson SD. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N. Engl. J. Med. 2013; 369: 1023–1034.
4. European Medicines Agency. Vectibix - Panitumumab. 2013.Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000741/human_med_001128.jsp&mid=WC0b01ac058001d124.
5. European Medicines Agency. Erbitux - Cetuximab. 2013.Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000558/human_med_000769.jsp&mid=WC0b01ac058001d124.
6. Querings S, Altmüller J, Ansén S, Zander T, Seidel D, Gabler F, Peifer M, Markert E, Stemshorn K, Timmermann B, Saal B, Klose S, Ernestus K, Scheffler M, Engel-Riedel W, Stoelben E, Brambilla E, Wolf J, Nürnberg P, Thomas RK. Benchmarking of mutation diagnostics in clinical lung cancer specimens. PLoS One 2011; 6: e19601.
7. Tsiatis AC, Norris-Kirby A, Rich RG, Hafez MJ, Gocke CD, Eshleman JR, Murphy KM. Comparison of Sanger sequencing, pyrosequencing, and melting curve analysis for the detection of KRAS mutations: diagnostic and clinical implications. J. Mol. Diagn. 2010; 12: 425–432.
8. Dijkstra JR, Heideman DAM, Meijer GA, Boers JE, ’t Hart NA, Diebold J, Hirschmann A, Hoefler G, Winter G, Miltenberger-Miltenyi G, Pereira S V, Richman SD, Quirke P, Rouleau EL, Guinebretiere JM, Tejpar S, Biesmans B, van Krieken JHJM. KRAS mutation analysis on low percentage of colon cancer cells: the importance of quality assurance. Virchows Arch. 2013; 462: 39–46.
9. Van Krieken JH, Siebers AG, Normanno N. European consensus conference for external quality assessment in molecular pathology. Ann. Oncol. 2013; 24: 1958–1963.
10. Ciardiello F, Normanno N, Maiello E, Martinelli E, Troiani T, Pisconti S, Giuliani F, Barone C, Cartenì G, Rachiglio a M, Montesarchio V, Tonini G, Rizzi D, Cinieri S, Bordonaro R, Febbraro A, De Vita F, Orditura M, Fenizia F, Lambiase M, Rinaldi A, Tatangelo F, Botti G, Colucci G. Clinical activity of FOLFIRI plus cetuximab according to extended gene mutation status by next generation sequencing: findings from the CAPRI-GOIM trial. Ann. Oncol. 2014; 25: 1756–1761.
11. Tembuyser L, Ligtenberg MJL, Normanno N, Delen S, van Krieken JH, Dequeker EMC. Higher quality of molecular testing, an unfulfilled priority: Results from external quality assessment for KRAS mutation testing in colorectal cancer. J. Mol. Diagn. 2014; 16: 371–377.
12. Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, Patterson SD, Chang DD. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J. Clin. Oncol. 2008; 26: 1626–1634.
13. Ciardiello F, Tejpar S, Normanno N, Mercadante D, Teague T, Wohlschlegel B, Van Cutsem E. Uptake of KRAS mutation testing in patients with metastatic colorectal cancer in Europe, Latin America and Asia. Target. Oncol. 2011; 6: 133–145.
14. De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V, Papamichael D, Laurent-Puig P, Penault-Llorca F, Rougier P, Vincenzi B, Santini D, Tonini G, Cappuzzo F, Frattini M, Molinari F, Saletti P, De Dosso S, Martini M, Bardelli A, Siena S, Sartore-Bianchi A, Tabernero J, Macarulla T, Di Fiore F, Gangloff AO, Ciardiello F, Pfeiffer P, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010; 11: 753–762.
15. Peeters M, Douillard J-Y, Van Cutsem E, Siena S, Zhang K, Williams R, Wiezorek J. Mutant KRAS codon 12 and 13 alleles in patients with metastatic colorectal cancer: assessment as prognostic and predictive biomarkers of response to panitumumab. J. Clin. Oncol. 2013; 31: 759–765.
16. Wong A, Ma BBY. Personalizing therapy for colorectal cancer. Clin. Gastroenterol. Hepatol. 2014; 12: 139–144.
17. Negru S, Papadopoulou E, Apessos A, Stanculeanu DL, Ciuleanu E, Volovat C, Croitoru A, Kakolyris S, Aravantinos G, Ziras N, Athanasiadis E, Touroutoglou N, Pavlidis N, Kalofonos HP, Nasioulas G. KRAS, NRAS and BRAF mutations in Greek and Romanian patients with colorectal cancer: a cohort study. BMJ Open 2014; 4: e004652.
18. Vaughn CP, Zobell SD, Furtado L V, Baker CL, Samowitz WS. Frequency of KRAS, BRAF, and NRAS mutations in colorectal cancer. Genes. Chromosomes Cancer 2011; 50: 307–312.
19. Peeters M, Oliner KS, Parker A, Siena S, Van Cutsem E, Huang J, Humblet Y, Van Laethem J-L, André T, Wiezorek J, Reese D, Patterson SD. Massively parallel tumor multigene sequencing to evaluate response to panitumumab in a randomized phase III study of metastatic colorectal cancer. Clin. Cancer Res. 2013; 19: 1902–1912.
20. Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012; 487: 330–337.
21. Thunnissen E, Bovée JVMG, Bruinsma H, van den Brule AJC, Dinjens W, Heideman DAM, Meulemans E, Nederlof P, van Noesel C, Prinsen CFM, Scheidel K, van de Ven PM, de Weger R, Schuuring E, Ligtenberg M. EGFR and KRAS quality assurance schemes in pathology: generating normative data for molecular predictive marker analysis in targeted therapy. J. Clin. Pathol. 2011; 64: 884–892.
22. Viray H, Li K, Long TA, Vasalos P, Bridge JA, Jennings LJ, Halling KC, Hameed M, Rimm DL. A prospective, multi-institutional diagnostic trial to determine pathologist accuracy in estimation of percentage of malignant cells. Arch. Pathol. Lab. Med. 2013; 137: 1545–1549.
23. Smits AJJ, Kummer JA, de Bruin PC, Bol M, van den Tweel JG, Seldenrijk KA, Willems SM, Offerhaus GJA, de Weger RA, van Diest PJ, Vink A. The estimation of tumor cell percentage for molecular testing by pathologists is not accurate. Mod. Pathol. 2014; 27: 168–174.
24. Viera AJ, Garrett JM. Understanding Interobserver Agreement : The Kappa Statistic. 2005; 37: 360–363.
25. Sie D, Snijders PJF, Meijer GA, Doeleman MW, van Moorsel MIH, van Essen HF, Eijk PP, Grünberg K, van Grieken NCT, Thunnissen E, Verheul HM, Smit EF, Ylstra B, Heideman DAM. Performance of amplicon-based next generation DNA sequencing for diagnostic gene mutation profiling in oncopathology. Cell. Oncol. (Dordr). 2014; 37: 353–361.