
INTRODUCTION
Despite advances in the treatments for breast cancer, effective treatments for brain metastases remain elusive. It is estimated that 30% of breast cancer patients will develop a brain metastasis, and with extended patient survival times due to improved drug management this rate is expected to increase [1, 2]. There are currently no biomarkers or tests to predict which patients will be afflicted and typically a brain tumor is only discovered when a patient exhibits debilitating neurological symptoms. Patients with metastatic brain tumors do not respond to chemotherapy leaving only surgical resection and radiotherapy as treatment options with an average survival of only 6–9 months. If left untreated, the median survival for a patient is 1 month.
The brain presents a unique and complex tissue microenvironment and the colonization and formation of metastatic tumors depends on interactions of the properties of the microenvironment and the phenotype of the colonizing metastatic breast cancer cells [3, 4]. This concept must now be reconciled with knowledge that cancer is ultimately a genetic disease with pathobiology driven by somatic gene mutations, i.e., gain-of-function mutations in oncogenes and loss-of-function mutations in tumor suppressors. Yet an examination of the biomedical literature reveals a relative lack of molecular genomic characterization of brain metastases [5], which could lead to effective therapeutic interventions and better patient outcomes as evidenced by advancements in therapies guided by molecular targets to treat primary breast tumors, e.g., drugs targeting estrogen receptor-alpha (ERα) or the epidermal growth factor receptor ERBB2/HER2 [6]. The projected feasibility of molecularly-targeted therapies for metastatic brain tumors is supported by a growing body of empirical evidence that small tyrosine kinase inhibitors and antigen-targeted drugs can traverse the blood-brain barrier and result in clinical benefit for patients harboring metastatic brain lesions [7–12].
By analyzing copy number aberrations in surgically-resected metastatic brain tumors and matched primary breast tumor specimens, the present work provides insights as to the nature of the genomic lesions that are likely molecular contributors to brain metastases. Furthermore, our bioinformatics analysis highlights various oncogene amplification events that are the focus of molecularly targeted FDA-approved drugs and drugs in clinical trials that might be redirected to treat metastatic brain tumors thus providing a shorter timeline for the design of new treatment strategies over the development of novel therapeutic agents.
RESULTS
Patient population
As shown in Table 1, all 10 study patients were female with an average age at primary breast cancer diagnosis of 51 ± 11 years. The average interval to the diagnosis of brain metastasis was 3.5 ± 2 years. Out of the 10 patients, 5 were hormone receptor positive (with 3 of those also ERBB2/HER2 positive), 4 were triple negative, and the hormone receptor profile was unknown in 1 patient.
Re-occurring cancer gene amplifications in breast metastatic brain tumors
Macro-colonization is described as the rate-limiting step in metastatic tumor formation, accounting for fewer than 0.1% of cancer cells that enter the circulatory system [13–16]. This suggests that the spectrum of mutation-activated oncogenes driving the processes of colonization and/or metastatic tumor outgrowth is limited, and that causative lesions would be recurrent in patient specimens. We investigated this using the copy number aberration data from our aCGH analysis of 10 metastatic brain tumors and identified recurring amplifications in 55 cancer-linked genes based on the minimum condition that gene amplification occurred in 40% of specimens at a log2 ratio greater or equal to +/-0.4 (Supplementary Table 2). According to these criteria there were no recurring gene losses. Among the recurrent gene amplifications were oncogenes PIK3CA and MYC, also known to be frequently amplified in breast cancers. Ingenuity Pathways Analysis (IPA) of the 55 gene set showed over-representation for canonical pathways categorized under Cellular Growth, Proliferation and Development descriptors; and the most over-represented was the Stem Cell Pluripotency pathway (p < 0.01; Figure 1); genes in this pathway showing copy number aberrations in the metastatic tumors were PIK3CA, SOX2 and the neurotrophin receptor NTRK1.
Cancer gene lesions with highest amplitude and potential for therapy
We then identified genes showing the highest levels of amplification in patient tumor specimens with the rationale that patients receiving the greatest benefit from molecularly targeted drugs have tumors with high target levels. We set the minimum amplitude threshold at a log2 ratio of +/-0.8 and producing a list of 109 genes (Supplementary Table 3). It was subjected to IPA and again the Stem Cell Pluripotency pathway rose to significance. By focusing on genes showing higher amplitude copy number aberrations as opposed to those that were recurrent, the numbers of analyzed genes mapping to this pathway increased, to include SOX2, PIK3CA, NTRK1 as well as GNAS, CTNNB1, and FGFR1 (Figure 2). In addition, among all the genes in this analyzed data set 13 amplified genes were identified as potential drug targets, or clinically informative biomarkers (Table 2).
Comparing brain metastases with matched primary cancer tissue
The relationship between cancer driver genes in primary breast cancer and those in metastatic brain tumors remains incompletely understood. Here, we pose the hypothesis that brain colonizing lesions pre-exist in the primary tumor specimen. It is an attractive hypothesis because, if true, this knowledge could allow development of lab tests to predict the propensity toward a metastatic event in the brain, or suggest therapeutic interventions to thwart the development of impending metastatic brain tumors. On the other hand, others put forth the possibility that important genomic lesions develop beyond the primary tumor or at the site of metastases [17]. Considering both concepts, we undertook a descriptive comparison of gene lesions for 4 sets of matched primary and metastatic specimens. Because only FFPE primary breast tumor specimens were available, we analyzed FFPE and fresh frozen metastatic brain tumor tissue samples and discovered the same cancer gene CNV results for the same patient specimens exposed to different preparation and storage. Graphs in Figure 3 display gene symbols and associated aberrant copy number (log2 ratio) identified in each FFPE metastatic or primary specimen; the graphs show if a particular gene lesion was identified in both the primary and metastatic tumor or if it was unique to either tumor in each set. Thus evidence from analysis of cancer gene copy number aberrations indicates that at a gene lesion level metastatic brain tumors can be highly similar or divergent from the cancer of origin.
Table 1: Study population demographics, breast tumor profiles and systemic treatments
Subject ID |
Age at Diagnosis (years) |
Interval to Brain Metastasis (years) |
Primary Tumor Receptor Profile |
Primary Tumor Histologic Subtype |
Systemic Treatment for Primary Tumor |
09–34 |
38 |
6.5 |
ER+PR+H2N- |
HR+ |
neoadjuvant AC x 4 cycles, no docetaxel, little response to chemotherapy, proceeded to MRM with T4N2 disease |
09–35 |
50 |
5 |
n/a |
n/a |
chemotherapy (unspecified) and breast RT after with primary disease |
12–16 |
65 |
3 |
ER/PR+ |
HR+ |
AC+T, post-mastectomy chest wall RT, anastrozole |
12–23 |
41 |
1 |
ER-PR-H2N- |
TNBC |
No chemotherapy because of multiple medical comorbidities (ESRD, DM) |
12–25 |
58 |
2.5 |
ER+PR-H2N+ |
HR+ HER2+ |
chemotherapy (unspecified), whole breast RT after lumpectomy |
12–33 |
39 |
5 |
ER+PR+H2N+ |
HR+ HER2+ |
AC x 6 cycles, TH, anastrozole, and zoledronic acid |
12–37 |
41 |
6 |
ER-PR-H2N- |
TNBC |
5-FU, epirubicin and cyclophosphamide x 6 cycles, whole breast RT then MRM 2 months later followed by post-mastectomy RT, then incisional recurrence treated with RT and capecitabine, then lung metastasis treated with surgery and docetaxel, then gemcitabine, then paclitaxel (each drug for disease progression while on previous chemotherapy agent) |
13–02 |
62 |
2 |
ER-PR-H2N- |
TNBC |
AC+T 2011, post-mastectomy RT |
13–03 |
60 |
1.5 |
ER-PR-H2N- |
TNBC |
neoadjuvant AC+T, whole breast RT after lumpectomy |
13–19 |
58 |
2.5 |
ER-PR+H2N+ |
HR+ HER2+ |
ACTH, whole breast RT after lumpectomy |
Abbreviations: n/a: not available; ER: estrogen receptor; PR: progesterone receptor; H2N: HER-2/neu; TNBC: triple negative breast cancer; AC: doxorubicin and cyclophosphamide; AC+T: doxorubicin, cyclophosphamide, and docetaxel; 5-FU: fluorouracil; MRM: modified-radical mastectomy; RT: radiation therapy; ESRD: end-stage renal disease; DM: diabetes mellitus; TH: docetaxel and trastuzumab (Herceptin); ACTH: doxorubicin and cyclophosphamide followed by paclitaxel and trastuzumab
Another important facet to our study of these paired specimens emerged that underscores the complexities of measuring oncogene mutations in tumor samples. Figure 4 provides an illustrative case. A graph of data from aCGH analysis of the metastatic tumor labeled 13–19 shows a high-level, focused amplification encompassing the entire ERBB2 gene on chromosome 17q12 (Figure 4a). Analysis of the matched primary tumor by aCGH showed that specimen was negative for ERBB2 amplification. This negative result concurs with the clinical pathology report for the original breast cancer biopsy specimen, where ERBB2 gene was not amplified according to FISH (Figure 4b). However, the original pathology report also describes the primary breast cancer specimen as being ERBB2 positive (HER2 positive score 3+) according to IHC testing, which we also demonstrated by repeating ERBB2 protein-level staining of the primary specimen (Figure 4c). In addition, IHC analysis of the 13–19 metastatic brain tumor specimen showed strong ERBB2 staining (Figure 4d).
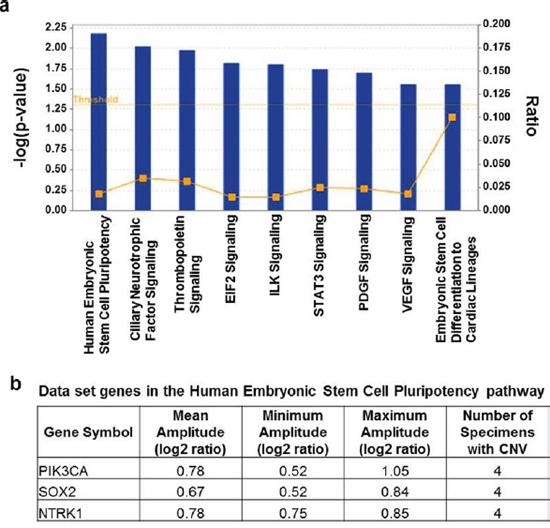
Figure 1: Result of pathways analysis of the set of genes showing repeated copy number gains in DNA from metastatic brain tumor tissue. a. Significant over-representation (p ≤ 0.05) is observed for each of the displayed canonical pathways and the most over-represented was the Stem Cell Pluripotency pathway (p < 0.01). Ratio on the right vertical axis refers to the number of genes from the analyzed data set mapping to each pathway divided by the total number of genes in the pathway. b. The genes mapping to the Stem Cell Pluripotency pathway from the analyzed data set that showed CNV ≥ 0.4 log2 ratio in ≥ 40% of the breast metastatic brain tumors included in the study. The complete list of 55 genes meeting these thresholds is in Supplementary Table 2.
DISCUSSION
Breast cancer patients who suffer metastatic brain tumors are in dire need of improved treatment options. There is mounting evidence that metastatic cancer patients will one day benefit from informed molecularly-targeted therapies based on each individual’s clinical and oncologic profile. There is growing literature on the molecular underpinnings of breast cancer metastatic brain tumor growth [5, 17–27], but even collectively from the literature the numbers of specimens reported on remain limited; also for many reports the number of features analyzed were limited. As such, what remains to be understood is the topic of this report: what are the mutated oncogenes or tumor suppressors found in breast metastatic brain tumor specimens? Our analyses describe 10 individual metastatic specimens and a subset of 4 available matched primary cancer specimens to appreciate the gene copy number lesions that are found in the metastatic tumors; honing in those that may present therapeutic options or important insights to tumor development.
The amplified oncogenes identified in metastatic tumor specimens included genes known to be frequently amplified in breast cancers such as PIK3CA and MYC. Pathway analysis of gene sets comprising amplifications showed significant enrichment for the Stem Cell Pluripotency pathway, highlighting our discovery of recurring significant amplification of SOX2, PIK3CA, NTRK1, GNAS, CTNNB1, and FGFR1 in the samples analyzed. This contributes genomic evidence to reinforce the importance of cancer stem-like cells in promoting metastatic brain tumors. Cancer stem-like cells represent a minor subset of cells within a tumor and similarly to stem cells, they maintain the capability for unlimited self-renewal and are a potential cell type from the primary tumor that is motile and invasive and initiates a metastatic lesion [28–30].
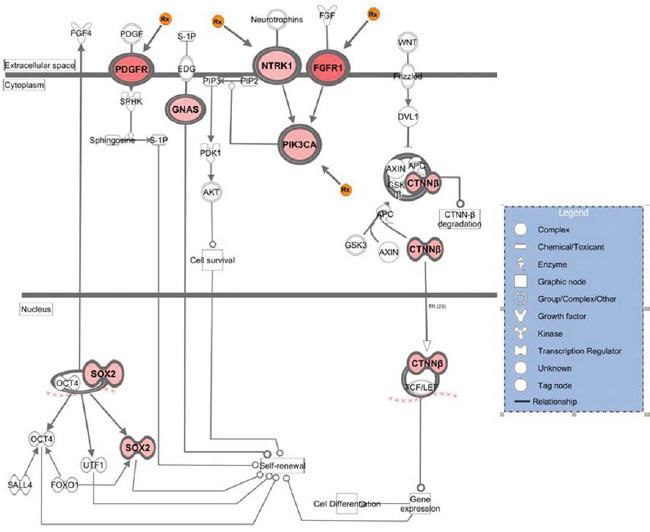
Figure 2: Genes that showed high-level amplification in the breast metastatic brain tumors that map to the stem cell pluripotency pathway. The figure is the output of Ingenuity Systems pathway analysis (IPA) tool. Genes included in the analysis demonstrated ≥ 0.8 log2 ratio in at least one specimen of 10. Intensity of color reflects relative amplitude of copy number gain detected, where darkest red indicates highest amplitude (the copy number log2 ratio data for genes here are in Supplementary Table 3). The orange Rx symbol points to those genes where targeted inhibitory drugs are available.
Table 2: Copy number variations detected in breast metastatic brain tumors for cancer genes that are the target of existing drugs
Gene |
Number Samples Identified |
Amplification Range (log2 ratio) |
Gene Function |
Inhibitor Drug Examples* |
CCND1 |
1 |
2.11 |
other |
daunorubicin, gemtuzumab |
DDR2 |
1 |
0.85 |
kinase |
regorafenib |
ERBB2 |
2 |
1.83–2.54 |
kinase |
trastuzumab, lapatinib, erlotinib |
FGFR1 |
1 |
1.75 |
kinase |
sorafenib, dexamethasone |
JAK2 |
1 |
2.17 |
kinase |
ruxolitinib |
JAK3 |
1 |
0.81 |
kinase |
tofacitinib, R-348 |
KDR |
1 |
0.88 |
kinase |
cabozantinib, bevacizumab |
KIT |
2 |
0.86–0.88 |
transmembrane receptor |
dasatinib, sunitinib |
MUC1 |
2 |
0.85–0.92 |
transcription regulator |
HuHMFG1 |
NTRK1 |
1 |
0.85 |
kinase |
regorafenib |
PDGFRA |
2 |
0.88–2.26 |
kinase |
sunitinib, pazopanib, imatinib |
PIK3CA |
2 |
0.84–1.05 |
kinase |
SF1126, PX-866 |
RAF1 |
1 |
0.86 |
kinase |
vemurafenib, sorafenib |
*Not a complete list.
Copy number gains are activating for oncogenes such as ERBB2 (identified in 2 of 10 metastatic tumor specimens analyzed) because they cause corresponding aberrant overexpression of gene transcripts. Among the more novel genes we uncovered in our analysis (amplified in 4 of 10 metastatic tumors analyzed) was the neurotrophin receptor NTRK1, a tyrosine kinase growth factor receptor. According to database queries NTRK1 is normally highly expressed in central and peripheral nervous system tissues and only the lowest levels of expression is detected in benign breast tissues [31, 32]. Also of interest to the outcomes of our study, according to data from The Cancer Genome Atlas (TCGA) — accessed and analyzed via the cBioPortal — NTRK1 copy number gains occur in 12% of invasive breast carcinomas (of 962 samples) [33, 34]. Altogether, the data provide early evidence to indicate that amplification and/or overexpression of NTRK1 has a potential role in the incidence of metastatic breast tumors and warrants further study.
The last topic of our discussion addresses whether or not primary breast cancers harbor the genomic lesions that are causal in the development of metastatic brain tumors; as opposed to a scenario where mutations evolve outside of the primary tumor, perhaps at the site of metastases prior to macro-colonization. The number of primary specimens available to us for this study was particularly limited because often patients with advanced or metastatic cancers come to the Karmanos Cancer Institute after having had their primary cancer diagnosed and treated elsewhere. However, based on our analyses of cancer gene CNV in 4 matched primary breast and metastatic brain tumors, we witnessed the full spectrum of what might be observed.
One primary and metastatic tumor set showed nearly identical cancer gene aberration profiles (Figure 3a). For this pair CCND1 specifically demonstrated a high amplification in the primary cancer (1.32 log2 ratio amplification spanning 1.5 Mb sequence and 13 genes); a similar amplified region containing CCND1 was more focused and of increased amplitude in the metastatic tumor (a 752,615 base pair, 2.2 log2 ratio amplification comprising only 6 genes, including CCND1, MRGPRD, MRGPRF, TPCN2, MYEOV and ORAOV1). AKT2 amplification was detected only in the metastatic tumor samples, as part of a broad low level amplification event (0.4 log2 ratio, 2.5Mb comprising more than 75 genes/transcripts). Together, the data suggest that AKT2 is not causal for the metastatic lesion, but that CCND1 likely has a role in the primary breast cancer that is reinforced in the metastatic cancer.
Two paired primary-metastatic tumor sets demonstrated a set of conserved cancer gene aberrations, though one set showed additional cancer gene lesions in the primary cancer specimen and the other had gains in the metastasis. In one instance, for only the primary tumor (set 13–02 Figure 3b) MET was part of a focal amplification event (1.1 log2 ratio spanning 287,678 base pairs and 2 genes: MET and CAV1); also, broad amplification events comprising MYC and DDR2 were identified in both primary and metastatic samples (≤ 0.8 log2 ratio, spanning from 5Mb to 40Mb of sequence and upwards of 100 genes; Figure 3b). Here the trend holds, cancer gene amplifications at relatively lower log2 ratios were associated with broad amplicons comprising many genes, and thus are less likely indicative of a driver lesion that would predict response to targeted treatment [35].
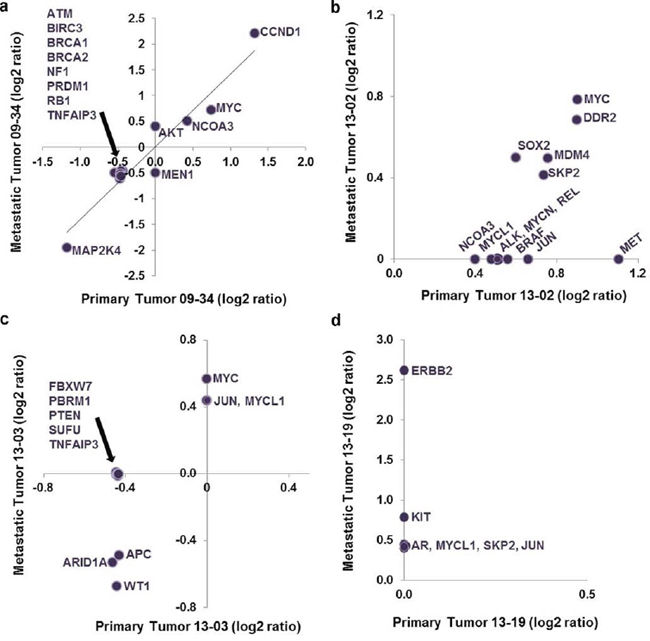
Figure 3: Plot of log2 ratio data for cancer gene aberrations discovered in matched primary and metastatic tumor specimens. Panels a–d. show the individual results of analysis of matched primary breast and metastatic cancer specimens from four patients. Representing one side of the possible outcomes spectrum, a displays outcomes where the log2 ratios for cancer gene aberrations were identical in the primary and metastatic tumors. d shows the extreme example where no lesions were discovered in the primary tumor, but 6 cancer gene aberrations were observed in the metastatic tumor with ERBB2 having the highest copy number gain.
The fourth and final matched tumor set showed no cancer gene aberrations in the primary specimen according to aCGH analysis, but significant copy number aberrations were detected for a number of genes in the metastatic tumor sample; including focused, high copy number gain for ERBB2. FISH analysis verified aCGH ERBB2-negative results in the cancer-of-origin specimen, but the alternative measure that detected high ERBB2 protein-levels in the specimen by IHC leads to a conclusion that the dependence of this patient’s cancer on ERBB2 was sustained in both the primary and metastatic sites by more than one mechanism. Of note, in the course of treatment for this primary breast cancer, the patient did receive anti-ERBB2 therapy as part of her treatment regimen, which a recent study has shown may actually contribute to the development of brain metastases [36]. This specific scenario challenges mutation or oncogene testing algorithms where the objective is to discover an actionable, activated oncogene in a patient specimen. While genomic approaches (e.g., aCGH, high-throughput sequencing of targeted exons or exome sequencing) conveniently allow for multi-gene testing and thus may provide benefit over single gene analysis such as by FISH, all efforts to analyze cancer specimen DNA genomic methods or by FISH can result in false negatives when an unforeseen mechanism upregulates an oncogene at the transcript or protein level. In line with the result we report here, signaling mechanisms are known to drive transcription and upregulation of ERBB2 in breast cancer absent any copy number gain [37].
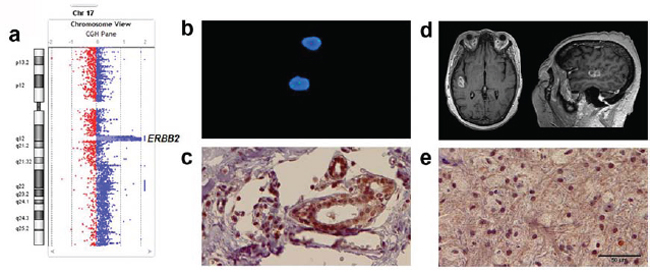
Figure 4: Various clinically viable approaches to measure ERBB2 in metastatic and primary tumors from a single patient both verify results and reveal complexities in interpreting outcomes. In this specific patient example, a. according to aCGH analysis high copy number gain for ERBB2 was observed in the metastatic brain tumor specimen. ERBB2 is the only translated gene that mapped to the focal amplification coordinates HG19: Chr17: 37863329–37866691. b. The result of FISH analysis of the original biopsied breast cancer specimen concluded that there was normal diploid copy number for ERBB2 in DNA, and this agrees with results of our aCGH analysis of the primary specimen that were also negative for ERBB2 amplification. c. IHC analysis of the primary breast cancer specimen indicates that although not DNA amplified, ERBB2 is upregulated at the protein level in the primary specimen. d. MRI scan of patient (13–19) with right temporal brain metastasis from primary breast cancer. e. IHC of the metastatic brain lesion shows relatively homogenous immunostaining for ERBB2. Scale bar is 50 μM and also applies to panel c.
To conclude, the results of this study demonstrate the potential to identify an array of known, druggable oncogenic lesions in breast metastatic brain tumor specimens – with links to the stem cell pluripotency pathway. All in all bolstering the expectation that continued molecular genomic study of primary breast cancer and metastatic brain tumors will lead to improved targeted, individualized therapies.
MATERIALS AND METHODS
Patients and tissues
This study was approved by the Wayne State University School of Medicine Institutional Review Board and written informed consent was obtained from all participants. Patients included in this study underwent craniotomy with microsurgical brain tumor resection as part of the standard-of-care for the treatment of breast cancer brain metastases. The study procurement period was 2009–2013. Fresh tissue specimens were collected and frozen in liquid nitrogen within 30 min post-resection and stored at −80°C until analysis. Formalin-fixed paraffin-embedded (FFPE) specimens were obtained from the Pathology archive. Patient demographic and clinical outcomes data collected included: date of birth, gender, age at diagnosis of primary breast cancer and metastatic brain tumor, and histopathological features of primary and metastatic tumors. A description of each specimen from all 10 patients who contributed to this study – including ER, ERBB2/HER2 and progesterone receptor (PR) status and treatments – is provided in Table 1.
Array Comparative Genomic Hybridization (aCGH) analysis
The analyses were done in a CLIA-certified laboratory and CGH was run using a methodology established in that laboratory that is reviewed and accredited by the College of American Pathologists (CAP). DNA was isolated from paraffin-embedded tissue or frozen tissue using the EZ1 Advanced XL magnetic bead technology, the EZ1 DNA de-paraffinization method (where appropriate), and the EZ1 DNA Tissue kit (Qiagen, Valencia, CA). Cytogenomic analysis was done by array comparative genomic hybridization (aCGH) method. Sample DNA was pre-assessed and selected based on Nanodrop spectrophotometer outcomes to estimate quantity and contaminating factors and TapeStation (Agilent Technologies, Santa Clara, CA) digital gel technology to estimate degree of degradation, where all FFPE samples analyzed and corresponding heat-treated reference DNA were observed to be similarly fragmented (range of 1000–2000bp). aCGH was performed with Agilent SurePrint G3 ISCA CGH+SNP 180K using a normal characterized female DNA reference (Agilent Technologies). Cancer and reference DNA (500 ng each) were labeled with fluorescent dyes, Cy5 and Cy3 respectively, using the SureTag Complete DNA Labeling Kit (Agilent Technologies) or Universal Linkage System labeling kit for DNA from FFPE specimens (Agilent Technologies) as in [38]. Slides were scanned using an Agilent G4900DA SureScan Microarray Scanner System, and Log2 ratios for coordinates showing copy number variation (CNV) were extracted using Agilent CytoGenomics Edition 2.5.8.1 using the ADM2 threshold set at 6; an 8 probe minimum; and an additional absolute log2 ratio threshold minimum set at 0.4 for analysis of reoccurring lesions or minimum of log2 ratio 0.8 to study high amplitude copy number aberrations. To identify genes with well-characterized roles in cancer, gene-level data were compared to a cancer gene list comprising known cancer promoting genes, oncogenes or tumor suppressor genes (Supplementary Table 1); the listed genes were compiled from the Cancer Gene Census (a cancer atlas project supported by the Wellcome Trust Sanger Institute, London, UK), and work by Lawrence et al. and Vogelstein et al. [39, 40]. Ingenuity Systems software and databases (Qiagen Silicon Valley, Redwood City, CA) were used for Pathways Enrichment Analysis and to highlight potentially druggable pathways and genes. Here the p-value associated with a pathway annotation is a measure of its statistical significance with respect to genes in the canonical pathway, eligible genes equal to the analyzed dataset of interest and a reference set equal to all human genes probed in the microarray. The p-value was calculated with the right-tailed Fisher's Exact Test. CGH data, raw and processed, are available at http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=ozkdeuiwjvkbbij&acc=GSE62009.
Immunohistochemistry (IHC) and Fluorescent In Situ Hybridization (FISH) assays
IHC was performed using standard protocols on 5 mM FFPE tissue sections after citrate buffer antigen retrieval. The ERBB2/HER2 antibody (rabbit clone 29D8, Cell Signaling Technology, Danvers, MA) was diluted 1:75 and incubated overnight at 4°C. Antibody binding was visualized with 3,3'-diaminobenzidine and sections were counterstained with Mayer’s hematoxylin before permanent mounting.
FISH was performed on FFPE tissue using the Vysis PathVysion ERBB2/HER2 DNA probe kit (Abbott Molecular Inc., Abbott Park, IL) following the manufacturer’s protocol. The LSI ERBB2/HER2 DNA probe (labeled with SpectrumOrange) is specific for the ERBB2/HER2 gene locus on 17q11.2–q12 while the CEP 17 DNA control probe (labeled with SpectrumGreen is specific for the centromeric region of chromosome 17 (17q11.1–q11.1). At least 20 non-overlapping cells in two separate areas of invasive cancer were scored following the American Society of Clinical Oncology (ASCO) and the College of American Pathologists (CAP) guidelines [41].
CONFLICTS OF INTEREST
The authors declare that they have no conflict of interest.
GRANT SUPPORT
This study was supported by a Strategic Research Initiative Grant awarded by the Karmanos Cancer Institute (ABF and SM). The Genomics Core and the Biobanking and Correlative Sciences Core are supported by NIH Center grant P30 CA022453 to the Karmanos Cancer Institute at Wayne State University.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
REFERENCES
1. Nathoo N, Chahlavi A, Barnett GH, Toms SA. Pathobiology of brain metastases. Journal of clinical pathology. 2005; 58:237–242.
2. Niwinska A, Murawska M, Pogoda K. Breast cancer subtypes and response to systemic treatment after whole-brain radiotherapy in patients with brain metastases. Cancer. 2010; 116:4238–4247.
3. Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer metastasis reviews. 1989; 8:98–101.
4. Hart IR, Fidler IJ. Role of organ selectivity in the determination of metastatic patterns of B16 melanoma. Cancer research. 1980; 40:2281–2287.
5. Bollig-Fischer A, Michelhaugh SK, Ali-Fehmi R, Mittal S. The molecular genomics of metastatic brain tumors. OA Molecular Oncology. 2013; 1:6.
6. Sharma SV, Settleman J. Oncogene addiction: setting the stage for molecularly targeted cancer therapy. Genes Dev. 2007; 21:3214–3231.
7. Heimberger AB, Learn CA, Archer GE, McLendon RE, Chewning TA, Tuck FL, Pracyk JB, Friedman AH, Friedman HS, Bigner DD, Sampson JH. Brain tumors in mice are susceptible to blockade of epidermal growth factor receptor (EGFR) with the oral, specific, EGFR-tyrosine kinase inhibitor ZD139 (iressa). Clinical cancer research: an official journal of the American Association for Cancer Research. 2002; 8:3496–3502.
8. Gluck S, Castrellon A. Lapatinib plus capecitabine resolved human epidermal growth factor receptor 2-positive brain metastases. American journal of therapeutics. 2009; 16:585–590.
9. Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, Pienkowski T, Jagiello-Gruszfeld A, Crown J, Chan A, Kaufman B, Skarlos D, Campone M, Davidson N, Berger M, Oliva C, Rubin SD, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006; 355:2733–2743.
10. Lin NU, Dieras V, Paul D, Lossignol D, Christodoulou C, Stemmler HJ, Roche H, Liu MC, Greil R, Ciruelos E, Loibl S, Gori S, Wardley A, Yardley D, Brufsky A, Blum JL, et al. Multicenter phase II study of lapatinib in patients with brain metastases from HER2-positive breast cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2009; 15:1452–1459.
11. Narayana A, Mathew M, Tam M, Kannan R, Madden KM, Golfinos JG, Parker EC, Ott PA, Pavlick AC. Vemurafenib and radiation therapy in melanoma brain metastases. Journal of neuro-oncology. 2013; 113:411–416.
12. Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, Baselga J, Norton L. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001; 344:783–792.
13. Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011; 147:275–292.
14. Nagrath S, Sequist LV, Maheswaran S, Bell DW, Irimia D, Ulkus L, Smith MR, Kwak EL, Digumarthy S, Muzikansky A, Ryan P, Balis UJ, Tompkins RG, Haber DA, Toner M. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature. 2007; 450:1235–1239.
15. Luzzi KJ, MacDonald IC, Schmidt EE, Kerkvliet N, Morris VL, Chambers AF, Groom AC. Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. The American journal of pathology. 1998; 153:865–873.
16. Cameron MD, Schmidt EE, Kerkvliet N, Nadkarni KV, Morris VL, Groom AC, Chambers AF, MacDonald IC. Temporal progression of metastasis in lung: cell survival, dormancy, and location dependence of metastatic inefficiency. Cancer research. 2000; 60:2541–2546.
17. Shah SP, Morin RD, Khattra J, Prentice L, Pugh T, Burleigh A, Delaney A, Gelmon K, Guliany R, Senz J, Steidl C, Holt RA, Jones S, Sun M, Leung G, Moore R, et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature. 2009; 461:809–813.
18. Kroigard AB, Larsen MJ, Laenkholm AV, Knoop AS, Jensen JD, Bak M, Mollenhauer J, Kruse TA, Thomassen M. Clonal expansion and linear genome evolution through breast cancer progression from pre-invasive stages to asynchronous metastasis. Oncotarget. 2015.
19. Moelans CB, van der Groep P, Hoefnagel LD, van de Vijver MJ, Wesseling P, Wesseling J, van der Wall E, van Diest PJ. Genomic evolution from primary breast carcinoma to distant metastasis: Few copy number changes of breast cancer related genes. Cancer letters. 2014; 344:138–146.
20. Ding L, Ellis MJ, Li S, Larson DE, Chen K, Wallis JW, Harris CC, McLellan MD, Fulton RS, Fulton LL, Abbott RM, Hoog J, Dooling DJ, Koboldt DC, Schmidt H, Kalicki J, et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature. 2010; 464:999–1005.
21. Da Silva L, Simpson PT, Smart CE, Cocciardi S, Waddell N, Lane A, Morrison BJ, Vargas AC, Healey S, Beesley J, Pakkiri P, Parry S, Kurniawan N, Reid L, Keith P, Faria P, et al. HER3 and downstream pathways are involved in colonization of brain metastases from breast cancer. Breast cancer research: BCR. 2010; 12:R46.
22. Wikman H, Lamszus K, Detels N, Uslar L, Wrage M, Benner C, Hohensee I, Ylstra B, Eylmann K, Zapatka M, Sauter G, Kemming D, Glatzel M, Muller V, Westphal M, Pantel K. Relevance of PTEN loss in brain metastasis formation in breast cancer patients. Breast cancer research: BCR. 2012; 14:R49.
23. Hohensee I, Lamszus K, Riethdorf S, Meyer-Staeckling S, Glatzel M, Matschke J, Witzel I, Westphal M, Brandt B, Muller V, Pantel K, Wikman H. Frequent genetic alterations in EGFR- and HER2-driven pathways in breast cancer brain metastases. The American journal of pathology. 2013; 183:83–95.
24. Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nature genetics. 2003; 33:49–54.
25. Weigelt B, Hu Z, He X, Livasy C, Carey LA, Ewend MG, Glas AM, Perou CM, Van't Veer LJ. Molecular portraits and 70-gene prognosis signature are preserved throughout the metastatic process of breast cancer. Cancer research. 2005; 65:9155–9158.
26. Salhia B, Kiefer J, Ross JT, Metapally R, Martinez RA, Johnson KN, DiPerna DM, Paquette KM, Jung S, Nasser S, Wallstrom G, Tembe W, Baker A, Carpten J, Resau J, Ryken T, et al. Integrated genomic and epigenomic analysis of breast cancer brain metastasis. PloS one. 2014; 9:e85448.
27. Ahmad A, Ginnebaugh KR, Sethi S, Chen W, Ali R, Mittal S, Sarkar FH. miR-20b is up-regulated in brain metastases from primary breast cancers. Oncotarget. 2015; In Press.
28. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008; 133:704–715.
29. Ocana OH, Corcoles R, Fabra A, Moreno-Bueno G, Acloque H, Vega S, Barrallo-Gimeno A, Cano A, Nieto MA. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer cell. 2012; 22:709–724.
30. Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer cell. 2012; 22:725–736.
31. Liu X, Yu X, Zack DJ, Zhu H, Qian J. TiGER: a database for tissue-specific gene expression and regulation. BMC bioinformatics. 2008; 9:271.
32. Uhlen M, Oksvold P, Fagerberg L, Lundberg E, Jonasson K, Forsberg M, Zwahlen M, Kampf C, Wester K, Hober S, Wernerus H, Bjorling L, Ponten F. Towards a knowledge-based Human Protein Atlas. Nature biotechnology. 2010; 28:1248–1250.
33. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery. 2012; 2:401–404.
34. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling. 2013; 6:l1.
35. Gomez-Martin C, Plaza JC, Pazo-Cid R, Salud A, Pons F, Fonseca P, Leon A, Alsina M, Visa L, Rivera F, Galan MC, Del Valle E, Vilardell F, Iglesias M, Fernandez S, Landolfi S, et al. Level of HER2 gene amplification predicts response and overall survival in HER2-positive advanced gastric cancer treated with trastuzumab. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2013; 31:4445–4452.
36. Kaplan MA, Ertugrul H, Firat U, Kucukoner M, Inal A, Urakci Z, Pekkolay Z, Isikdogan A. Brain metastases in HER2-positive metastatic breast cancer patients who received chemotherapy with or without trastuzumab. Breast cancer. 2014.
37. Ithimakin S, Day KC, Malik F, Zen Q, Dawsey SJ, Bersano-Begey TF, Quraishi AA, Ignatoski KW, Daignault S, Davis A, Hall CL, Palanisamy N, Heath AN, Tawakkol N, Luther TK, Clouthier SG, et al. HER2 drives luminal breast cancer stem cells in the absence of HER2 amplification: implications for efficacy of adjuvant trastuzumab. Cancer research. 2013; 73:1635–1646.
38. Salawu A, Ul-Hassan A, Hammond D, Fernando M, Reed M, Sisley K. High quality genomic copy number data from archival formalin-fixed paraffin-embedded leiomyosarcoma: optimisation of universal linkage system labelling. PloS one. 2012; 7:e50415.
39. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science. 2013; 339:1546–1558.
40. Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014; 505:495–501.
41. Wolff AC, Hammond ME, Hicks DG, Dowsett M, McShane LM, Allison KH, Allred DC, Bartlett JM, Bilous M, Fitzgibbons P, Hanna W, Jenkins RB, Mangu PB, Paik S, Perez EA, Press MF, et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. Archives of pathology & laboratory medicine. 2014; 138:241–256.