
INTRODUCTION
Oral squamous cell carcinoma (OSCC) is the most common malignancy of the oral cavity, accounting for more than 90% of all oral neoplasms [1]. Approximately 300,000 new oral cavity cancer cases and 145,000 oral cancer-related deaths were registered in 2012 (GLOBOCAN 2012, http://globocan.iarc.fr/), showing increasing trends from 2008 [2]. Risky oral habits (including smoking, alcohol drinking, and betel quid chewing) are major risk factors for OSCC development [2]. The 5-year overall survival (OS) and disease-free survival (DFS) rates for OSCC patients are approximately 70% and 67%, respectively [3, 4]. However, the prognosis of patients with advanced disease (stage III/IV) remains dismal [5–7]. Different prognostic factors have been identified in OSCC patients (e.g., tumor and nodal stages, tumor differentiation, AJCC stage, tumor invasiveness, treatment modalities, and surgical margins) [3, 7]. Although extracapsular spread (ECS) is generally considered as the main predictor of disease progression, alone it does not provide sufficient information on the patient’s clinical course [7]. Approximately 36% of OSCC patients with ECS are able to achieve an acceptable survival, whereas poor OS rates can be observed in 40% of OSCC patients without ECS [6]. In this scenario, an improved molecular characterization of OSCC may be helpful for informing prognosis and devising tailored treatment strategies.
Mutations of the TP53, CDKN2A, HRAS, and PIK3CA genes have been frequently reported in OSCC samples [8, 9]. Moreover, mutations in NOTCH1, FAT1, and CASP8 may promote the initiation and progression of OSCC [10–12]. However, the question as to whether such mutations may improve the prognostic stratification of OSCC remains open. Notably, previous studies mainly identified mutations in tumor suppressor genes (TSG) rather than oncogenes, the only exceptions being HRAS and PIK3CA. However, most small molecules and/or biological agents under clinical development can directly engage activating mutations in oncogenes [13, 14].
Herein, we sought to identify genetic alterations that may improve the prognostic stratification of advanced OSCC patients, being also actionable as potential treatment targets. To this end, we conducted ultra-deep sequencing of the mutational hotspot in 45 cancer-related genes using formalin-fixed paraffin-embedded (FFPE) primary tumor samples (n = 345). A total of 29 oncogenes and 16 tumor suppressor genes [TSG] were examined, including ABL1, AKT1, ALK, APC, ATM, BRAF, CDH1, CDKN2A, CSF1R, CTNNB1, EGFR, ERBB2, ERBB4, FBXW7, FGFR1, FGFR2, FGFR3, FLT3, GNAS, HNF1A, HRAS, IDH1, JAK3, KDR, KIT, KRAS, MET, MLH1, MPL, NOTCH1, NPM1, NRAS, PDGFRA, PIK3CA, PTEN, PTPN11, RB1, RET, SMAD4, SMARCB1, SMO, SRC, STK11, TP53, and VHL. The high-sequencing depth allowed a highly sensitive mutation detection and the availability of long-term follow-up data was useful for improving molecular prognostic stratification, ultimately favoring tailored treatment approaches.
RESULTS
Patient characteristics
We obtained a total of 355 tumor samples from pN+ patients with previously untreated stage III/IV OSCC. NGS sequencing data were available for 345 patients (97.2%; 325 males and 20 females). Among the remaining 10 patients, 6 (1.7%) had insufficient DNA and 4 (1.1%) inadequate DNA quality (Figure 1). Tumor sites were as follows: bucca (n = 132, 38.3%), tongue (n = 130, 37.7%), alveolar ridge (n = 44, 12.8%), retromolar trigone (n = 16, 4.6%), mouth floor (n = 15, 4.3%), hard palate (n = 6, 1.7%), and lip (n = 2, 0.6%). The prevalence rates of pre-operative alcohol drinking, betel quid chewing, and cigarette smoking use were 71% (n = 246), 82% (n = 282), and 92% (n = 313), respectively. The pathological stage was p-stage III in 85 patients (25%) and p-stage IV in 260 patients (75%). After a minimum follow-up of 30 months (or censoring at the date of death), there were 172 cases (49.9%) of tumor relapse, with a median DFS of 65 months. A total of 201 patients (58.3%) had ECS. Table 1 summarizes the general characteristics of the study participants.
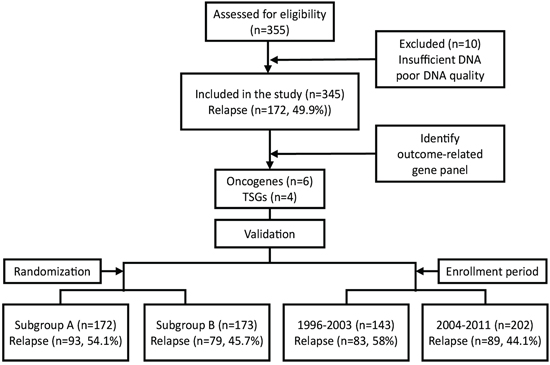
Figure 1: Selection of the study population.
Table 1: Characteristics of OSCC patients in the entire cohort and subsets used for internal validation
Characteristics |
Entire cohort |
Randomization |
Enrollment period |
||
Subgroup A |
Subgroup B |
1996–2003 |
2004–2011 |
||
N (%) |
345 (100) |
172 (100) |
173 (100) |
143 (100) |
202 (100) |
Age at onset (years) |
|||||
< 65 |
307 (89.0) |
157 (91.3) |
150 (86.7) |
132 (92.3) |
175 (86.6) |
≥ 65 |
38 (11.0) |
15 (8.7) |
23 (13.3) |
11 (7.7) |
27 (13.4) |
Range |
27 – 89 |
27 – 89 |
27 – 83 |
27 – 74 |
29 – 89 |
Mean ± SD |
49.6 ± 11.0 |
49.3 ± 11.1 |
50.0 ± 11.9 |
48.3 ± 10.8 |
50.6 ± 10.9 |
Sex |
|||||
Male |
325 (94.2) |
166 (96.5) |
159 (91.9) |
136 (95.1) |
189 (93.6) |
Female |
20 (5.8) |
6 (3.5) |
14 (8.1) |
7 (4.9) |
13 (6.4) |
Alcohol drinking |
|||||
No |
99 (28.7) |
51 (29.7) |
48 (27.7) |
51 (35.7) |
48 (23.8) |
Yes |
246 (71.3) |
121 (70.3) |
125 (72.3) |
92 (64.3) |
154 (76.2) |
Betel quid chewing |
|||||
No |
63 (18.3) |
25 (14.5) |
38 (22.0) |
21 (14.7) |
42 (20.8) |
Yes |
282 (81.7) |
147 (85.5) |
135 (78.0) |
122 (85.3) |
160 (79.2) |
Cigarette smoking |
|||||
No |
32 (9.3) |
14 (8.1) |
18 (10.4) |
10 (7.0) |
22 (10.9) |
Yes |
313 (90.7) |
158 (91.9) |
155 (89.6) |
133 (93.0) |
180 (89.1) |
Pathological T status |
|||||
pT1–2 |
153 (44.4) |
75 (43.6) |
78 (45.1) |
59 (41.3) |
94 (46.5) |
pT3–4 |
192 (55.6) |
97 (56.4) |
95 (54.9) |
84 (58.7) |
108 (53.5) |
Pathological N status |
|||||
pN1 |
123 (35.7) |
59 (34.3) |
64 (37.0) |
50 (35.0) |
73 (36.1) |
pN2 |
222 (64.3) |
113 (65.7) |
109 (63.0) |
93 (65.0) |
129 (63.9) |
Pathological stage |
|||||
III |
85 (24.6) |
39 (22.7) |
46 (26.6) |
34 (23.8) |
51 (25.2) |
IV |
260 (75.4) |
133 (77.3) |
127 (73.4) |
109 (76.2) |
151 (74.8) |
Extracapsular spread |
|||||
No |
144 (41.7) |
68 (39.5) |
76 (43.9) |
53 (37.1) |
91 (45.0) |
Yes |
201 (58.3) |
104 (60.5) |
97 (56.1) |
90 (62.9) |
111 (55.0) |
Margin status |
|||||
≤4 mm |
43 (12.5) |
25 (14.5) |
18 (10.4) |
20 (14.0) |
23 (11.4) |
>4 mm |
298 (86.4) |
144 (83.7) |
154 (89.0) |
119 (83.2) |
179 (88.6) |
unknown |
4 (1.2) |
3 (1.8) |
1 (0.6) |
4 (100) |
0 (0) |
Treatment modality |
|||||
Surgery |
25 (7.2) |
12 (7.0) |
13 (7.5) |
10 (7.0) |
15 (7.2) |
Surgery + RT/CCRT |
320 (92.8) |
160 (93.0) |
160 (92.5) |
133 (93.0) |
202 (92.8) |
Relapse status |
|||||
No |
173 (50.1) |
79 (45.9) |
94 (54.3) |
60 (42.0) |
113 (55.9) |
Yes |
172 (49.9) |
93 (54.1) |
79 (45.7) |
83 (58.0) |
89 (44.1) |
5-yr survival rate (%) |
50.8 |
46.3 |
54.7 |
43.2 |
56.4 |
Abbreviations: RT, radiotherapy; CCRT, concurrent chemoradiation.
Sequencing results
We were able to achieve a 2410-fold mean sequence coverage for the targeted regions (97.5% of them were covered at >100 folds). The complete coverage details are provided in the Supplement (Data Supplement, Supplementary Table S1). A total of 1,269 non-synonymous (missense, nonsense, indel and splicing site) mutations with an allele frequency ≥ 3% were detected in 276 (80%) samples (Supplementary Figure S2). The average number of non-synonymous mutation per tumor was 4.6. However, the mutation rate was highly dependent on the specimen (from 1 to 166 mutations per sample). Sixty-nine samples (20%) had no detectable non-synonymous mutations. Based on the total number of mutations per tumor, the 276 specimens were divided into three different mutation groups, as follows: ultra-mutators (>50 mutations/tumor, n = 5), hyper-mutators (17−46 mutations/tumor, n = 7), and mutators (1−11 mutations/tumor, n = 265). The number of detected mutations was not significantly influenced by the sample storage time (Supplementary Figure S1), suggesting that even long-time stored FFPE specimens were suitable for NGS analysis. Using the remaining genomic DNA specimens, a total of 120 detected mutations were validated by means of Sanger sequencing or pyrosequencing. Of them, 99 (82.5%) were successfully validated (Data Supplement, Supplementary Table S2). The validation rate further increased to 96.1% (73 of 76) for genetic variants with an allele frequency >7.5%, suggesting that alternative sequencing methods may not be sufficiently sensitive for the detection of low-frequency mutations [15]. Missense mutations accounted for the majority (73.6%) of the identified variants, followed by nonsense mutations (14.6%). Insertions/deletions (indels) accounted for only 2.3% of all variants. We also analyzed the association between the number of mutations in each sample and risky oral habits (alcohol drinking, betel nut chewing, and cigarette smoking) (Supplementary Figure S3). The number of genetic mutations in tumor samples was significantly higher in smokers than in non-smokers (3.9 ± 14.8 vs. 1.6 ± 1.5, P = 0.0093) as well as in betel nut users compared with non-users (4.1 ± 15.5 vs. 1.9 ± 3.8, P = 0.0375). However, the mean number of sequence variants did not differ significantly in alcohol users compared with non-users (3.7 ± 16.8 vs. 4.37 ± 12.9, P = 0.9769).
Mutational landscape in the targeted regions
The 1,269 non-synonymous mutations identified in the current study were located in 44 genes (Figure 2). The most frequently mutated genes were TP53 (65%), PIK3CA (16.8%), CDKN2A (12.8%), HRAS (9.3%), BRAF (9.0%), EGFR (6.7%) and FGFR3 (5.8%). Genetic mutations in the ten most frequently mutated genes were identified in 263 (76.2%) samples (Figure 2). As only the hotspot regions of 45 cancer-related genes were sequenced in our study, we analyzed whether our targeted sequencing approach could distort the observed mutation spectra. We therefore compared our findings with the mutational patterns reported in The Cancer Genome Atlas (TCGA) head and neck squamous cell carcinoma (HNSCC) dataset (containing the whole-exome sequencing data of 279 tumors). The frequency of genetic variations in TSG detected in our study was largely similar to that observed in TCGA dataset, the only exceptions being CDKN2A (12.8% vs. 22.6% in the TCGA data) and NOTCH1 (3.2% vs. 18.6% in the TCGA data) which showed a significantly lower degree of sequence variation in our study (Figure 3A). In contrast, mutations in several oncogenes (including potential drug targets) were more commonly observed in our study than in the TCGA (Figure 3B). Notably, several oncogenes had a 3-fold higher mutation rates in the current report compared with the TCGA data, including AKT1 (3.2% vs. 0.7%), BRAF (9% vs. 1.4%), CTNNB1 (2.3% vs. 0.7%), FGFR1 (1.4% vs. 0.4%), FGFR2 (4.3% vs. 0.7%), KIT (4.1% vs. 1.1%), KRAS (2.3% vs. 0.4%), and MET (4.3% vs. 1.1%; Figure 3B). Moreover, we also detected 11 samples with ABL1 (3.2%) mutations and 11 cases with SMO (3.2%) mutations. No such mutations were reported in the TCGA dataset. Mutations in the PI3K pathway, including PIK3CA, AKT1 and PTEN, were identified in 68 (19.7%) tumors, suggesting that these patients may benefit from AKT-PI3K-mTOR inhibitors.
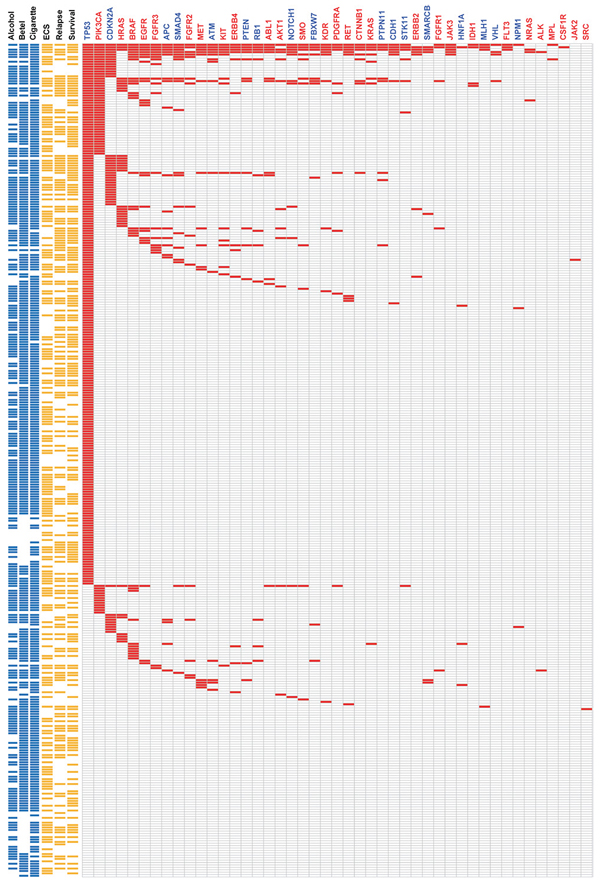
Figure 2: Heat-map representation of individual non-silent variants identified in 345 specimens (rows). (Left) Risk behaviors of individual patients. Individual highlighted in blue were exposed to alcohol drinking, betel nut chewing, and cigarette smoking, respectively. (Center) Clinical characteristics of individual patients. Individual highlighted in yellow were positive for ECS, had tumor relapse, or died. (Right) Mutation metrix for individual genes in each patient. Tumor suppressor genes are labeled in blue, whereas oncogenes are indicated in red.
Molecular characterization of the detected mutations
The four most commonly mutated genes were selected for further analysis. TP53 showed sequence mutations in 224 (65%) samples (324 mutations distributed across 142 loci). The majority of TP53 mutations were missense mutations (80.2%), followed by nonsense mutations (14.2%), and splice-site mutations (2.5%). The distribution of mutation types in our study is similar to the TCGA data. Most (67.6%) of the TP53 mutations were disruptive mutations (46 nonsense mutations, 7 indels, 8 splice-site mutations, and 158 missense mutations leading to changes in charged amino acids) [16]. We also identified 13 tumors harboring two mutations in TP53, likely due to the presence of multiple subclones in the sequenced specimens. Although our targeted region covers only 69% of the entire coding region, the mutational spectrum of TP53 observed in our study is highly similar to the TP53 mutation spectrum observed in the TCGA HNSCC dataset (Figure 3C). PIK3CA was the most frequently mutated oncogene in our study, with a total of 65 genetic variants (63 missense and 2 nonsense mutations) identified in 58 (16.8%) tumors. Both of the two nonsense mutations were identified in tumors characterized by a hypermutator phenotype. The most common PIK3CA mutational hotspots detected in our study were located in exon 20 (H1047, G1049), whereas the most frequently mutated hotspots in the TCGA HNSCC study were located in the exon 9 (E542 and E545; Figure 3D). We also identified 53 CDKN2A mutations in 44 (12.8%) tumors; of them, 40 were definitely inactivating mutations (29 nonsense mutations, 3 indels, and 8 splice site mutations). The most commonly identified CDKN2A variant was the R58X nonsense mutations (identified in 20 tumors). According to the COSMIC database, the R58X mutation is frequently detected in cancers from upper aerodigestive trait, skin, esophagus, pancreas, and lung. HRAS mutations were identified in 32 tumors; all of them were well-known missense mutations located on three specific amino acid residues, G12 (n = 13), G13 (n = 9), and Q61 (n = 6). Notably, the allele-specific G12S mutation on HRAS was detected at a higher frequency (n = 9 in 345 subjects) in our cohort than in the TCGA HNSCC study (n = 2 in 279 subjects). The G12S mutation in KRAS has been previously associated with radiation resistance in cultured cell lines [17]. Overall, the frequency and type of mutations observed in the TP53, CDKN2A, PIK3CA, and HRAS genes were consistent with both previous genetic studies of OSCC as well as the TCGA HNSCC dataset [18–21]. The subtle difference in specific alleles may reflect difference in etiology, ethnicity, or tumor stage between the two cohorts.
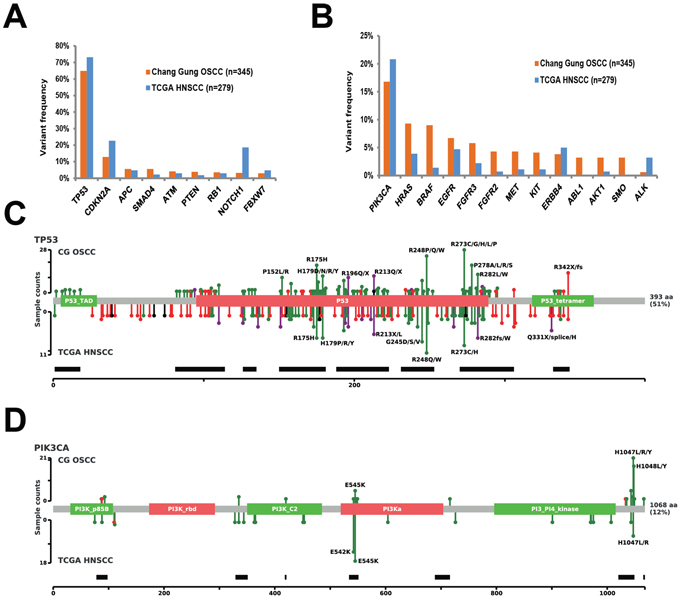
Figure 3: Extent of genetic disruption in advanced OSCC. A. Prevalence of tumors harboring tumor suppressor gene variants in the Chang Gung cohort and in the TCGA HNSCC cohort. B. Prevalence of tumors harboring oncogenic variants in the Chang Gung cohort and in the TCGA HNSCC cohort. C. Distribution of TP53 mutations in the Chang Gung cohort and in the TCGA HNSCC cohort. D. Distribution of PIK3CA mutations in the Chang Gung cohort and in the TCGA cohort.
Genetic mutations associated with survival rates
We used Cox proportional hazard regression to identify the genetic variants independently associated with survival outcomes. Genetic mutations located in 14 genes (ABL1, AKT1, BRAF, CTNNB1, FGFR3, HRAS, KIT, MPL, NOTCH1, PTEN, PTPN11, SMAD4, STK11, and VHL) were significantly associated with DFS in univariate Cox regression analysis (Table 2); among them, mutations in 11 genes (AKT1, BRAF, CTNNB1, FGFR3, HRAS, KIT, NOTCH1, PTEN, PTPN11, SMAD4 and STK11) were found to be high-risk factors for poorer survival (hazard ratio > 2 and P < 0.01; Table 2). Kaplan-Meier analyses confirmed the presence of statistically significant differences in terms of survival according to the mutational status of individual genes (Table 2), with two genes in the EGFR signaling pathway, HRAS and BRAF, showing the most significant difference. The median DFS for patients with and without HRAS mutation was 6.5 and 94 months (p < 0.0001) (Figure 4A). The median DFS rates for patients with and without BRAF mutations were 11 months and 94 months, respectively (P = 0.0008; Figure 4B). Other variables found to be significantly associated with DFS in the entire cohort were pT3-4 tumor status, pN2 nodal status, pathological stage IV, positive ECS status, close margins (≤ 4 mm), and treatment based solely on surgery (Table 2).
Table 2: Univariate analyses of the prognostic gene panel and traditional risk factors in relation to disease-free survival in 345 OSCC patients
Variable |
N (%) |
HR |
95% CI |
p-value |
q-valuea |
p-valueb |
Markerc |
Traditional risk factor |
|||||||
Age (≥ 65 vs < 65 years) |
38 (11.0) |
0.88 |
0.54 – 1.46 |
0.626 |
0.227 |
0.620 |
|
Gender (male vs female) |
325 (94.2) |
1.23 |
0.61 – 2.51 |
0.562 |
0.189 |
0.555 |
|
Extracapsular spread (Y vs N) |
201 (58.3) |
1.90 |
1.38 – 2.61 |
< 0.001 |
< 0.001 |
< 0.001 |
|
Stage (IV vs III) |
260 (75.4) |
2.26 |
1.50 – 3.41 |
< 0.001 |
< 0.001 |
< 0.001 |
|
T-status (pT34 vs pT12) |
192 (55.6) |
1.91 |
1.39 – 2.61 |
< 0.001 |
< 0.001 |
< 0.001 |
|
N-status (pN2 vs pN1) |
222 (64.3) |
2.04 |
1.45 – 2.88 |
< 0.001 |
< 0.001 |
< 0.001 |
|
Margin (≤ 4 vs > 4 mm) |
43 (12.5) |
2.03 |
1.37 – 3.01 |
< 0.001 |
< 0.001 |
< 0.001 |
|
Perineural invasion (Y vs N) |
177 (51.3) |
1.22 |
0.91 – 1.65 |
0.186 |
0.087 |
0.179 |
|
Vascular invasion (Y vs N) |
18 (5.2) |
1.04 |
0.53 – 2.04 |
0.901 |
0.277 |
0.899 |
|
Lymphatic invasion (Y vs N) |
44 (12.7) |
1.85 |
1.25 – 2.73 |
0.002 |
0.003 |
0.001 |
|
Treatment (surgery only vs surgery plus RT/CCRT) |
25 (7.2) |
1.78 |
1.06 – 2.98 |
0.028 |
0.022 |
0.024 |
|
Genotype (Mut vs Wt) |
|||||||
ABL1 |
11 (3.2) |
2.06 |
1.01 – 4.19 |
0.046 |
0.031 |
0.039 |
|
AKT1 |
11 (3.2) |
3.04 |
1.55 – 5.97 |
0.001 |
0.003 |
< 0.001 |
Y |
BRAF |
31 (9.0) |
2.07 |
1.33 – 3.22 |
0.001 |
0.003 |
< 0.001 |
Y |
CTNNB1 |
8 (2.3) |
2.83 |
1.33 – 6.03 |
0.007 |
0.009 |
0.004 |
Y |
FGFR3 |
15 (4.3) |
2.25 |
1.34 – 3.77 |
0.002 |
0.004 |
0.001 |
Y |
HRAS |
32 (9.3) |
2.74 |
1.79 – 4.19 |
< 0.001 |
< 0.001 |
< 0.001 |
Y |
KIT |
14 (4.1) |
2.21 |
1.23 – 3.99 |
0.008 |
0.009 |
0.006 |
Y |
MPL |
2 (0.6) |
4.37 |
1.08 – 17.67 |
0.039 |
0.003 |
0.021 |
|
NOTCH1 |
11 (3.2) |
2.57 |
1.31 – 5.03 |
0.006 |
0.009 |
0.004 |
Y |
PTEN |
13 (3.8) |
2.33 |
1.23 – 4.42 |
0.009 |
0.009 |
0.007 |
Y |
PTPN11 |
8 (2.3) |
2.72 |
1.27 – 5.79 |
0.010 |
0.009 |
0.006 |
Y |
SMAD4 |
19 (5.5) |
2.50 |
1.47 – 4.26 |
0.001 |
0.002 |
< 0.001 |
Y |
STK11 |
7 (2.0) |
3.25 |
1.43 – 7.35 |
0.005 |
0.008 |
0.002 |
|
VHL |
4 (1.2) |
2.74 |
1.01 – 7.40 |
0.047 |
0.031 |
0.035 |
Abbreviations: HR, hazard ratio; CI, confidence interval; RT, radiotherapy; CCRT, concurrent chemoradiation.
aq-value calculated with the Storey method
bKaplan-Meier anaysis, log-rank test p-value
cSelected in the gene signature
Prognostic mutation-based biomarker panel
As patients harboring specific gene mutations represented only a small fraction of the entire cohort, we sought to generate a mutation-based prognostic signature through the combined evaluation of multiple genetic mutations. To this aim, we selected 10 statistically significant (P < 0.01; false discovery rate-adjusted p-value < 0.05) genes with adequate population frequency (>2%). A similar criterion was used to identify mutated genes significantly associated with survival by Kandoth et al. [22]. The mutation-based gene signature consisted of HRAS, BRAF, FGFR3, SMAD4, KIT, PTEN, NOTCH1, AKT1, CTNNB1, and PTPN11 (Figure 4C). A patient was considered signature-positive if one or more genes in the signature panel were mutated. A total of 77 patients in our cohort harbored at least one mutation included in the gene signature. There were 53 (68.8%) relapse cases among the 77 patients with the mutated signatures. In contrast, only 119 (44.4%) relapsed cases were identified among the 268 patients with wild-type signatures. Kaplan-Meier curves confirmed the presence of statistically significant differences in terms of survival according to the gene signature status (Figure 4D). The median DFS periods for patients with and without the mutated signature were 12 months and > 180 months, respectively (p < 0.0001) (Figure 4D). The hazard ratio for disease relapse for patients with a mutated gene signature (compared with those harboring wild-type gene signatures) was 2.03 (95% confidence interval: 1.47−2.81, Table 3).
Subgroup validation of the prognostic gene signature
We further analyzed the reproducibility of the identified gene signature using two different resampling approaches. First, we randomly divided the cohort into two test subgroups − subgroup A (n = 172) and subgroup B (n = 173) − with a similar sample size and a comparable distribution of clinicopathological risk factors (Figure 1, Table 1). We identified 93 (54.1%) and 79 (45.7%) relapsed in subgroup A and subgroup B, respectively. We then estimated the hazard ratios associated with the presence of the prognostic gene signature for DFS in each subgroup using Cox regression. In both subgroups, patients with mutations in the gene signature showed significantly poorer DFS compared with wild-type patients (Table 3). The median DFS periods for patients with and without mutations in the gene signature for subgroup A and subgroup B were 11 vs. 94 months (P = 0.0016) and 12 vs. >180 months (P = 0.0033), respectively (Figures 5A, 5B). In the second resampling approach, we divided the entire study cohort into two different subgroups based on the time of patient enrollment. The 1996−2003 group consisted of 143 samples collected between 1996 and 2003, whereas the 2004−2011 group consisted of 202 samples collected between 2004 and 2011 (Figure 1, Table 1). In each group, we identified a total of 83 (58%) and 89 (44.1%) relapsed cases, respectively. We also analyzed the hazard ratios of the prognostic gene signature for DFS in each subgroup. In both subgroups, the prognostic gene signature was significantly associated with an increased risk of disease relapse (Table 3). Kaplan-Meier curves confirmed the presence of statistically significant differences in terms of DFS according to the prognostic gene signature in both subgroups (Table 3). The median DFS periods for patients with and without the prognostic gene signature in subgroup 1996–2003 and subgroup 2004–2011 were 10.5 vs. 56 months (P = 0.016) and 12 vs. >180 months (P = 0.0003), respectively (Figure 5C, 5D).
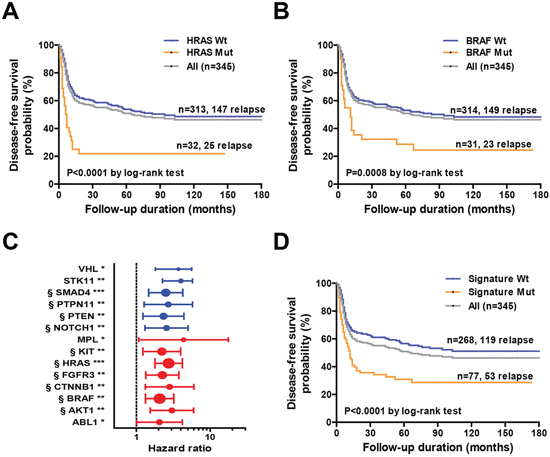
Figure 4: A. Kaplan-Meier analysis of disease-free survival in the entire cohort (n = 345) based on HRAS mutation status. B. Kaplan-Meier analysis of disease-free survival in the entire cohort (n = 345) based on BRAF mutation status. C. Univariate hazard ratios and 95% confidence intervals for disease relapse according to specific genetic alterations in individual genes. The strength of statistical significance identified in Cox proportional hazard models is reported in brackets (***P < 0.001, **P < 0.01, *P < 0.05). § denotes genes selected in the genetic signature. D. Kaplan-Meier analysis of disease-free survival of the entire cohort (n = 345) according to the genetic signature mutation status.
Table 3: Univariate analyses of the prognostic gene panel in relation to disease-free survival in the entire cohort and different patient subgroups
Group |
Signature mutant |
Signature Wt |
HR |
95% CI |
p-value |
q-valuea |
p-valueb |
||
N |
Event (%) |
N |
Event (%) |
||||||
Entire cohort (345, 172, 49.9) |
77 |
53 (68.8) |
268 |
119 (44.4) |
2.03 |
1.47–2.81 |
< 0.001 |
< 0.001 |
< 0.0001 |
Subgroup A (172, 93, 54.1) |
43 |
31 (72.1) |
129 |
62 (48.1) |
1.96 |
1.27–3.02 |
0.002 |
0.031 |
0.0016 |
Subgroup B (173, 79, 45.7) |
34 |
22 (64.7) |
139 |
57 (41.0) |
2.04 |
1.25–3.34 |
0.005 |
0.008 |
0.0033 |
1996–2003 (143, 83, 58) |
38 |
27 (71.1) |
105 |
56 (53.3) |
1.73 |
1.09–2.75 |
0.020 |
0.168 |
0.0158 |
2004–2011 (202, 89, 44.1) |
39 |
26 (66.7) |
163 |
63 (38.7) |
2.26 |
1.43–3.57 |
< 0.001 |
0.002 |
0.0003 |
Abbreviations: Wt, wild-type; HR, hazard ratio; CI, confidence interval.
aq-value calculated with the Storey method
bKaplan-Meier anaysis, log-rank test p-value
Associations between the prognostic gene signature and standard prognostic factors
The associations between the prognostic gene signature and standard prognostic factors are illustrated in Table 4. The prognostic gene signature was significantly associated with the pathological T status (P = 0.038). Although pT1-2 tumors were signature-positive in only 17% of cases, 26.6% of all pT3-4 tumors were found to be positive. We did not identify other significant relationships between the prognostic gene signature and other known prognostic factors, including age, sex, alcohol, betel nut chewing, cigarette smoking, pathological N status, pathological staging, and ECS status (Table 4).
Multivariate analysis
In the entire study cohort, the 5-year DFS was 51%. We then examined the entire study cohort (n = 345) with respect to the ability of the prognostic gene signature and other clinicopathological risk factors (pT3–4 vs. pT1–2, pN2 vs. pN1, p-Stage IV vs. p-Stage III, ECS [yes vs. no], perineural invasion [yes vs. no], lymphatic invasion [yes vs. no], vascular invasion [yes vs. no], margin status [positive margins/close margins vs. clear margins, i.e. ≤ 4 mm vs. > 4 mm], surgery alone vs. surgery plus RT or CCRT) to predict DFS. Table 5 shows the results of multivariate analyses of 5-year DFS in the entire study cohort. The presence of ECS (P = 0.021), pT3–4 disease (P = 0.001), pN2 (P = 0.005), close margins (P = 0.01), and surgery alone (P < 0.001) were independent risk factors for DFS. The hazard ratio for disease relapse for patients with a mutated gene signature (compared with those harboring wild-type gene signatures) was 1.62 (95% confidence interval = 1.16−2.28, P = 0.005, Table 5). These results indicated that the prognostic gene signature was independently associated with the DFS even after allowance for traditional risk factors.
DISCUSSION
Previous whole-exome sequencing studies of tumor specimens have shed more light on the mutation landscape of OSCC and identified several novel pathogenic mutations. However, due to limitations in sequencing depth, such reports have often failed to identify low-frequency mutations located in known causative genes [10–12]. Moreover, previous studies lacked long-term survival data useful to investigate the clinical impact of such mutations [10–12]. This study specifically aimed at detecting actionable mutations for targeted therapy and investigating the prognostic significance of genetic mutations in a large series of OSCC samples. To this aim, we used ultra-deep NGS to target hotspot regions of genes that may act as oncogenic drivers and/or serve as drug targets.
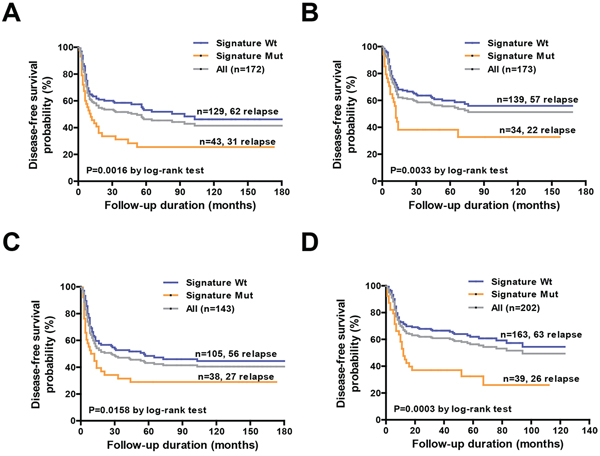
Figure 5: Association of the mutation-based genetic signature with disease-free survival in specific subgroups. A, B. Kaplan-Meier analysis of disease-free survival in subgroup A (n = 172) and subgroup B (n = 173) according to the genetic signature mutational status. C, D. Kaplan-Meier analysis of disease-free survival in the year 1996–2003 cohort (n = 143) and year 2004–2011 cohort (n = 202) according to the genetic signature mutation status.
Table 4: Association between the prognostic gene signature and the general characteristics of the 345 OSCC patients
Variable |
Signature mutant |
Signature wild-type |
p-value |
N (%) |
77 (22.3) |
268 (77.7) |
|
Age at onset (years) |
0.409 |
||
< 65 |
71 (23.1) |
236 (76.9) |
|
≥65 |
6 (15.8) |
32 (84.2) |
|
Sex |
1.0 |
||
Male |
73 (22.5) |
252 (77.5) |
|
Female |
4 (20.0) |
16 (80.0) |
|
Alcohol drinking |
0.777 |
||
No |
23 (23.2) |
76 (76.8) |
|
Yes |
54 (22.0) |
192 (78.0) |
|
Betel quid chewing |
1.0 |
||
No |
14 (22.2) |
49 (77.8) |
|
Yes |
63 (22.3) |
219 (77.7) |
|
Cigarette smoking |
0.263 |
||
No |
10 (31.3) |
22 (68.7) |
|
Yes |
67 (21.4) |
246 (78.6) |
|
Pathological T status |
0.038 |
||
pT1-2 |
26 (17.0) |
127 (83.0) |
|
pT3-4 |
51 (26.6) |
141 (73.4) |
|
Pathological N status |
0.105 |
||
pN1 |
21 (17.1) |
102 (82.9) |
|
pN2 |
56 (25.2) |
166 (74.8) |
|
Pathological stage |
0.294 |
||
III |
15 (17.7) |
70 (82.3) |
|
IV |
62 (23.9) |
198 (76.1) |
|
Extracapsular spread |
0.794 |
||
No |
31 (21.5) |
113 (78.5) |
|
Yes |
46 (22.9) |
155 (77.1) |
|
Perineural invasion |
0.898 |
||
No |
38 (22.6) |
130 (77.4) |
|
Yes |
39 (22.0) |
138 (78.0) |
|
Lymphatic invasion |
0.698 |
||
No |
66 (21.9) |
235 (78.1) |
|
Yes |
11 (25.0) |
33 (75.0) |
|
Vascular invasion |
0.383 |
||
No |
75 (22.9) |
252 (77.1) |
|
Yes |
2 (11.1) |
16 (88.9) |
|
Margin statusa |
0.333 |
||
≤ 4 mm |
12 (27.9) |
31 (72.1) |
|
> 4 mm |
64 (21.5) |
234 (78.5) |
|
Treatment modality |
0.004 |
||
Surgery only |
12 (48) |
13 (52) |
|
Surgery + RT/CCRT |
65 (20) |
255 (80) |
|
Relapse status |
< 0.001 |
||
No |
24 (13.9) |
149 (86.1) |
|
Yes |
53 (30.8) |
119 (69.2) |
Abbreviations: RT, radiotherapy; CCRT, concurrent chemoradiation.
aResults available for 341 patients.
Table 5: Multivariate analyses of the prognostic gene panel and traditional risk factors in relation to disease-free survival in 345 OSCC patients
Variable |
N |
HR |
95% CI |
p-value |
Mutation signature (Mutant vs Wild type) |
77 |
1.624 |
1.157 – 2.279 |
0.005 |
Extracapsular spread (Yes vs No) |
201 |
1.537 |
1.066 – 2.216 |
0.021 |
T-status (pT34 vs pT12) |
192 |
1.750 |
1.260 – 2.431 |
0.001 |
N-status (pN2 vs pN1) |
222 |
1.775 |
1.194 – 2.638 |
0.005 |
Stage (IV vs III) |
260 |
ns |
- |
< 0.001a |
Margin (≤ 4 vs > 4 mm) |
43 |
1.707 |
1.139 – 2.559 |
0.010 |
Treatment (surgery only vs surgery plus RT/CCRT) |
25 |
3.127 |
1.772 – 5.516 |
< 0.001 |
Abbreviation: HR, hazard ratio; RT, radiotherapy; CCRT, concurrent chemoradiation; CI, confidence interval; ns, not significant.
ap-value obtained in univariate analyses
Our study yielded at least four important results. First, we demonstrated that FFPE samples are suitable for ultra-deep NGS and that storage time of tumor specimens does not have affect sequencing results [23]. The ability to uncover genetic alteration in well-annotated FFPE samples thus allows studying the prognostic significance of common genetic alterations even under a retrospective study design. Second, our ultra-deep NGS approach had a high sensitivity for the detection of cancer-related mutations [24]. The high sensitivity of ultra-deep NGS can be attributed to its deeper coverage (> 2400-fold) compared with whole-exome sequencing. Third, at least 3% of our samples harbored sequence variants in genes that were not previously implicated in OSCC pathogenesis, including BRAF, ABL1, AKT1, KIT, MET, FGFR2, and FGFR3 [25, 26]. Notably, therapeutic agents targeting such genes are currently available or under development [27–29]. Finally, we identified a prognostic gene signature that predicts both DFS and OS.
Although we specifically focused on target regions with known mutation hotspots, our findings indicate that ultra-deep NGS allows a reliable broad mutation testing for most oncogenes and tumor suppressor genes. For example, while we only covered 68% of the TP53 coding region (including most of the DNA-binding region), the mutation frequency and spectrum of TP53 observed in our study were in line with those reported in the TCGA HNSCC dataset [10]. Although the number of genetic variations observed in NOTCH1 was significantly lower in our study [10, 11], this may be caused by the limited targeted sequence of NOTCH1 (that covered only 2.1% of the entire transcript). Preliminary sequencing data covering the entire NOTCH1 coding region revealed a mutation frequency similar to that reported in TCGA (data not shown).
Although the tumor proportion varied from 15 to 90% and tissue microdissection was not performed, we were able to confirm >90% of the identified mutations by orthogonal sequencing techniques. Oncogenic mutations are typically heterozygous and can sometimes co-occur with amplification of the wild-type allele in the same tumor, which can further complicate their detection in tumor specimens [30]. In order to overcome these issues, sequencing depth should be increased as much as possible [24]. Using an average depth greater than 2000×, herein we were able to confidently identify mutations with an allele frequency as low as 3% in a single sample. We detected 12 frequently mutated oncogenes (frequency ≥ 3%) in this cohort. Compared to the TCGA HNSCC data, 8 oncogenes were observed at 3-fold higher population frequency and 2 oncogenes were detected only in our study. Taken together, these findings suggest that ultra-deep NGS of hotspot regions is a highly sensitive method for detecting mutations in actionable cancer-related genes.
Slight differences in the mutation landscape were evident between the current study and the TCGA HNSCC dataset. For example, herein we detected the HRAS p.G12S allele in 9 cases (2.6%) compared with 2 cases (0.7%) in the TCGA data. The germline p.G12S HRAS mutation has been causally linked to Costello syndrome, a complex developmental disorder characterizing by facial dysmorphism [31, 32]. Moreover, the p.G12S HRAS somatic mutation has been mainly observed in upper aerodigestive tract malignancies [33]. Similar differences are evident for PIK3CA mutations. We observed 45 cases (13%) with mutations in the kinase domain (M1043, A1046, H1047, H1048, and G1049 mutations on exon 20) and 7 cases (2%) with mutations in the helical domain (E542, E545, and D549 mutations on exon 9). In contrast, the TCGA HNSCC dataset identified only 10 cases (3.6%) with kinase domain mutations (M1043 and H1047 on exon 20) and 32 cases (11.5%) with helical domain mutations (E542 and E545 on exon 9). Such differences in mutational hotspots can be attributed to differences in exposure to known carcinogens, disease stages, ethnicity, or genetic background. Previous studies have shown that helical domain mutations (e.g., p.E545K) activate the pathway by disrupting the p85-mediated inhibition [34], whereas kinase domain mutations (e.g., p.H1047R) promote an association with lipids to enhance the kinase activity [34]. A recent meta-analysis reported that PIK3CA exon 20 mutations are associated with a significantly shorter DFS in KRAS wild-type metastatic colorectal cancer patients treated with anti-EGFR antibody cetuximab [35]. A total of 68 cases (19%) in this study were found to harbor mutations in genes involved in the AKT-PI3K-mTOR pathway, suggesting that its specific inhibitors may warrant further studies in advanced OSCC patients.
In summary, ultra-deep NGS was clinically useful for identifying specific gene variants predicting prognosis in patients with advanced OSCC. The identified genetic panel needs independent validation before being introduced in clinical practice.
METHODS
Tumor samples
We retrospectively analyzed tumor samples from 345 nodal-positive, stage III/IV OSCC patients who were treated in Chang Gung Memorial Hospital between 1996 and 2011. The patients had not been previously treated, had no proven metastatic disease at the time of diagnosis and no synchronous or metachronous-occurring cancer. The primary inclusion criterion was amplifiable DNA from FFPE tissue. All patients were followed at least 30 months or until death. The estimated median follow-up time (calculated by the reverse Kaplan-Meier method) was 107 months. The pathological diagnosis of each OSCC case was confirmed using hematoxylin-eosin staining. Patients were staged according to the 2010 (7th) American Joint Committee on Cancer (AJCC) staging criteria. All of the study patients were treated by surgery, radiotherapy, or combined chemoradiotherapy at the Chang Gung Memorial Hospital between 1996 and 2011. Primary tumors were excised with safety margins of 1 cm or greater. Patients with cN+ disease received level I-V neck dissection, whereas patients with cN- disease received level I-III neck dissection. Post-operative radiotherapy (60 Gy) was given to patients with pathological risk factors according to the NCCN guidelines (before 2008) or the Chang Gung guidelines (as of 2008). Concomitant chemoradiation (CCRT) with cisplatin-based regimens were administered to patients with ECS, multiple lymph node metastases, or positive margins. Because the entire cohort was considered as a single test-set, positive findings required validated in an independent patient population. The number of patients, frequencies of sequences, and the study power depended on individual genetic mutations. Under the assumption of a 5-year DFS rate of 50% for patients with a positive mutation signature, a sample size of 345 patients will have 87% power at a significance level of 5% to detect an absolute difference of 15% (e.g. 42.5% vs 57.5%) in DFS associated with a positive signature. The study protocol followed the tenets of the Helsinki declaration and was approved by the Institutional Review Board of Chang Gung Memorial Hospital (CGMH 101-4457B). Patient consent was waived due to the retrospective nature of the study.
Sample preparation, DNA sequencing and data processing
Tumor specimens were obtained at the time of surgery and fixed with formalin following standard protocols. Archived FFPE blocks were stored at ambient temperature for up to 16 years. All samples used for DNA extraction contained a minimum of 15% DNA derived from tumor cells. For each tumor, one roll of ten-micrometer FFPE sections was used for genomic DNA extraction and library preparation. Of the 355 independent tumors, 345 tumors had amplifiable DNA. Targeted regions of 45 cancer-related genes (Data Supplementary Table S1 and Table S2) were amplified by PCR, barcoded, and sequenced using the Ion Torrent PGM system with the Ion 318 chip. Raw reads were mapped to the hg19 reference genome using Torrent Suite Server (version 3.2) and variants were identified using the Torrent Variant Caller plug-in (version 3.2). Common variants in dbSNP 138 and 1000 Genome project were filtered out. The remaining genetic variants were annotated using the ANNOVAR [36] and CPAP [37] pipelines. Selected mutations were confirmed using Sanger sequencing or pyrosequencing. Details regarding sequencing and data processing are provided in the Data Supplement. All sequencing and variant annotation were performed by the Genomic Core (HL) and the Bioinformatics Core (PJH) of the Molecular Medicine Research Center at Chang Gung University without knowledge of the clinical data.
Statistical analysis
Differences between the study groups were analyzed using Student’s t-tests or Fisher’s exact tests, as appropriate. Samples classified as unknown for any variable were excluded. The primary endpoint, DFS, was defined as the period of time in months from the date of surgery to the date of local or distant progression, death from any causes, or the date of last follow-up. Relative risk of relapse associated with mutations in the 45 genes tested were estimated from univariate Cox proportional hazards model for the entire cohort. Multiple test analyses was adjusted by using the q-value method to control for the false discovery rate. All genes with an adequate population frequency (> 2%) associated with DFS in the univariate analysis with p < 0.01 and q-value < 0.05 were selected to construct a mutation-based gene signature. A similar criterion was used to identify mutated genes significantly associated with survival by Kandoth et al. [22]. The mutation-based gene signature involved the HRAS, BRAF, FGFR3, SMAD4, KIT, PTEN, NOTCH1, AKT1, CTNNB1, and PTPN11 genes. A patient was considered signature-positive if one or more genes in the signature panel were mutated. The prognostic significance of the gene signature was investigated using univariate Cox regression model in the entire cohort and each subgroup. To identify additional factors associated with DFS, we evaluated the following clinicopathological variables in a univariate Cox regression model: age (≥ 65 vs. < 65 years ), sex (male vs. female), tumor status (pT3–4 vs pT1–2), nodal status (pN2 v pN1), AJCC stage (p-Stage IV vs. p-Stage III), ECS status (positive vs. negative), perineural invasion status (positive vs. negative), lymphatic invasion status (positive vs negative), vascular invasion status (positive vs negative), margin status (≤ 4 mm vs > 4 mm), treatment (only surgery vs. surgery plus RT or CCRT). All variables associated with DFS with p < 0.05 in univariate analysis and the gene signature mutation status were entered into a multivariate Cox regression models with a forced entry (or backward elimination) method. Disease-free survival in relation to gene signature mutation status was assessed with Kaplan-Meier survival curves (log-rank test). All analyses were performed using IBM SPSS Statistics v17. A two-tailed p-value < 0.05 was considered statistically significant.
FUNDING
This work was supported by grants NMRPG3B0403, CIRPG1E0012, and CMRPG3D1071 from the Chang Gung Memorial Hospital at Linkou.
ACKNOWLEDGMENTS
We appreciate the contribution and the valuable assistance of the Linkou Chang Gung Memorial Hospital Cancer Center databank and the Bioinformatics Core and Genomic Core of the Molecular Medicine Research Center at Chang Gung University.
REFERENCES
1. Neville BW, Day TA. Oral cancer and precancerous lesions. CA: a cancer journal for clinicians. 2002; 52:195–215.
2. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA: a cancer journal for clinicians. 2011; 61:69–90.
3. Amit M, Yen TC, Liao CT, Binenbaum Y, Chaturvedi P, Agarwal JP, Kowalski LP, Ebrahimi A, Clark JR, Cernea CR, Brandao SJ, Kreppel M, Zoller J, Fliss D, Bachar G, Shpitzer T, et al. Clinical nodal stage is a significant predictor of outcome in patients with oral cavity squamous cell carcinoma and pathologically negative neck metastases: results of the international consortium for outcome research. Annals of surgical oncology. 2013; 20:3575–3581.
4. Amit M, Yen TC, Liao CT, Chaturvedi P, Agarwal JP, Kowalski LP, Ebrahimi A, Clark JR, Kreppel M, Zoller J, Fridman E, Bolzoni VA, Shah JP, Binenbaum Y, Patel SG, Gil Z, et al. Improvement in survival of patients with oral cavity squamous cell carcinoma: An international collaborative study. Cancer. 2013; 119:4242–4248.
5. Liao CT, Lee LY, Hsueh C, Lin CY, Fan KH, Wang HM, Ng SH, Lin CH, Tsao CK, Chen IH, Chang KP, Huang SF, Kang CJ, Fang KH, Wang YC, Chang YL, et al. Comparative outcomes in oral cavity cancer with resected pT4a and pT4b. Oral oncology. 2013; 49:230–236.
6. Liao CT, Lee LY, Huang SF, Chen IH, Kang CJ, Lin CY, Fan KH, Wang HM, Ng SH, Yen TC. Outcome analysis of patients with oral cavity cancer and extracapsular spread in neck lymph nodes. International journal of radiation oncology, biology, physics. 2011; 81:930–937.
7. de Juan J, Garcia J, Lopez M, Orus C, Esteller E, Quer M, Leon X. Inclusion of extracapsular spread in the pTNM classification system: a proposal for patients with head and neck carcinoma. JAMA otolaryngology–head & neck surgery. 2013; 139:483–488.
8. Murugan AK, Munirajan AK, Tsuchida N. Ras oncogenes in oral cancer: the past 20 years. Oral oncology. 2012; 48:383–392.
9. Pitiyage G, Tilakaratne WM, Tavassoli M, Warnakulasuriya S. Molecular markers in oral epithelial dysplasia: review. Journal of oral pathology & medicine: official publication of the International Association of Oral Pathologists and the American Academy of Oral Pathology. 2009; 38:737–752.
10. Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, Kryukov GV, Lawrence MS, Sougnez C, McKenna A, Shefler E, Ramos AH, Stojanov P, Carter SL, Voet D, Cortes ML, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011; 333:1157–1160.
11. Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, Fakhry C, Xie TX, Zhang J, Wang J, Zhang N, El-Naggar AK, Jasser SA, Weinstein JN, Trevino L, Drummond JA, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011; 333:1154–1157.
12. Pickering CR, Zhang J, Yoo SY, Bengtsson L, Moorthy S, Neskey DM, Zhao M, Ortega Alves MV, Chang K, Drummond J, Cortez E, Xie TX, Zhang D, Chung W, Issa JP, Zweidler-McKay PA, et al. Integrative genomic characterization of oral squamous cell carcinoma identifies frequent somatic drivers. Cancer discovery. 2013; 3:770–781.
13. Takeuchi K, Ito F. Receptor tyrosine kinases and targeted cancer therapeutics. Biological & pharmaceutical bulletin. 2011; 34:1774–1780.
14. Pytel D, Sliwinski T, Poplawski T, Ferriola D, Majsterek I. Tyrosine kinase blockers: new hope for successful cancer therapy. Anti-cancer agents in medicinal chemistry. 2009; 9:66–76.
15. Tsiatis AC, Norris-Kirby A, Rich RG, Hafez MJ, Gocke CD, Eshleman JR, Murphy KM. Comparison of Sanger sequencing, pyrosequencing, and melting curve analysis for the detection of KRAS mutations: diagnostic and clinical implications. The Journal of molecular diagnostics: JMD. 2010; 12:425–432.
16. Poeta ML, Manola J, Goldwasser MA, Forastiere A, Benoit N, Califano JA, Ridge JA, Goodwin J, Kenady D, Saunders J, Westra W, Sidransky D, Koch WM. TP53 mutations and survival in squamous-cell carcinoma of the head and neck. The New England journal of medicine. 2007; 357:2552–2561.
17. Affolter A, Drigotas M, Fruth K, Schmidtmann I, Brochhausen C, Mann WJ, Brieger J. Increased radioresistance via G12S K-Ras by compensatory upregulation of MAPK and PI3K pathways in epithelial cancer. Head & neck. 2013; 35:220–228.
18. Murugan AK, Munirajan AK, Tsuchida N. Genetic deregulation of the PIK3CA oncogene in oral cancer. Cancer letters. 2013; 338:193–203.
19. Hassan NM, Tada M, Hamada J, Kashiwazaki H, Kameyama T, Akhter R, Yamazaki Y, Yano M, Inoue N, Moriuchi T. Presence of dominant negative mutation of TP53 is a risk of early recurrence in oral cancer. Cancer letters. 2008; 270:108–119.
20. Ohta S, Uemura H, Matsui Y, Ishiguro H, Fujinami K, Kondo K, Miyamoto H, Yazawa T, Danenberg K, Danenberg PV, Tohnai I, Kubota Y. Alterations of p16 and p14ARF genes and their 9p21 locus in oral squamous cell carcinoma. Oral surgery, oral medicine, oral pathology, oral radiology, and endodontics. 2009; 107:81–91.
21. Loyo M, Li RJ, Bettegowda C, Pickering CR, Frederick MJ, Myers JN, Agrawal N. Lessons learned from next-generation sequencing in head and neck cancer. Head & neck. 2013; 35:454–463.
22. Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, Leiserson MD, Miller CA, Welch JS, Walter MJ, Wendl MC, Ley TJ, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013; 502:333–339.
23. Kerick M, Isau M, Timmermann B, Sultmann H, Herwig R, Krobitsch S, Schaefer G, Verdorfer I, Bartsch G, Klocker H, Lehrach H, Schweiger MR. Targeted high throughput sequencing in clinical cancer settings: formaldehyde fixed-paraffin embedded (FFPE) tumor tissues, input amount and tumor heterogeneity. BMC medical genomics. 2011; 4:68.
24. Jiang L, Huang J, Morehouse C, Zhu W, Korolevich S, Sui D, Ge X, Lehmann K, Liu Z, Kiefer C, Czapiga M, Su X, Brohawn P, Gu Y, Higgs BW, Yao Y. Low frequency KRAS mutations in colorectal cancer patients and the presence of multiple mutations in oncogenic drivers in non-small cell lung cancer patients. Cancer genetics. 2013; 206:330–339.
25. Agada FO, Patmore H, Alhamarneh O, Stafford ND, Greenman J. Genetic profile of head and neck squamous cell carcinoma: clinical implications. The Journal of laryngology and otology. 2009; 123:266–272.
26. McBride SM, Rothenberg SM, Faquin WC, Chan AW, Clark JR, Ellisen LW, Wirth LJ. Mutation frequency in 15 common cancer genes in high-risk head and neck squamous cell carcinoma (HNSCC). Head & neck. 2013.
27. Smith JK, Mamoon NM, Duhe RJ. Emerging roles of targeted small molecule protein-tyrosine kinase inhibitors in cancer therapy. Oncology research. 2004; 14:175–225.
28. Montagut C, Settleman J. Targeting the RAF-MEK-ERK pathway in cancer therapy. Cancer letters. 2009; 283:125–134.
29. Hamakawa H, Nakashiro K, Sumida T, Shintani S, Myers JN, Takes RP, Rinaldo A, Ferlito A. Basic evidence of molecular targeted therapy for oral cancer and salivary gland cancer. Head & neck. 2008; 30:800–809.
30. Albertson DG. Gene amplification in cancer. Trends in genetics: TIG. 2006; 22:447–455.
31. Gripp KW, Lin AE. Costello syndrome: a Ras/mitogen activated protein kinase pathway syndrome (rasopathy) resulting from HRAS germline mutations. Genetics in medicine: official journal of the American College of Medical Genetics. 2012; 14:285–292.
32. Rauen KA. HRAS, and the Costello syndrome. Clinical genetics. 2007; 71:101–108.
33. Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer research. 2012; 72:2457–2467.
34. Miled N, Yan Y, Hon WC, Perisic O, Zvelebil M, Inbar Y, Schneidman-Duhovny D, Wolfson HJ, Backer JM, Williams RL. Mechanism of two classes of cancer mutations in the phosphoinositide 3-kinase catalytic subunit. Science. 2007; 317:239–242.
35. Mao C, Yang ZY, Hu XF, Chen Q, Tang JL. PIK3CA exon 20 mutations as a potential biomarker for resistance to anti-EGFR monoclonal antibodies in KRAS wild-type metastatic colorectal cancer: a systematic review and meta-analysis. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO. 2012; 23:1518–1525.
36. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research. 2010; 38:e164.
37. Huang PJ, Yeh YM, Gan RC, Lee CC, Chen TW, Lee CY, Liu H, Chen SJ, Tang P. CPAP: Cancer Panel Analysis Pipeline. Human mutation. 2013; 34:1340–1346.