
Introduction
Glaucoma is a family of irreversible blinding diseases that are projected to affect 79.6 million people worldwide by the year 2020 [1]. The most common form of glaucoma, primary open angle glaucoma, is an aging associated disease (AAD) often characterized by elevated intraocular pressure induced by increased outflow resistance of the aqueous humor [2]. The human trabecular meshwork (HTM), a complex three-dimensional structure comprised of cells, interwoven collagen beams and perforated sheets, is believed to provide the majority of outflow resistance in both normal and glaucomatous eyes [3-7]. HTM cells, depending on the region of the HTM, either form sheets covering extracellular matrix (ECM) structures or are scattered throughout the ECM [8-11]. What changes in the HTM resulting in increased resistance is poorly understood, but our recent study showed the HTM is ~20 fold stiffer in glaucoma [12], suggesting a prominent role of HTM mechanobiology. This tissue-scale stiffening is likely a result of biophysical changes to both the ECM and constituent cells, as structural changes to both the cytoskeleton [13, 14] and ECM [15-19] have long been associated with glaucoma.
Building upon these findings, further research has led to a growing body of evidence that these biophysical changes are not epiphenomena, but upstream of factors important in the progression of the disease. In vitro studies by our lab and others have shown that primary HTM cells have alterations in expression of genes associated with glaucoma, in biophysical properties, and in responsiveness to potential therapeutics when grown on hydrogels of varying stiffness [20-25]. Importantly, when cultured on hydrogels mimicking the stiffness of glaucomatous HTM, HTM cells increased expression of genes known to be associated with glaucoma progression [26-34], including myocilin [21, 23], secreted protein acidic and rich in cysteine (SPARC) [23], and secreted frizzled related protein-1 (SFRP1) [21]. These studies suggest a mechanism by which altered HTM mechanobiology reinforces the biological mediators of the glaucomatous phenotype. However, it remains unclear what processes induce stiffening.
A prime candidate for this process is cellular senescence, the irreversible arrest of cellular proliferation. Senescence is thought to contribute to many of the physiological changes associated with aging as well as AAD [35-40]. Induction of senescence generally occurs either due to telomere shortening after repeated mitosis (“replicative senescence”) or presentation of physiological stress (“stress-induced senescence”), although the distinction is blurry at best [35, 38, 41-47]. Both telomere shortening and cellular senescence are correlated with aging [48-52], and this is hypothesized as a prime driver of aging and associated diseases [53, 54]. Increased cell senescence is observed in the HTM of glaucoma patients [55], and glaucoma is likewise one among many AADs associated with increased rigidity of the tissue. Other AADs known to be associated with increased tissue stiffness include atherosclerosis [56-58], age-related macular degeneration [59-62], and cancer microenvironments [63-65]. Improved understanding of the causes of HTM stiffening in glaucoma will likely provide insight into other AADs as well.
Senescence is associated with increased expression of vimentin [66, 67] and filamentous actin (F-actin) [68-70], both of which are key determinants of cellular mechanics [71-74]. Both cytoskeletal elements are expressed in HTM cells [75-77], and altered F-actin morphology has been associated with HTM dysfunction [13, 14, 78-80]. Additionally, we have recently shown that exogenous SFRP1 induces pronounced and long-lasting stiffening of HTM cells [81]. SFRP1 has been shown to be necessary and sufficient for the induction of the senescent phenotype [82], suggesting SFRP1 induced stiffening may be related to senescence as well. In aggregate, there is strong support for a hypothesis of cellular senescence contributing to the glaucoma phenotype by increasing cellular stiffening associated with cytoskeletal changes. However, senescence has yet to be directly linked to HTM mechanobiology.
In this study, primary HTM cells were serially passaged until senescence and atomic force microscopy (AFM) was used to measure the intrinsic mechanical properties of senescent cells compared to normally proliferating controls. We found that stiffness was significantly increased in high passage HTM cells, and this was associated with increased staining of vimentin and F-actin. Further, SFRP1 expression was also elevated in senescent cultures. In aggregate, these results demonstrate HTM cellular senescence profoundly alters HTM mechanobiology and suggest a causal link between HTM cell senescence, altered cell mechanics and glaucoma progression.
Results
Confirmation of senescence
For all experiments, we serially passaged primary HTM cells until a complete loss of proliferative response was observed. Failure of proliferation was defined as having equal to or fewer viable cells one week after plating of a given passage. In these cultures, the cells took on an enlarged, flattened morphology, typical of senescent cells [83-88]. To confirm this method resulted in senescence, we plated cells of three donors (HTM553, HTM667, HTM631) on glass coverslips and assayed for senescence associated β-galactosidase (SAβGal) activity, a known marker of senescence [52, 89, 90]. In terminally passaged HTM cells, prominent blue staining indicates SAβGal activity which is minimal at earlier passages (Figure 1). Under these culture conditions, HTM cells typically senesced at passages 12-15, although cells isolated from some donors senesced earlier.
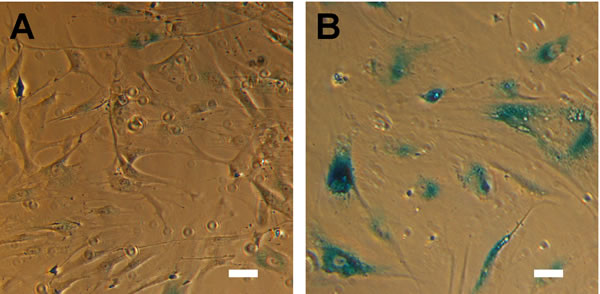
Figure 1: HTM cells exhibit hallmarks of senescence at advanced passage. (A) HTM cells under routine culture are relatively small and elongated, and have minimal SAβGal activity. (B) After serial passaging, HTM cells assume a senescent phenotype included an enlarged, flattened morphology, and pronounced SAβGal activity. Images are of HTM728 (passages 5 and 7) and are representative of cells from other donors. Scale bars are 50 μm.
Measurement of stiffness with increasing passage
Having established an in vitro model of HTM senescence, we turned to the primary objective of our study, determining if senescent HTM cells are intrinsically stiffer. We serially passaged primary HTM cells from 7 donors and measured cellular mechanics using AFM at each passage. Values of representative donor 517 are shown for passage 7 through senescence at passage 12 (Figure 2A). Minimal variation or serum dependence was observed at lower passages; the calculated elastic modulus of proliferating cells were similar to previously published values for HTM cells [25, 81, 91]. At terminal passage cells were substantially stiffer in full media, while the last three passages of cells in serum free media exhibited substantial stiffening. One way ANOVA and Bonferroni comparison to the earliest passage revealed these differences were significant to at least the p < 0.05 level, demonstrating a passage effect with cells from this donor. We performed similar experiments with cells derived from 6 other donors, observing similar results (Figure 2B-2G). Stiffness at or immediately before terminal passage was typically elevated in full media and/or serum free media, although the response in serum free media was more robust. Despite the apparent trend, there was substantial donor variability both in baseline stiffness and effect of passage. To control for donor-to-donor variability, we normalized the measurements of the senescent cells at terminal passage to proliferative cells at the earliest passage (Figure 2H). Senescent cell populations were 1.88±0.14 (n=7 donors; p < 0.05) or 2.57±0.14 (n=7 donors; p < 0.01) fold stiffer than proliferative controls in the presence or absence of serum, respectively.
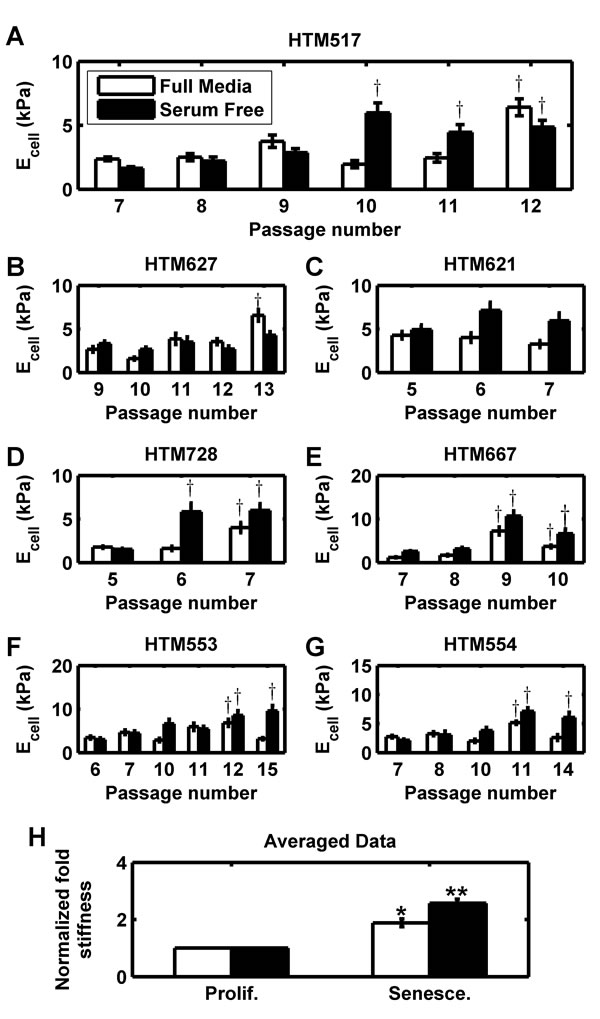
Figure 2: Intrinsic stiffness of HTM cells is increased in senescent populations. (A) HTM cells from donor 517 underwent serial passaging and stiffness (Ecell) was measured each passage for both full and serum free media conditions. At terminal passage, stiffness is substantially upregulated. (B-G) HTM cells from 6 other donors (627, 621, 728, 667, 553, and 554) similarly were serially passaged to senescence, and similarly exhibited stiffening at terminal passage. Data are mean±SEM. † p < 0.05 with respect to earliest passage. (H) For all 7 donors, the average elastic modulus at terminal passage (‘Senesce.’) was normalized to average elastic modulus at the earliest passage (‘Prolif.’). For each donor and each condition, 5-7 cells were each indented 5-7 times and averaged. Data are mean±SEM. * p < 0.05; ** p < 0.01.
Staining of actin and vimentin with passage
As both intermediate filaments and the actin cytoskeleton have been linked to cell stiffness and senescence [66-74], we fixed and stained proliferative and senescent cultures for vimentin and F-actin (Figure 3A-3B). In proliferative cultures, vimentin was minimally expressed in the cell body but the expression was greatly increased in senescent cells. Similarly, proliferative HTM cultures exhibited numerous stress fibers as previously reported [14, 75, 79, 80], however, after senescence the stress fibers became more prominent. To quantify these changes, we averaged staining intensity of vimentin and F-actin for both senescent and proliferative cultures from 4 different donors (Figure 3C-3D). Senescent cultures exhibited significantly brighter staining intensity of vimentin, both in full (1.46±0.19 fold; p < 0.05) and serum free (1.57±0.18 fold; p < 0.05) media. Similar results were obtained with F-actin staining for full (1.52±0.25 fold; p = 0.086) and serum free (1.53±0.17 fold; p < 0.05) culture, although the results did not rise to significance in full media cultures.
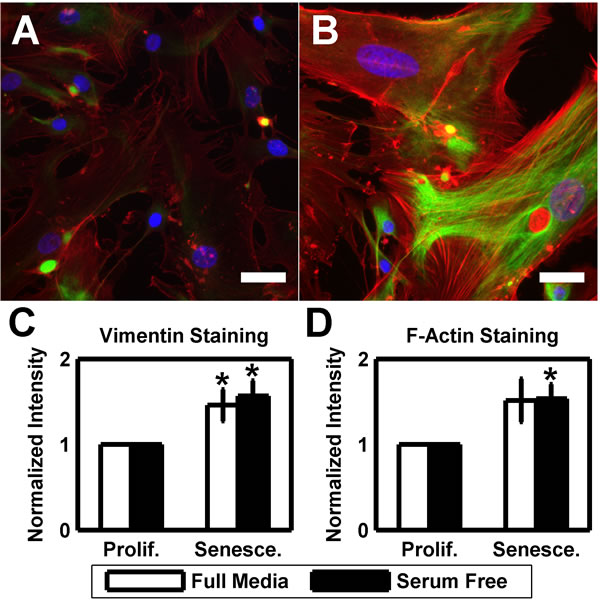
Figure 3: HTM cells exhibit more pronounced cytoskeleton at senescence. (A) HTM cells under routine culture stain positively for both F-actin (red) and vimentin (green). DAPI used as nuclear counterstain (blue). (B) After serial passaging, HTM cells exhibit more pronounced stress fibers and brighter vimentin staining. Images are of HTM728 (passages 5 and 7) and are representative of cells from other donors. Scale bars are 50 μm. (C) Quantification of the average vimentin staining intensity reveals a significantly increased signal in senescent cultures when compared earlier passages (n=4). (D) Similar results are observed with average F-actin staining intensity, however, only serum free cultures demonstrate statistical significance (n=4). Data are mean±SEM. * p < 0.05.
Expression of SFRP1 with passage
Finally, as we have previously linked exogenous SFRP1 to increased HTM cellular stiffness, we used immunofluorescence and qPCR to assay the expression of SFRP1 in proliferative and senescent cultures of 3 different donors. Proliferative HTM cultures stained positive for SFRP1 (Figure 4A), but this was greatly increased in senescent cells (Figure 4B). SFRP1 staining intensity was increased 1.6131±0.15 fold (p<0.05) in full media and 1.7211±0.31 fold (p = 0.080) in serum free cultures (Figure 4C). Additionally, we used qPCR to quantify SFRP1 mRNA expression. In serum free cultures of 3 different donors, SFRP1 was upregulated 4.18±1.07 fold (p < 0.05) with senescence.
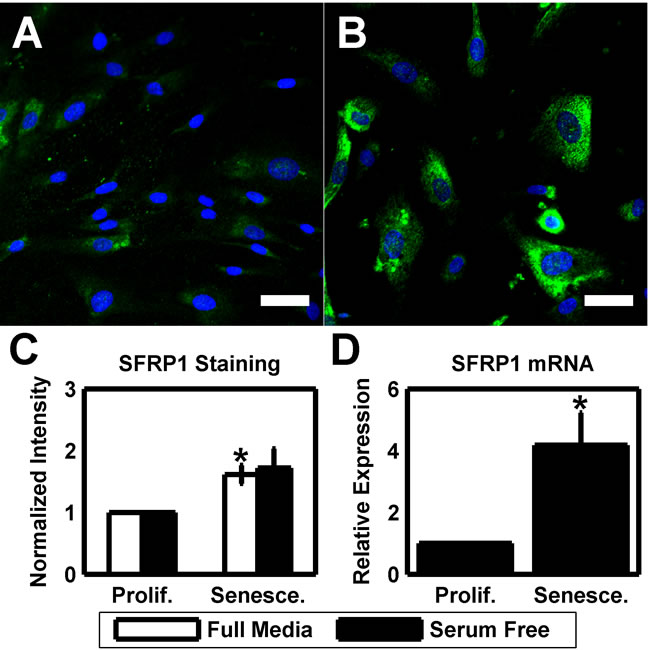
Figure 4: Elevated SFRP1 expression is a phenotype of HTM senescence. (A) HTM cells under routine culture (HTM667; passage 6) stain positive for SFRP1 (green). DAPI is used as a nuclear counterstain. Scale bar is 50 μm. (B) After serial passaging, senescent HTM cells (HTM667; passage 10) exhibit a substantial increase in SFRP1 staining intensity. Images are of HTM667 (passages 6 and 10) and are representative of cells from other donors. Scale bars are 50 μm. (C) Quantification of the average SFRP1 staining intensity reveals a significantly increased signal in senescent cultures when compared earlier passages for full media cultures (n=3). While elevated, serum free cultures are not significant. (D) SFRP1 is elevated at the message level in serum free senescent cultures relative to earlier passages (n=3). Data are mean±SEM. * p < 0.05.
Discussion
This study demonstrates senescence plays a prominent role in HTM mechanobiology. Utilizing serial passage of primary cells as an in vitro means of inducing senescence, this study demonstrates profound increases in cellular stiffness in senescent HTM cell populations. The increase occurred in the presence of serum but appeared more robust in serum free culture. This difference was an unexpected finding, and the role of serum signaling in senescence associated stiffening requires further study. The increase is correlated with, and likely related to, increases in vimentin, F-actin, and SFRP1 expression. Importantly, these data fill in crucial knowledge gaps in our understanding of HTM mechanobiology and build on previous work.
Our lab has shown that the HTM in glaucoma is ~20 fold stiffer [12], and available data suggests this is due to changes in both cytoskeletal dynamics and extracellular matrix [13-19]. Further, it is likely that these changes are coupled, as we have previously shown that HTM cells grown on stiffer matrices will have increased intrinsic stiffness [25] and elevated SFRP1 expression [21]. Despite the correlation of glaucoma with HTM stiffness, there is a paucity of data directly linking biological phenotypes of glaucoma with stiffening of the cells or matrix. Recently, we have shown that transforming growth factor β signaling, implicated in glaucoma, causes HTM cells to deposit a physically stiffer matrix with elevated SFRP1 secretion [92]. In a separate study, we demonstrated SFRP1, also associated with glaucoma, induces intrinsic stiffening of HTM cells [81]. Adding to these recent studies, we have now identified a third potential contributor to the overall stiffness of the HTM in glaucoma: cellular senescence.
It is instructive to consider the magnitude of the baseline HTM cell stiffness and the stiffening associated with senescence. As mentioned in the results, the baseline cellular stiffness is consistent with previous data on HTM cells [25, 81, 91] and is within the range previously reported for various cell types [93]. Further, the 2-3 fold change in HTM cellular stiffness with senescence is comparable to differences observed between metastatic and non-metastatic cancer cells [94-99], suggesting this difference could be coupled to dramatic functional changes. However, it is important to note that a 2-3 fold change is substantially smaller than the ~20 fold stiffening observed with glaucoma [12], suggesting senescence is but one of the factors contributing to HTM stiffening in vivo. The consequence that stiffening of cells has on ECM deposition and remodeling, particularly as it senesces, is poorly understood and warrants further investigations.
While the mechanisms underlying senescence-associated stiffening remain incompletely defined, it is likely cytoskeletal reorganization plays a key role. In numerous other studies, senescent cells or those from aged organisms exhibited marked changes to their cytoskeleton [66-70]. In this study, we stained for vimentin and F-actin, as both are known to be expressed in HTM [75-77] and relevant to cell stiffness [71-74]. Microtubules, while expressed in the HTM and altered in senescence [100-103], have been shown to have minimal impact on cellular mechanics [104, 105]. Another likely contributor, not explored in this study, is changes to the nuclear lamins, which contribute to nuclear stiffness [106, 107]. There is a well understood connectivity between the cytoskeleton and the lamina [108-110] and we feel it likely that the cytoskeletal alterations we have described are coupled with changes in lamin morphology. Indeed, the premature aging disease Hutchinson-Gilford progeria syndrome is linked to a splice variant of Lamin A, progerin [111, 112]. Progerin accumulation in cells leads to severe functional impairments [113-120], is associated with in vitro senescence [121], and has been identified in normal aging [122, 123]. As our studies utilize the nucleus as a consistent landmark for AFM indentation, the role of lamins is especially relevant. Future studies will investigate the role of lamin isoform expression in senescence associated stiffness.
We determined that SFRP1 is upregulated in HTM senescence (Figure 4), consistent with a previous report in different cell types [82]. SFRP1 is a potent inhibitor of Wnt signaling [124, 125], a key pathway that regulates numerous processes, including proliferation [126-129]. The findings of the current study serve to link findings from numerous previous reports by our lab and others. First, SFRP1 itself has been implicated in increased resistance to aqueous humor outflow in the HTM [32, 33], suggesting that senescence associated SFRP1 expression could directly hinder HTM function. Second, we have recently shown that SFRP1 induces HTM cell stiffening [81], providing a mechanism for a subpopulation of SFRP1 secreting cells to spread stiffening throughout the tissue. Third, HTM cells grown on hydrogels that mimic the stiffness of the glaucomatous HTM have increased stiffness as well as increased expression of SFRP1 [21, 25], suggesting that SFRP1 will be upregulated in the presence of tissue stiffening. Finally, high expression of SFRP1 has been shown to be sufficient to induce senescence in otherwise normal cells [82]. In aggregate, these data point to a feedback loop, where exposure to SFRP1 induces stiffening and senescence, which in turn induces further stiffening and SFRP1 expression/secretion to the extracellular milieu capable of affecting adjacent cells, compromising HTM function, and ultimately leading to glaucoma progression.
It is important to note that the above studies were performed on cells isolated from a range of donor ages (51-72 y.o.). It is possible that some of the observed donor variability is due to age, however, limited availability of tissue from a wide range of ages hinders the ability to properly explore these effects. As such, the above findings were not analyzed with respect to donor age due to the limited diversity of tissues.
Finally, while this study was performed specifically on HTM cells in the context of glaucoma, it potentially has broad applicability. Glaucoma exhibits many of the hallmarks of AADs, including fibrosis [15, 27, 130-133], oxidative stress [134-139], and loss of cellularity [55, 140-142]. It is quite possible the finding of increased stiffness with senescence is an aspect of many AADs. If so, further research into the causes and effects of this stiffening may identify novel therapeutic targets for treatment of a broad number of aging-associated diseases.
This study demonstrates a direct link between cellular senescence, intrinsic cell stiffening, and SFRP1 (a potent inhibitor of canonical Wnt signaling) expression in the HTM, all phenotypes associated with glaucoma. Taken with previous findings that SFRP1 can induce senescence and stiffening, these results suggest cellular senescence and resulting SFRP1 expression would induce further senescence and stiffening in neighboring cells, reinforcing and spreading the senescent phenotype and potentially leading to ocular hypertension and glaucoma. The identification of this feedback loop could have important ramifications for the design of effective therapeutics. Further, as glaucoma has many of the hallmarks of AADs, there is substantial potential that these findings may provide insight into other AADs.
materials and Methods
Culture of HTM cells and serial passaging
All experiments were performed in compliance with the Declaration of Helsinki. Primary HTM cells were isolated from eight donor corneoscleral rims (Saving Sight Eye Bank, St. Louis, MO) as described previously [143]. Cells derived from each donor are given a unique 3 digit identifier. For this study, HTM cells HTM517, HTM553, HTM554, HTM621, HTM627, HTM631, HTM667, and HTM728 were isolated from donors of age 51, 55, 55, 62, 62, 63, 66, and 72. HTM cells were cultured in DMEM/F12 (Hyclone, Logan, UT) with 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA) and 2 mM penicillin, streptomycin, amphotericin-B (Life Technologies, Carlsbad, CA). HTM cells were plated on glass coverslips at a 25,000 cells/cm2 and allowed to attach overnight. In order to isolate the influence of serum, cells were washed the following day and incubated in either serum-containing media (full media) or serum free media for three days before analysis.
Cells were routinely passaged at approximately 90% confluence. Cultures were continued at 1x106 cells per 75 cm2 tissue culture flask. Cells were determined to be senescent when they failed to proliferate and exhibited a flattened, enlarged morphology. Failure of proliferation was determined by cell count (Cellometer Vision; Nexcelom, Lawrence, MA) as having equal to or fewer viable cells one week after plating of a given passage. A similar methodology has previously been successful at inducing a senescent phenotype in porcine trabecular meshwork cells [144]. This methodology was confirmed by the prominent expression of senescence associated β-galactosidase, assayed with a commercial kit (5 donors; Cell Signaling Technology, Danvers, MA). Cultures typically become predominantly senescent at passages 10 to 15, although 2 sets of donor cells senesced as early as passage 7.
Atomic force microscopy analysis
Cell mechanics were determined as described previously using the Asylum MFP-3D-Bio AFM [25, 91]. Cells were rinsed in Hanks’ Balanced Salt Solution (Hyclone, Logan, Utah), equilibrated on the AFM stage to minimize thermal drift, and indented in contact mode with silicon nitride cantilevers with square pyramidal tips (PNP-TR-50, Nano World, Switzerland). Prior to each experiment, the spring constant and deflection sensitivity of the cantilever were determined using thermal tuning and constant compliance methods. The elastic modulus (E) of each sample was obtained by fitting indentation force versus indentation depth to the Hertz model as shown in Eq. 1, where F is the force applied by indenter, α is the tip half angle (35o), ν is Poisson’s ratio, and δ is indentation depth.
![]() (1)
(1)
The Hertz model assumes that the samples were linearly elastic, homogenous, and infinitely thick. However, in the limit of small deformations, the Hertz model can be used for materials (such as cells) which are viscoelastic, heterogeneous, and finite [145]. Additionally, we assume the cells are incompressible (ν = 0.5), a good approximation for biological materials with high water content [146-149]. For each sample, approximately 5-7 cells were indented 5-7 times at 2 µm/s. All indentations were centered above the cell nucleus to minimize variability.
Immunocytochemical analysis
Cells were washed in HBSS and fixed for 20 minutes in 4% formaldehyde and 0.25% Triton-X 100 (Fisher Scientific, Waltham, MA) in phosphate buffered saline (PBS) and blocked with 2% bovine serum albumin (Fisher), 0.2% gelatin (cold-fish; Sigma-Aldrich), 0.1% Tween-20 (Fisher) in PBS for 1 hr. Following blocking, cells were stained overnight using rabbit anti-SFRP1 (Abcam, Cambridge, UK) or rabbit anti-vimentin (Cell Signaling Technology, Danvers, MA) with a secondary of DyLight 488 conjugated goat anti-rabbit (Fisher). Nuclei and filamentous actin were counterstained with DAPI and phalloidin conjugated to Alexafluor 594, respectively. Cells were rinsed in PBS, mounted, and imaged on a Zeiss Axiovert 200 inverted microscope. All acquisition settings were kept constant for documentation of the same stain type (e.g. all phalloidin imaging utilized the same acquisition parameters). A minimum of 8 random fields were acquired for each case (approximately 100 cells). Cell borders were traced using semi-automated thresholding algorithm and mean gray scale intensity was averaged across the cell covered area across all images. For accurate quantification of the SFRP1 intensity, rolling ball background subtraction was employed. All analysis was conducted using custom image analysis scripts in MATLAB 2008b (Mathworks, Natick, MA). Thresholding and background subtraction parameters were manually checked for fidelity and applied uniformly across all images of a stain.
Quantitative real-time polymerase chain reaction (qPCR)
Cells were cultured in 60 mm dishes under SF conditions as described above. After 3 days, the cells were lysed and mRNA was isolated using an RNeasy kit (Qiagen, Venlo, Netherlands) and the relative expression of SFRP1 was quantified from the mRNA by qPCR using SensiFAST Hi-ROX One-Step mastermix (Bioline, Taunton, MA) and Taqman primers (Hs00610060_m1; Life Technologies, Carlsbad, CA) using 60 ng of mRNA per reaction. Gene expression was normalized to that of the endogenous control, 18S rRNA (Hs99999901_s1; Life Technologies). Expression of the senescent cultures was determined relative to proliferative controls.
Statistics
All experiments were with HTM cells isolated from multiple donors. The number of replicates is noted for each experimental set. For comparison AFM results within an individual donor cells, significance was assessed for full and serum free conditions separately using one-way ANOVA followed by Bonferroni’s post-hoc test. Significant differences (p<0.05) from the earliest passage are indicated with †. For all other data, fold change of senescent cultures was determined in reference to control cells (proliferative cells of earlier passage) and significance was assessed using Student’s t-test and significance denoted by ** = p<0.01, and * = p<0.05. All quantification is displayed as mean ± SEM and the number of biological replicates is noted.
Acknowledgements
The authors would like to thank Lola Davis for immunofluorescent image collection. This work was funded by grants from the National Institutes of Health R01EY019475, R01EY019970 and P30EY12576, and an unrestricted grant from Research to Prevent Blindness. The authors have no conflicts of interest related to this publication.
Conflicts of interest
There is no conflict of interest to declare.
References
1. Quigley HA and Broman AT. The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol. 2006; 90:262-267.
2. Quigley HA. Open-angle glaucoma. N Engl J Med. 1993; 328:1097-1106.
3. Johnson M. ‘What controls aqueous humour outflow resistance?’. Exp Eye Res. 2006; 82:545-557.
4. Johnstone MA and Grant WG. Pressure-dependent changes in structures of the aqueous outflow system of human and monkey eyes. Am J Ophthalmol. 1973; 75:365-383.
5. Maepea O and Bill A. Pressures in the juxtacanalicular tissue and Schlemm’s canal in monkeys. Exp Eye Res. 1992; 54:879-883.
6. Acott TS and Kelley MJ. Extracellular matrix in the trabecular meshwork. Exp Eye Res. 2008; 86(4):543-561.
7. Keller KE and Acott TS. The Juxtacanalicular Region of Ocular Trabecular Meshwork: A Tissue with a Unique Extracellular Matrix and Specialized Function. Journal of ocular biology. 2013; 1(1):3.
8. Bhatt K, Gong H and Freddo TF. Freeze-fracture studies of interendothelial junctions in the angle of the human eye. Invest Ophthalmol Vis Sci. 1995; 36:1379-1389.
9. Gong H, Ruberti J, Overby D, Johnson M and Freddo TF. A new view of the human trabecular meshwork using quick-freeze, deep-etch electron microscopy. Exp Eye Res. 2002; 75:347-358.
10. Grierson I, Lee WR, Abraham S and Howes RC. Associations between the cells of the walls of Schlemm’s canal. Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie Albrecht von Graefe’s archive for clinical and experimental ophthalmology. 1978; 208:33-47.
11. Inomata H, Bill A and Smelser GK. Aqueous humor pathways through the trabecular meshwork and into Schlemm‘s canal in the cynomolgus monkey (Macaca irus). An electron microscopic study. Am J Ophthalmol. 1972; 73:760-789.
12. Last JA, Pan T, Ding Y, Reilly CM, Keller K, Acott TS, Fautsch MP, Murphy CJ and Russell P. Elastic modulus determination of normal and glaucomatous human trabecular meshwork. Invest Ophthalmol Vis Sci. 2011; 52:2147-2152.
13. Hoare MJ, Grierson I, Brotchie D, Pollock N, Cracknell K and Clark AF. Cross-linked actin networks (CLANs) in the trabecular meshwork of the normal and glaucomatous human eye in situ. Invest Ophthalmol Vis Sci. 2009; 50:1255-1263.
14. Clark AF, Miggans ST, Wilson K, Browder S and McCartney MD. Cytoskeletal changes in cultured human glaucoma trabecular meshwork cells. Journal of glaucoma. 1995; 4:183-188.
15. Fuchshofer R and Tamm ER. The role of TGF-beta in the pathogenesis of primary open-angle glaucoma. Cell Tissue Res. 2012; 347:279-290.
16. Saika S. TGFbeta pathobiology in the eye. Lab Invest. 2006; 86:106-115.
17. Gottanka J, Johnson DH, Martus P and Lutjen-Drecoll E. Severity of optic nerve damage in eyes with POAG is correlated with changes in the trabecular meshwork. Journal of glaucoma. 1997; 6:123-132.
18. Lutjen-Drecoll E. Morphological changes in glaucomatous eyes and the role of TGFbeta2 for the pathogenesis of the disease. Exp Eye Res. 2005; 81:1-4.
19. Rohen JW, Lutjen-Drecoll E, Flugel C, Meyer M and Grierson I. Ultrastructure of the trabecular meshwork in untreated cases of primary open-angle glaucoma (POAG). Exp Eye Res. 1993; 56:683-692.
20. Schlunck G, Han H, Wecker T, Kampik D, Meyer-ter-Vehn T and Grehn F. Substrate rigidity modulates cell matrix interactions and protein expression in human trabecular meshwork cells. Invest Ophthalmol Vis Sci. 2008; 49:262-269.
21. Raghunathan VK, Morgan JT, Dreier B, Reilly CM, Thomasy SM, Wood JA, Ly I, Tuyen BC, Hughbanks M, Murphy CJ and Russell P. Role of substratum stiffness in modulating genes associated with extracellular matrix and mechanotransducers YAP and TAZ. Invest Ophthalmol Vis Sci. 2013; 54:378-386.
22. Thomasy SM, Morgan JT, Wood JA, Murphy CJ and Russell P. Substratum stiffness and latrunculin B modulate the gene expression of the mechanotransducers YAP and TAZ in human trabecular meshwork cells. Exp Eye Res. 2013; 113:66-73.
23. Thomasy SM, Wood JA, Kass PH, Murphy CJ and Russell P. Substratum stiffness and latrunculin B regulate matrix gene and protein expression in human trabecular meshwork cells. Invest Ophthalmol Vis Sci. 2012; 53:952-958.
24. Wood JA, McKee CT, Thomasy SM, Fischer ME, Shah NM, Murphy CJ and Russell P. Substratum compliance regulates human trabecular meshwork cell behaviors and response to latrunculin B. Invest Ophthalmol Vis Sci. 2011; 52:9298-9303.
25. McKee CT, Wood JA, Shah NM, Fischer ME, Reilly CM, Murphy CJ and Russell P. The effect of biophysical attributes of the ocular trabecular meshwork associated with glaucoma on the cell response to therapeutic agents. Biomaterials. 2011; 32:2417-2423.
26. Haddadin RI, Oh DJ, Kang MH, Filippopoulos T, Gupta M, Hart L, Sage EH and Rhee DJ. SPARC-null mice exhibit lower intraocular pressures. Invest Ophthalmol Vis Sci. 2009; 50:3771-3777.
27. Oh DJ, Kang MH, Ooi YH, Choi KR, Sage EH and Rhee DJ. Overexpression of SPARC in human trabecular meshwork increases intraocular pressure and alters extracellular matrix. Invest Ophthalmol Vis Sci. 2013; 54:3309-3319.
28. Swaminathan SS, Oh D, Kang M, Shepard AR, Pang IH and Rhee DJ. (2014). In vivo study of the role of SPARC in TGFb2-mediated ocular hypertension. The Association for Research in Vision and Ophthalmology Annual Meeting. (Orlando, Florida.
29. Swaminathan SS, Oh DJ, Kang MH, Ren R, Jin R, Gong H and Rhee DJ. Secreted protein acidic and rich in cysteine (SPARC)-null mice exhibit more uniform outflow. Invest Ophthalmol Vis Sci. 2013; 54:2035-2047.
30. Tamm ER. Myocilin and glaucoma: facts and ideas. Prog Retin Eye Res. 2002; 21:395-428.
31. Polansky JR, Fauss DJ, Chen P, Chen H, Lutjen-Drecoll E, Johnson D, Kurtz RM, Ma ZD, Bloom E and Nguyen TD. Cellular pharmacology and molecular biology of the trabecular meshwork inducible glucocorticoid response gene product. Ophthalmologica. 1997; 211:126-139.
32. Wang WH, McNatt LG, Pang IH, Millar JC, Hellberg PE, Hellberg MH, Steely HT, Rubin JS, Fingert JH, Sheffield VC, Stone EM and Clark AF. Increased expression of the WNT antagonist sFRP-1 in glaucoma elevates intraocular pressure. J Clin Invest. 2008; 118:1056-1064.
33. Mao W, Millar JC, Wang WH, Silverman SM, Liu Y, Wordinger RJ, Rubin JS, Pang IH and Clark AF. Existence of the canonical Wnt signaling pathway in the human trabecular meshwork. Invest Ophthalmol Vis Sci. 2012.
34. Fingert JH, Stone EM, Sheffield VC and Alward WL. Myocilin glaucoma. Surv Ophthalmol. 2002; 47:547-561.
35. Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013; 75:685-705.
36. Ohtani N and Hara E. Roles and mechanisms of cellular senescence in regulation of tissue homeostasis. Cancer Sci. 2013; 104:525-530.
37. Hayflick L. The Limited in Vitro Lifetime of Human Diploid Cell Strains. Exp Cell Res. 1965; 37:614-636.
38. Takahashi A, Ohtani N and Hara E. Irreversibility of cellular senescence: dual roles of p16INK4a/Rb-pathway in cell cycle control. Cell division. 2007; 2:10.
39. Campisi J and d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007; 8:729-740.
40. Hayflick L and Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961; 25:585-621.
41. Ben-Porath I and Weinberg RA. When cells get stressed: an integrative view of cellular senescence. J Clin Invest. 2004; 113:8-13.
42. von Zglinicki T, Petrie J and Kirkwood TB. Telomere-driven replicative senescence is a stress response. Nat Biotechnol. 2003; 21:229-230.
43. von Zglinicki T, Serra V, Lorenz M, Saretzki G, Lenzen-Grossimlighaus R, Gessner R, Risch A and Steinhagen-Thiessen E. Short telomeres in patients with vascular dementia: an indicator of low antioxidative capacity and a possible risk factor? Lab Invest. 2000; 80(11):1739-1747.
44. Henle ES, Han Z, Tang N, Rai P, Luo Y and Linn S. Sequence-specific DNA cleavage by Fe2+-mediated fenton reactions has possible biological implications. J Biol Chem. 1999; 274:962-971.
45. Serra V, von Zglinicki T, Lorenz M and Saretzki G. Extracellular superoxide dismutase is a major antioxidant in human fibroblasts and slows telomere shortening. J Biol Chem. 2003; 278:6824-6830.
46. Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S and Campisi J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol. 2003; 5:741-747.
47. Chen Q, Fischer A, Reagan JD, Yan LJ and Ames BN. Oxidative DNA damage and senescence of human diploid fibroblast cells. Proc Natl Acad Sci U S A. 1995; 92:4337-4341.
48. Jeyapalan JC, Ferreira M, Sedivy JM and Herbig U. Accumulation of senescent cells in mitotic tissue of aging primates. Mech Ageing Dev. 2007; 128:36-44.
49. Takubo K, Aida J, Izumiyama-Shimomura N, Ishikawa N, Sawabe M, Kurabayashi R, Shiraishi H, Arai T and Nakamura K. Changes of telomere length with aging. Geriatrics & gerontology international. 2010; 10 Suppl 1:S197-206.
50. Herbig U, Ferreira M, Condel L, Carey D and Sedivy JM. Cellular senescence in aging primates. Science. 2006; 311:1257.
51. Waaijer ME, Parish WE, Strongitharm BH, van Heemst D, Slagboom PE, de Craen AJ, Sedivy JM, Westendorp RG, Gunn DA and Maier AB. The number of p16INK4a positive cells in human skin reflects biological age. Aging Cell. 2012; 11:722-725.
52. Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O and et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995; 92:9363-9367.
53. Campisi J and Sedivy J. How does proliferative homeostasis change with age? What causes it and how does it contribute to aging? J Gerontol A Biol Sci Med Sci. 2009; 64:164-166.
54. Tchkonia T, Zhu Y, van Deursen J, Campisi J and Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. 2013; 123:966-972.
55. Liton PB, Challa P, Stinnett S, Luna C, Epstein DL and Gonzalez P. Cellular senescence in the glaucomatous outflow pathway. Exp Gerontol. 2005; 40:745-748.
56. Gotschy A, Bauer E, Schrodt C, Lykowsky G, Ye YX, Rommel E, Jakob PM, Bauer WR and Herold V. Local arterial stiffening assessed by MRI precedes atherosclerotic plaque formation. Circ Cardiovasc Imaging. 2013; 6:916-923.
57. Peloquin J, Huynh J, Williams RM and Reinhart-King CA. Indentation measurements of the subendothelial matrix in bovine carotid arteries. J Biomech. 2011; 44:815-821.
58. Tracqui P, Broisat A, Toczek J, Mesnier N, Ohayon J and Riou L. Mapping elasticity moduli of atherosclerotic plaque in situ via atomic force microscopy. J Struct Biol. 2011; 174:115-123.
59. Fisher RF. The influence of age on some ocular basement membranes. Eye (Lond). 1987; 1:184-189.
60. Spraul CW, Lang GE, Grossniklaus HE and Lang GK. Histologic and morphometric analysis of the choroid, Bruch’s membrane, and retinal pigment epithelium in postmortem eyes with age-related macular degeneration and histologic examination of surgically excised choroidal neovascular membranes. Surv Ophthalmol. 1999; 44 Suppl 1:S10-32.
61. Zarbin MA. Current concepts in the pathogenesis of age-related macular degeneration. Arch Ophthalmol. 2004; 122:598-614.
62. Castle WD and Gow BS. Changes in the microindentation properties of aortic intimal surface during cholesterol feeding of rabbits. Atherosclerosis. 1983; 47:251-261.
63. Schedin P and Keely PJ. Mammary gland ECM remodeling, stiffness, and mechanosignaling in normal development and tumor progression. Cold Spring Harb Perspect Biol. 2011; 3:a003228.
64. Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, Hammer DA and Weaver VM. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005; 8:241-254.
65. Au FW, Ghai S, Moshonov H, Kahn H, Brennan C, Dua H and Crystal P. Diagnostic performance of quantitative shear wave elastography in the evaluation of solid breast masses: determination of the most discriminatory parameter. AJR Am J Roentgenol. 2014; 203:W328-336.
66. Nishio K and Inoue A. Senescence-associated alterations of cytoskeleton: extraordinary production of vimentin that anchors cytoplasmic p53 in senescent human fibroblasts. Histochem Cell Biol. 2005; 123:263-273.
67. Nishio K, Inoue A, Qiao S, Kondo H and Mimura A. Senescence and cytoskeleton: overproduction of vimentin induces senescent-like morphology in human fibroblasts. Histochem Cell Biol. 2001; 116:321-327.
68. Garcia GG and Miller RA. Age-related defects in the cytoskeleton signaling pathways of CD4 T cells. Ageing Res Rev. 2011; 10:26-34.
69. Sanchez-Perez Y, Chirino YI, Osornio-Vargas AR, Herrera LA, Morales-Barcenas R, Lopez-Saavedra A, Gonzalez-Ramirez I, Miranda J and Garcia-Cuellar CM. Cytoplasmic p21(CIP1/WAF1), ERK1/2 activation, and cytoskeletal remodeling are associated with the senescence-like phenotype after airborne particulate matter (PM10) exposure in lung cells. Toxicol Lett. 2014; 225:12-19.
70. Chen QM, Tu VC, Catania J, Burton M, Toussaint O and Dilley T. Involvement of Rb family proteins, focal adhesion proteins and protein synthesis in senescent morphogenesis induced by hydrogen peroxide. J Cell Sci. 2000; 113:4087-4097.
71. Plodinec M, Loparic M, Suetterlin R, Herrmann H, Aebi U and Schoenenberger CA. The nanomechanical properties of rat fibroblasts are modulated by interfering with the vimentin intermediate filament system. J Struct Biol. 2011; 174:476-484.
72. Chahine NO, Blanchette C, Thomas CB, Lu J, Haudenschild D and Loots GG. Effect of age and cytoskeletal elements on the indentation-dependent mechanical properties of chondrocytes. PLoS One. 2013; 8:e61651.
73. Lee SM, Nguyen TH, Na K, Cho IJ, Woo DH, Oh JE, Lee CJ and Yoon ES. Nanomechanical measurement of astrocyte stiffness correlated with cytoskeletal maturation. J Biomed Mater Res A. 2015; 103:365-370.
74. Lulevich V, Yang HY, Isseroff RR and Liu GY. Single cell mechanics of keratinocyte cells. Ultramicroscopy. 2010; 110:1435-1442.
75. Ryder MI, Weinreb RN, Alvarado J and Polansky J. The cytoskeleton of the cultured human trabecular cell. Characterization and drug responses. Invest Ophthalmol Vis Sci. 1988; 29:251-260.
76. Iwamoto Y and Tamura M. Immunocytochemical study of intermediate filaments in cultured human trabecular cells. Invest Ophthalmol Vis Sci. 1988; 29:244-250.
77. Weinreb RN and Ryder MI. In situ localization of cytoskeletal elements in the human trabecular meshwork and cornea. Invest Ophthalmol Vis Sci. 1990; 31:1839-1847.
78. Tian B, Geiger B, Epstein DL and Kaufman PL. Cytoskeletal involvement in the regulation of aqueous humor outflow. Invest Ophthalmol Vis Sci. 2000; 41:619-623.
79. Clark AF, Brotchie D, Read AT, Hellberg P, English-Wright S, Pang IH, Ethier CR and Grierson I. Dexamethasone alters F-actin architecture and promotes cross-linked actin network formation in human trabecular meshwork tissue. Cell Motil Cytoskeleton. 2005; 60:83-95.
80. Clark AF, Wilson K, McCartney MD, Miggans ST, Kunkle M and Howe W. Glucocorticoid-induced formation of cross-linked actin networks in cultured human trabecular meshwork cells. Invest Ophthalmol Vis Sci. 1994; 35:281-294.
81. Morgan JT, Raghunathan VK, Chang YR, Murphy CJ and Russell P. Wnt inhibition induces persistent increases in intrinsic stiffness of human trabecular meshwork cells. Exp Eye Res. 2015; In Press.
82. Elzi DJ, Song M, Hakala K, Weintraub ST and Shiio Y. Wnt Antagonist SFRP1 Functions as a Secreted Mediator of Senescence. Mol Cell Biol. 2012; 32:4388-4399.
83. Cristofalo VJ and Sharf BB. Cellular senescence and DNA synthesis. Thymidine incorporation as a measure of population age in human diploid cells. Exp Cell Res. 1973; 76:419-427.
84. Yanishevsky R, Mendelsohn ML, Mayall BH and Cristofalo VJ. Proliferative capacity and DNA content of aging human diploid cells in culture: a cytophotometric and autoradiographic analysis. J Cell Physiol. 1974; 84:165-170.
85. Kumazaki T, Robetorye RS, Robetorye SC and Smith JR. Fibronectin expression increases during in vitro cellular senescence: correlation with increased cell area. Exp Cell Res. 1991; 195:13-19.
86. Angello JC, Pendergrass WR, Norwood TH and Prothero J. Proliferative potential of human fibroblasts: an inverse dependence on cell size. J Cell Physiol. 1987; 132:125-130.
87. Sherwood SW, Rush D, Ellsworth JL and Schimke RT. Defining cellular senescence in IMR-90 cells: a flow cytometric analysis. Proc Natl Acad Sci U S A. 1988; 85:9086-9090.
88. Kulju KS and Lehman JM. Increased p53 protein associated with aging in human diploid fibroblasts. Exp Cell Res. 1995; 217:336-345.
89. Itahana K, Campisi J and Dimri GP. Methods to detect biomarkers of cellular senescence: the senescence-associated beta-galactosidase assay. Methods Mol Biol. 2007; 371:21-31.
90. de Jesus BB and Blasco MA. Assessing cell and organ senescence biomarkers. Circ Res. 2012; 111:97-109.
91. Murphy KC, Morgan JT, Wood JA, Sadeli A, Murphy CJ and Russell P. The formation of cortical actin arrays in human trabecular meshwork cells in response to cytoskeletal disruption. Exp Cell Res. 2014.
92. Raghunathan VK, Morgan JT, Chang YR, Weber D, Phinney B, Murphy CJ and Russell P. Transforming growth factor beta 3 modifies mechanics and composition of extracellular matrix deposited by human trabecular meshwork cells. ACS Biomaterials Science & Engineering. 2015; In Press.
93. Kuznetsova TG, Starodubtseva MN, Yegorenkov NI, Chizhik SA and Zhdanov RI. Atomic force microscopy probing of cell elasticity. Micron. 2007; 38:824-833.
94. Cross SE, Jin YS, Tondre J, Wong R, Rao J and Gimzewski JK. AFM-based analysis of human metastatic cancer cells. Nanotechnology. 2008; 19:384003.
95. Li QS, Lee GY, Ong CN and Lim CT. AFM indentation study of breast cancer cells. Biochem Biophys Res Commun. 2008; 374:609-613.
96. Lekka M, Laidler P, Gil D, Lekki J, Stachura Z and Hrynkiewicz AZ. Elasticity of normal and cancerous human bladder cells studied by scanning force microscopy. Eur Biophys J. 1999; 28:312-316.
97. Cross SE, Jin YS, Rao J and Gimzewski JK. Nanomechanical analysis of cells from cancer patients. Nat Nanotechnol. 2007; 2:780-783.
98. Xu W, Mezencev R, Kim B, Wang L, McDonald J and Sulchek T. Cell stiffness is a biomarker of the metastatic potential of ovarian cancer cells. PLoS One. 2012; 7:e46609.
99. Watanabe T, Kuramochi H, Takahashi A, Imai K, Katsuta N, Nakayama T, Fujiki H and Suganuma M. Higher cell stiffness indicating lower metastatic potential in B16 melanoma cell variants and in (-)-epigallocatechin gallate-treated cells. J Cancer Res Clin Oncol. 2012.
100. Raes M, Geuens G, de Brabander M and Remacle J. Microtubules and microfilaments in ageing hamster embryo fibroblasts in vitro. Exp Gerontol. 1983; 18:241-254.
101. Wang E and Gundersen D. Increased organization of cytoskeleton accompanying the aging of human fibroblasts in vitro. Exp Cell Res. 1984; 154:191-202.
102. Van Gansen P, Siebertz B, Capone B and Malherbe L. Relationships between cytoplasmic microtubular complex, DNA synthesis and cell morphology in mouse embryonic fibroblasts (effects of age, serum deprivation, aphidicolin, cytochalasin B and colchicine). Biol Cell. 1984; 52:161-174.
103. Raes M. Involvement of microtubules in modifications associated with cellular aging. Mutat Res. 1991; 256:149-168.
104. Pesen D and Hoh JH. Micromechanical architecture of the endothelial cell cortex. Biophys J. 2005; 88:670-679.
105. Haga H, Sasaki S, Kawabata K, Ito E, Ushiki T and Sambongi T. Elasticity mapping of living fibroblasts by AFM and immunofluorescence observation of the cytoskeleton. Ultramicroscopy. 2000; 82:253-258.
106. Lammerding J, Schulze PC, Takahashi T, Kozlov S, Sullivan T, Kamm RD, Stewart CL and Lee RT. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest. 2004; 113:370-378.
107. Lammerding J, Fong LG, Ji JY, Reue K, Stewart CL, Young SG and Lee RT. Lamins A and C but not lamin B1 regulate nuclear mechanics. J Biol Chem. 2006; 281:25768-25780.
108. Lee JS, Hale CM, Panorchan P, Khatau SB, George JP, Tseng Y, Stewart CL, Hodzic D and Wirtz D. Nuclear lamin A/C deficiency induces defects in cell mechanics, polarization, and migration. Biophys J. 2007; 93:2542-2552.
109. Stewart-Hutchinson PJ, Hale CM, Wirtz D and Hodzic D. Structural requirements for the assembly of LINC complexes and their function in cellular mechanical stiffness. Exp Cell Res. 2008; 314:1892-1905.
110. Maniotis AJ, Chen CS and Ingber DE. Demonstration of mechanical connections between integrins, cytoskeletal filaments, and nucleoplasm that stabilize nuclear structure. Proc Natl Acad Sci U S A. 1997; 94:849-854.
111. Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, Dutra A, Pak E, Durkin S, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003; 423:293-298.
112. De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, Lyonnet S, Stewart CL, Munnich A, Le Merrer M and Levy N. Lamin a truncation in Hutchinson-Gilford progeria. Science. 2003; 300:2055.
113. Verstraeten VL, Ji JY, Cummings KS, Lee RT and Lammerding J. Increased mechanosensitivity and nuclear stiffness in Hutchinson-Gilford progeria cells: effects of farnesyltransferase inhibitors. Aging Cell. 2008; 7:383-393.
114. Dahl KN, Scaffidi P, Islam MF, Yodh AG, Wilson KL and Misteli T. Distinct structural and mechanical properties of the nuclear lamina in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2006; 103:10271-10276.
115. Snow CJ, Dar A, Dutta A, Kehlenbach RH and Paschal BM. Defective nuclear import of Tpr in Progeria reflects the Ran sensitivity of large cargo transport. J Cell Biol. 2013; 201:541-557.
116. Kelley JB, Datta S, Snow CJ, Chatterjee M, Ni L, Spencer A, Yang CS, Cubenas-Potts C, Matunis MJ and Paschal BM. The defective nuclear lamina in Hutchinson-Gilford progeria syndrome disrupts the nucleocytoplasmic Ran gradient and inhibits nuclear localization of Ubc9. Mol Cell Biol. 2011; 31:3378-3395.
117. Goldman RD, Shumaker DK, Erdos MR, Eriksson M, Goldman AE, Gordon LB, Gruenbaum Y, Khuon S, Mendez M, Varga R and Collins FS. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2004; 101:8963-8968.
118. Cao K, Capell BC, Erdos MR, Djabali K and Collins FS. A lamin A protein isoform overexpressed in Hutchinson-Gilford progeria syndrome interferes with mitosis in progeria and normal cells. Proc Natl Acad Sci U S A. 2007; 104:4949-4954.
119. Capell BC, Erdos MR, Madigan JP, Fiordalisi JJ, Varga R, Conneely KN, Gordon LB, Der CJ, Cox AD and Collins FS. Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2005; 102:12879-12884.
120. Glynn MW and Glover TW. Incomplete processing of mutant lamin A in Hutchinson-Gilford progeria leads to nuclear abnormalities, which are reversed by farnesyltransferase inhibition. Hum Mol Genet. 2005; 14:2959-2969.
121. Cao K, Blair CD, Faddah DA, Kieckhaefer JE, Olive M, Erdos MR, Nabel EG and Collins FS. Progerin and telomere dysfunction collaborate to trigger cellular senescence in normal human fibroblasts. J Clin Invest. 2011; 121:2833-2844.
122. Scaffidi P and Misteli T. Lamin A-dependent nuclear defects in human aging. Science. 2006; 312:1059-1063.
123. Rodriguez S, Coppede F, Sagelius H and Eriksson M. Increased expression of the Hutchinson-Gilford progeria syndrome truncated lamin A transcript during cell aging. Eur J Hum Genet. 2009; 17:928-937.
124. Satoh W, Matsuyama M, Takemura H, Aizawa S and Shimono A. Sfrp1, Sfrp2, and Sfrp5 regulate the Wnt/beta-catenin and the planar cell polarity pathways during early trunk formation in mouse. Genesis. 2008; 46:92-103.
125. Kawano Y and Kypta R. Secreted antagonists of the Wnt signalling pathway. J Cell Sci. 2003; 116:2627-2634.
126. Yu J and Virshup DM. Updating the Wnt pathways. Biosci Rep. 2014.
127. Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006; 127:469-480.
128. Freese JL, Pino D and Pleasure SJ. Wnt signaling in development and disease. Neurobiol Dis. 2010; 38:148-153.
129. Macdonald BT and He X. Frizzled and LRP5/6 Receptors for Wnt/beta-Catenin Signaling. Cold Spring Harb Perspect Biol. 2012; 4.
130. Wordinger RJ, Sharma T and Clark AF. The Role of TGF-beta2 and Bone Morphogenetic Proteins in the Trabecular Meshwork and Glaucoma. J Ocul Pharmacol Ther. 2014; 30:154-162.
131. Rhee DJ, Haddadin RI, Kang MH and Oh DJ. Matricellular proteins in the trabecular meshwork. Exp Eye Res. 2009; 88:694-703.
132. Tamm ER and Fuchshofer R. What increases outflow resistance in primary open-angle glaucoma? Surv Ophthalmol. 2007; 52 Suppl 2:S101-104.
133. Fuchshofer R and Tamm ER. Modulation of extracellular matrix turnover in the trabecular meshwork. Exp Eye Res. 2009; 88:683-688.
134. Sacca SC and Izzotti A. Focus on molecular events in the anterior chamber leading to glaucoma. Cell Mol Life Sci. 2013.
135. Ghanem AA, Arafa LF and El-Baz A. Oxidative stress markers in patients with primary open-angle glaucoma. Curr Eye Res. 2010; 35:295-301.
136. Izzotti A, Sacca SC, Longobardi M and Cartiglia C. Mitochondrial damage in the trabecular meshwork of patients with glaucoma. Arch Ophthalmol. 2010; 128:724-730.
137. Sacca SC and Izzotti A. Oxidative stress and glaucoma: injury in the anterior segment of the eye. Prog Brain Res. 2008; 173:385-407.
138. Sacca SC, Izzotti A, Rossi P and Traverso C. Glaucomatous outflow pathway and oxidative stress. Exp Eye Res. 2007; 84:389-399.
139. Sacca SC, Pascotto A, Camicione P, Capris P and Izzotti A. Oxidative DNA damage in the human trabecular meshwork: clinical correlation in patients with primary open-angle glaucoma. Arch Ophthalmol. 2005; 123:458-463.
140. Alvarado J, Murphy C and Juster R. Trabecular meshwork cellularity in primary open-angle glaucoma and nonglaucomatous normals. Ophthalmology. 1984; 91:564-579.
141. Alvarado J, Murphy C, Polansky J and Juster R. Age-related changes in trabecular meshwork cellularity. Invest Ophthalmol Vis Sci. 1981; 21:714-727.
142. Grierson I and Howes RC. Age-related depletion of the cell population in the human trabecular meshwork. Eye. 1987; 1:204-210.
143. Morgan JT, Wood JA, Walker NJ, Raghunathan VK, Borjesson DL, Murphy CJ and Russell P. Human Trabecular Meshwork Cells Exhibit Several Characteristics of, but Are Distinct from, Adipose-Derived Mesenchymal Stem Cells. J Ocul Pharmacol Ther. 2014; 30:254-266.
144. Yamazaki Y, Matsunaga H, Nishikawa M, Ando A, Kaneko S, Okuda K, Wada M, Ito S and Matsumura M. Senescence in cultured trabecular meshwork cells. Br J Ophthalmol. 2007; 91:808-811.
145. Mahaffy RE, Shih CK, MacKintosh FC and Kas J. Scanning probe-based frequency-dependent microrheology of polymer gels and biological cells. Phys Rev Lett. 2000; 85:880-883.
146. Dimitriadis EK, Horkay F, Maresca J, Kachar B and Chadwick RS. Determination of elastic moduli of thin layers of soft material using the atomic force microscope. Biophys J. 2002; 82:2798-2810.
147. Ahearne M, Yang Y, El Haj AJ, Then KY and Liu KK. Characterizing the viscoelastic properties of thin hydrogel-based constructs for tissue engineering applications. Journal of the Royal Society, Interface / the Royal Society. 2005; 2:455-463.
148. Vinckier A and Semenza G. Measuring elasticity of biological materials by atomic force microscopy. FEBS Lett. 1998; 430:12-16.
149. Anseth KS, Bowman CN and Brannon-Peppas L. Mechanical properties of hydrogels and their experimental determination. Biomaterials. 1996; 17:1647-1657.