
INTRODUCTION
Reactive oxygen species (ROS) are oxygen-containing reactive chemical species which include such biologically important molecules as superoxide, nitric oxide, hydroxyl radical and hydrogen peroxide (H2O2). Endogenous H2O2 is a by-product of mitochondrial respiration [1]. In addition, various signaling molecules, including growth factors, cytokines, hormones and neurotransmitters induce increases in intracellular H2O2 through the activation of NADPH oxidases (Nox). H2O2-dependent signaling has been implicated in diverse processes such as regulation of cell proliferation, differentiation, migration and apoptosis [2, 3, 4]. The production of a cytotoxic molecule has obvious potential risks to the cells as H2O2 is a major contributor to DNA damage, protein oxidation and lipid peroxidation [5, 6]. Cells therefore show a biphasic response to increased H2O2 levels; this is due to its role as a second messenger in intracellular signaling cascades (proliferative effect) and at high concentrations as an oxidant of proteins, lipids and nucleic acids (apoptotic and/or necrotic effect).
Cancer cells typically express higher levels of ROS compared to normal cells which gives them both a proliferative advantage and also promotes malignant progression [7, 8]. Ionizing radiation (IR) and chemotherapeutic drugs that elevate ROS levels are toxic to cancer cells because the same dosage of drug or radiation will cause death of the cancer cell due to ROS overload, but will be less toxic to neighboring normal cells which contain lower endogenous levels of ROS.
Redox-sensitive cysteines are the primary target for protein oxidation by H2O2. The amino acid cysteine can exist in proteins as a Cys sulfhydryl group (Cys-SH) or a Cys thiolate anion group (Cys-S-). Since the pKa value of most protein Cys residues is about 8.5, at neutral pH most Cys residues are in the Cys-SH form. However, a very small subset of proteins contain Cys residue(s) in the Cys-S- form at neutral pH typically due to the stabilization of this form by salt bridges to positively charged amino acids. Only the Cys-S- form is readily susceptible to oxidation by ROS and for this reason is referred to as a redox-sensitive cysteine. The redox-sensitive cysteines function as sensors of H2O2 and the oxidation of these residues affects the biological activity of the protein. For example, certain transcription factors such as JunD have a redox sensitive cysteine strategically positioned within their DNA binding domains such that oxidation of this cysteine by H2O2 blocks DNA-binding activity. Alternatively, antioxidant proteins utilize their redox sensitive cysteine(s) catalytically to reduce H2O2 to H2O.
Cells have developed several antioxidant systems to avoid ROS induced damage. These include catalase, superoxide dismutase, glutathione peroxidase and thioredoxin peroxidase (peroxiredoxins). The primary redox sensitive cysteine reductases are thioredoxin (Trx) and glutathione (GSH). Trx regulates signaling molecules, transcription factors, peroxidases and mediates redox-regulated gene expression [9]. During reduction of target proteins, Trx is oxidized and subsequently reduced/regenerated by thioredoxin reductase and NADPH. The Trx, Trx reductase and NADPH, collectively called the Trx redox system, constitute a fundamental antioxidant system.
Annexins are a structurally related family of calcium and phospholipid-binding proteins that are involved in the regulation of a range of molecular and cellular processes [10, 11]. The de-regulated expression of annexins has been shown to constitute a valuable marker of cancer progression [12, 13]. Annexin A2 (ANXA2) in particular has been positively associated with malignant progression [14] and resistance to chemotherapy [15, 16]. ANXA2 is unique among the annexins in that it possesses redox sensitive cysteine(s). Our laboratory has shown that extracellular ANXA2 binds to and reduces the enzyme plasmin and that ANXA2 oxidized during this reaction is subsequently reduced by the thioredoxin (Trx) redox system [17]. The presence of redox sensitive cysteine(s) on ANXA2 that can participate in cycles of oxidation and reduction, coupled with its ubiquitous and abundant expression in cells led us to hypothesize that this protein could play a role in cellular redox regulation.
Here, we report a novel role for ANXA2 as a redox regulatory protein. We show that upon oxidative stress, ANXA2 depleted cells display higher levels of ROS, increased activation of the ROS-induced pro-apoptotic kinases: p38, JNK and Akt and higher sensitivity to oxidative stress-mediated cell death compared to control cells. We further identify the Cys8 residue as the redox sensitive cysteine of ANXA2 and show that this cysteine is reversibly oxidized by H2O2 and reduced by the thioredoxin redox system. In vivo studies show a significant increase in protein oxidation in the liver and lung tissues of ANXA2-null mice compared to WT mice. Furthermore, the growth of tumors resulting from the subcutaneous injection of ANXA2-depleted human cancer cell lines, HT1080 and A549, in mice showed severe growth impairment compared to control cells. Intraperitoneal injection of the antioxidant, N-acetyl cysteine (NAC) enabled these tumors to grow at a similar rate as the control tumors. We also observed enhanced protein oxidation in the ANXA2 depleted HT1080 tumors compared to control tumors, which was prevented by NAC treatment. These results show that replacement of ANXA2 by another antioxidant, such as NAC, reverses the tumor growth deficit phenotype observed in the ANXA2 depleted cells, indicating that ANXA2 is a redox regulatory protein that plays a key role in tumorigenesis. Ex-vivo human cancer studies showed that in general, cancer cells express significantly higher levels of the reduced form of ANXA2 compared to normal tissue and that the up-regulation of the levels of reduced ANXA2 correlate with protection from oxidation of the proteins in these tumors, indicating that ANXA2 may function as a redox regulatory protein in human tumors.
RESULTS
ANXA2 protein is a redox sensor
In order to test if ANXA2 plays a role in cellular redox regulation we investigated if this protein is sensitive to H2O2-induced oxidative stress. We first examined ANXA2 sensitivity to a physiological stimulus that produced small localized increases in intracellular H2O2. The interaction of epidermal growth factor (EGF) with its receptor stimulates the activity of the NADPH oxidase, Nox resulting in a rapid and transient increase in intracellular H2O2 levels to nanomolar concentrations [18]. In order to determine if ANXA2 was oxidized by EGF-generated H2O2 we took advantage of the selective reactivity of Biotin-conjugated iodoacetamide (BIAM) with the reactive thiols of proteins. BIAM has been commonly used to quantify redox sensitive cysteine oxidation by ROS since it selectively reacts with redox sensitive cysteine(s) (Cys-S-) at physiological pH, but not with Cys-SH or oxidized cysteine residues (Cys-SOH or Cys-S-S-Cys) [19, 20]. A decrease in BIAM labeling of proteins, as monitored by streptavidin blot analysis, indicates oxidation of the redox sensitive cysteine(s) by ROS. Accordingly, TIME cells were incubated with EGF and cellular extracts were either analyzed by SDS-PAGE followed by western blotting for ANXA2 or incubated with BIAM and the labeled/ biotinylated proteins purified by incubation with streptavidin beads, followed by SDS-PAGE and western blotting for ANXA2. Our results showed that ANXA2 was highly oxidized by 30 minutes after EGF stimulation. However, by 1 hour after treatment, we observed an up-regulation in the levels of reduced ANXA2. Pre-incubation of cells with the antioxidant agent N-acetyl cysteine (NAC) prevented the EGF-dependent oxidation of ANXA2 (Figure 1A), confirming that the EGF-dependent loss in the labeling of ANXA2 by BIAM was due to oxidation of ANXA2.
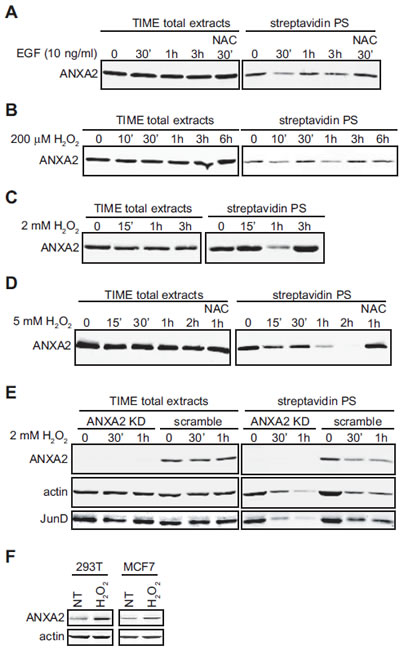
Figure 1: Cellular ANXA2 is responsive to reactive oxygen species. (A) TIME cells were treated with 10 ng/ml EGF in the absence or presence of NAC for the times indicated; (B) TIME cells were treated with 200 μM H2O2 for the times indicated; (C) TIME cells were treated with 2 mM H2O2 for the times indicated; (D) TIME cells were treated with 5 mM H2O2 in the absence or presence of 10 mM NAC for the times indicated; (E) TIME ANXA2 shRNA2 (KD) or TIME ANXA2 scramble cells were treated with 2 mM H2O2 for the times indicated. (A-E) Lysates were labeled with 10 μM BIAM and 200 µg of each labeled lysate was incubated with streptavidin beads. Total cell extracts and streptavidin purified samples (streptavidin PS) were subjected to SDS-PAGE followed by western blotting with the antibodies indicated. (F) 293T and MCF7 cells were treated with 100 μM H2O2 for two weeks. 20 μg of each cell lysate was subjected to SDS-PAGE followed by western blotting with the antibodies indicated.
Next, we induced a slight increase in intracellular H2O2 levels by adding 200 μM H2O2 to the medium of TIME cells (see Figure 2A, grey bar), and observed that ANXA2 was transiently and reversibly oxidized by H2O2 (Figure 1B). Three hours after treatment with H2O2, the levels of reduced ANXA2 were similar to untreated cells. Importantly, the total levels of ANXA2 were unchanged during the time course of the experiment. This result suggested that ANXA2 was oxidized by H2O2 and this oxidation occurred concomitantly with small changes of intracellular H2O2. We also examined the redox status of ANXA2 during more extreme oxidative stress conditions. We observed ANXA2 oxidation one hour after adding 2 mM H2O2 to the medium of TIME cells. However, by three hours, the levels of reduced ANXA2 were similar to untreated cells (Figure 1C). These results show that high levels of H2O2 can also result in the reversible oxidation of ANXA2.
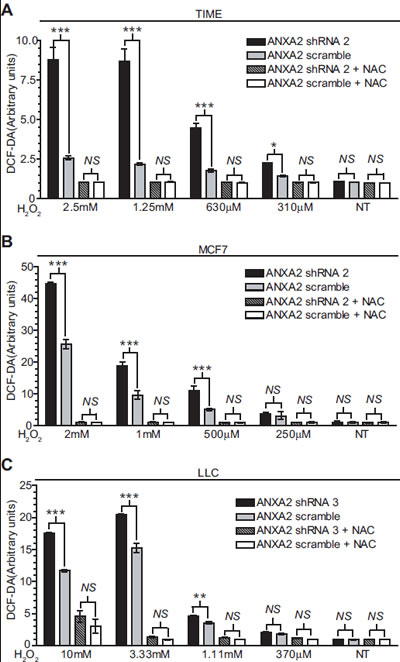
Figure 2: ANXA2 depleted cells accumulate higher levels of ROS upon oxidative stress. (A) TIME ANXA2 shRNA 2 or TIME ANXA2 scramble cells, (B) MCF7 ANXA2 shRNA 2 or MCF7 ANXA2 scramble cells and (C) LLC ANXA2 shRNA 3 or LLC ANXA2 scramble cells were incubated with 50 µM DCF-DA reagent. Cells were treated with different concentrations of H2O2 in the absence or presence of 10 mM NAC. Fluorescence was measured using a fluorometer plate reader. Data are represented as ± StDev (N=4).
We investigated the redox status of ANXA2 under oxidative conditions that result in cell death. For these experiments, 5 mM H2O2 was added to the medium of TIME cells. The observed H2O2-dependent loss of BIAM labeling demonstrated that ANXA2 was oxidized by lethal concentrations of H2O2, however under these conditions, ANXA2 was irreversibly oxidized (Figure 1D). Pre-incubation of cells with the antioxidant NAC prevented the oxidation of ANXA2 (Figure 1D), again confirming that oxidation of ANXA2 by H2O2 was responsible for the loss of BIAM reactivity.
Although our results suggested that ANXA2 was a target for oxidation by H2O2 it was unclear if ANXA2 was involved in redox signaling or in redox regulation. Therefore, we generated stable ANXA2 knockdown cell lines using a retroviral vector-based shRNA system as shown in Figures 1E and S1 and confirmed that other annexins were not down-regulated in the ANXA2 depleted cells by western blot analysis (Figure S1). Next we increased intracellular H2O2 by addition of 2 mM H2O2 to the medium of ANXA2 depleted TIME and control cells. The cell extracts were incubated with BIAM and the BIAM-labeled proteins were collected by incubation with streptavidin beads, followed by SDS-PAGE and western blot analysis. We examined the redox status of two well described redox-sensitive proteins, namely JunD, a major oxidative stress responsive transcription factor and actin which is also highly sensitive to changes in intracellular ROS levels. As shown in Figure 1E, oxidation of both JunD and actin was more rapid and pronounced in ANXA2 depleted TIME cells compared to control cells suggesting that the intracellular levels of ROS were increased in ANXA2 depleted cells during oxidative stress. This result suggested that ANXA2 might regulate ROS levels during oxidative stress i.e. play a role in cellular redox regulation.
Several reports have shown that cells up-regulate antioxidant proteins in response to oxidative stress in order to maintain redox equilibrium and avoid cellular damage/death [21]. We investigated ANXA2 expression in response to a long term treatment with sub-lethal doses of H2O2. For this experiment we used cell lines that express ANXA2 at low levels, 293T cells, or moderate levels, MCF7 cells. These results showed up-regulation of ANXA2 expression in both cell lines under oxidative stress conditions, further supporting a role for ANXA2 as a redox regulatory protein (Figure 1F).
ANXA2 depleted cells accumulate higher levels of ROS upon oxidative stress
Since our results suggested that ANXA2 might regulate ROS levels during oxidative stress we measured intracellular ROS levels using the probe, 2’,7’ dichlorodihydrofluorescein diacetate (DCF-DA) in either untreated or H2O2 treated cells. We noted that depletion of ANXA2 did not seem to significantly affect the basal levels of ROS in normal (TIME) or cancer cells (MCF7 and LLC) (Figure 2A-C). Strikingly, we observed that ROS levels were significantly higher in ANXA2 depleted cells after exposure to H2O2. Pre-incubation of cells with NAC blocked H2O2-induced increase in intracellular ROS, as expected. The levels of other redox regulatory proteins, including catalase, glutathione peroxidase, SOD-1, SOD-2, peroxiredoxins (I-IV) and thioredoxin were not altered in the ANXA2 depleted cells or in cells over-expressing ANXA2 as examined by western blotting (Figure S2). We also did not detect increases in the levels of glutathione (data not shown). Together these results demonstrate that ANXA2 plays a role in the regulation of ROS levels during oxidative stress.
ANXA2 protects cells from oxidative stress induced death
When oxidative stress exceeds the capacity of the cellular ROS scavenging system, cells succumb to the cytotoxic effects of the accumulated ROS and die [22, 23]. Since we observed increased levels of ROS in ANXA2 depleted versus control cells following oxidative stress, we hypothesized that ANXA2 might play a role protecting cells from ROS induced cell death. In order to investigate this possibility we analyzed the viability of ANXA2 depleted and control cells upon treatment with different concentrations of H2O2. We observed that cells depleted of ANXA2 were significantly more susceptible to H2O2-induced death compared to control cells (Figure 3A-C, left panels). The concentration of H2O2 that resulted in 50% loss in cell viability (EC50), decreased significantly in ANXA2 depleted cells compared to control cells (Figure 3D and table S1). Incubation of cells with NAC prior to treatment with H2O2 reversed this effect, as expected (Figure 3A-C, middle panels).
Many chemotherapeutic agents kill cancer cells through mechanisms that involve production of ROS. We observed that depletion of ANXA2 resulted in a significant increase in susceptibility to cell death induced by the ROS producing chemotherapeutic agent, etoposide (Figure 3A-C, right panels) [24]. The EC50 values were significantly lower in ANXA2 depleted cells compared to control cells (Figure 3E and table S1). We also observed a significant decrease in the EC50 values for ANXA2 depleted TIME and LLC cells treated with the chemotherapeutic doxorubicin and ANXA2 depleted MCF7 cells treated with tamoxifen (Figure S3 and Table S1).
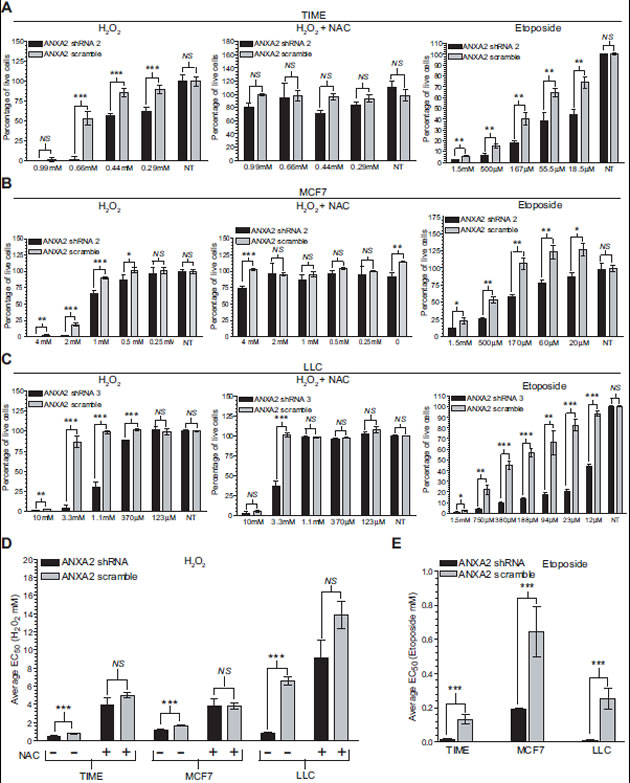
Figure 3: ANXA2 depleted cells are more sensitive to oxidative stress induced death. (A) TIME ANXA2 shRNA2 or TIME ANXA2 scramble cells, (B) MCF7 ANXA2 shRNA2 or MCF7 ANXA2 scramble cells and (C) LLC ANXA2 shRNA 3 or LLC ANXA2 scramble cells were treated with different concentrations of H2O2 in the absence (left panel) or presence of 10 mM NAC (middle panel) for 24 hours or treated with etoposide (right panel) for 48 hours. Cell viability was measured using the CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay. Average 490 nm absorbance for non-treated (NT) cells was set as 100% viability. (D) Ec50 values for the cells treated with H2O2 in the absence or presence of NAC as indicated. (E) Ec50 values for the cells treated with etoposide. Data was analysed using the two tailed Student’s t test and represented as ± StDev (N=4).
ANXA2 depleted cells show enhanced activation of oxidative stress responsive signaling proteins
After observing that ANXA2 depletion resulted in increased levels of ROS and loss of cell viability during oxidative stress, we investigated which oxidative stress responsive pathways were sensitive to the increased ROS levels observed in ANXA2 depleted cells. Cells typically use two main signaling pathways to respond to oxidative stress; the MAPK pathway that includes activation of the pro-apoptotic stress kinases c-Jun N-terminal kinases (JNKs) and p38 and the PI3K pathway, where activation of Akt by oxidative stress has been shown to be associated with apoptosis [25, 26].
We treated ANXA2 depleted and control cells with H2O2 and subjected the cell extracts to SDS-PAGE followed by western blot analysis. The levels of phospho-p38 (P-p38), phospho-JNK (P-JNK) and phospho-Akt (P-Akt) prior to oxidative insult were similar in ANXA2 depleted and control cells, indicating that these pathways were not being activated simply by depletion of ANXA2. Interestingly, upon oxidative stress ANXA2 depleted cells showed significantly higher levels of P-p38, P-Akt and slightly elevated P-JNK compared to control cells (Figure 4), consistent with increased intracellular ROS levels and susceptibility of these cells to oxidative stress induced death. As predicted, pre-incubation of cells with NAC prevented the phosphorylation of these redox-responsive kinases, confirming that their activation was due to oxidative stress (Figure 4B, C).
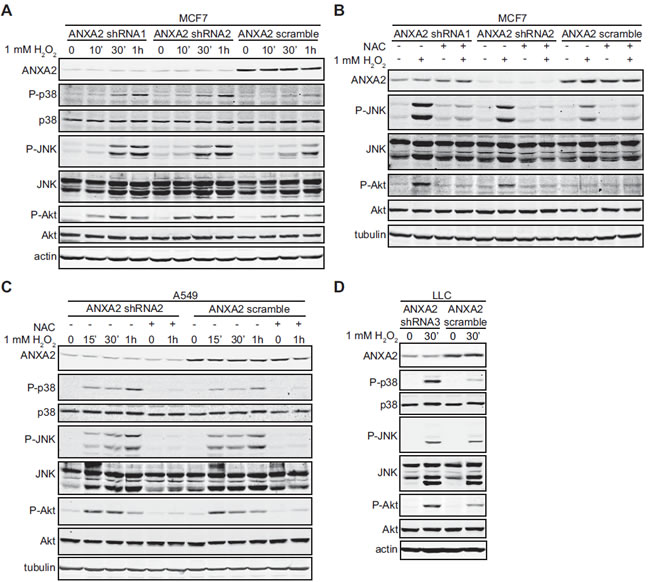
Figure 4: ANXA2 depleted cells show enhanced activation of oxidative stress responsive signaling proteins. (A, B) MCF7 ANXA2 shRNA1, MCF7 ANXA2 shRNA2 or MCF7 ANXA2 scramble cells were treated with (A) 1 mM H2O2 for the times indicated or (B) 1 mM H2O2 in the absence or presence of 10 mM NAC for 30 min; (C) A549 ANXA2 shRNA2 or A549 ANXA2 scramble cells were treated with 1 mM H2O2 in the absence or presence of 10 mM NAC for the times indicated; (D) LLC ANXA2 shRNA3 or LLC ANXA2 scramble cells were treated with 1 mM H2O2 for 30 minutes. (A-D) 25 µg of each cell lysate was subjected to SDS-PAGE followed by western blotting with the antibodies indicated.
H2O2 oxidized ANXA2 is reversibly reduced by the Trx system
Collectively, our data indicated that ANXA2 acted as a cellular redox regulatory protein; however the mechanism by which ANXA2 interacted with ROS was unclear. Therefore, we investigated which cysteine residue(s) of cellular ANXA2 were oxidized by ROS. Previous work from our laboratory has identified two cysteine residues of ANXA2, Cys8 and Cys132, as potential reactive residues based on their modification by glutathione in vitro. In order to further investigate which of these residues are redox sensitive cysteine(s) in vivo, we over-expressed ANXA2 wild-type (WT), and several cysteine mutants in 293T cells. These cells were chosen because they have low levels of endogenous ANXA2. The levels of over-expression of ANXA2 in the 293T cells were comparable to the endogenous levels of this protein in A549 breast cancer cells (Figure S4A) and lower than in the TIME cells (data not shown), indicating that ANXA2 over-expression in the 293T cells resulted in intracellular levels of ANXA2 were similar to the levels of ANXA2 observed in other cell lines. We incubated cell lysates from the 293T cells with BIAM, followed by SDS-PAGE and western blotting with a streptavidin probe, in order to identify BIAM labeled proteins. We observed that both ANXA2C8S and ANXA2C8A proteins were expressed in the 293T cells at comparable levels to ANXA2 WT, ANXA2C132S and ANXA2C132A. However, the ANXA2 Cys8 mutant proteins were not labeled by BIAM. In contrast, the labeling of ANXA2C132S and ANXA2C132A was similar to that of ANXA2 WT (Figure 5A). This result shows that only Cys8 and not Cys132 of ANXA2 is a redox sensitive cysteine in vivo. Furthermore, we also observed that ANXA2 was one of the main BIAM labeled bands in the whole cell extracts, indicating that the Cys8 residue of ANXA2 is highly reactive/ sensitive to oxidation. Next, we repeated this experiment, but collected the BIAM labeled proteins with streptavidin beads and performed a western blotting for ANXA2. As shown in Figure 5B we were unable to detect the ANXA2 Cys8 mutants. This established that Cys8 of ANXA2 is the redox sensitive cysteine.
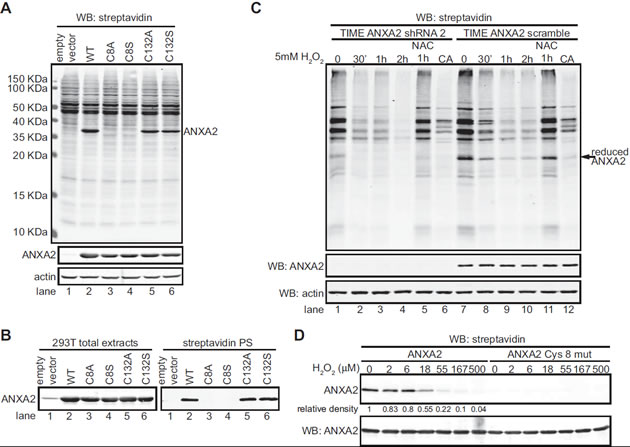
Figure 5: Cys8 residue of cellular ANXA2 is a redox sensitive cysteine. (A) 293T cells were transiently transfected with pcDNA3 empty vector (lane 1), pcDNA3-ANXA2 WT (lane 2), or a series of ANXA2 cysteine mutant plasmids: pcDNA3-ANXA2-Cys-8-Ala (lane 3), pcDNA3-ANXA2-Cys-8-Ser (lane 4), pcDNA3-ANXA2-Cys-132-Ala (lane 5) or pcDNA3-ANXA2-Cys-132-Ser (lane 6) for 48 hours. 20 μg of each cell extract was labeled with 20 μM BIAM and subjected to SDS-PAGE followed by western blotting with a streptavidin probe and the antibodies indicated. (B) 200 μg of each cell lysate was incubated with 20 μM BIAM, labeled proteins were purified with streptavidin beads. Cell extracts and streptavidin purified samples (streptavidin PS) were subjected to SDS-PAGE followed by western blotting for ANXA2. (C) TIME ANXA2 shRNA2 or TIME ANXA2 scramble cells were treated with 5 mM H2O2 in the absence or presence of 10 mM NAC for the times indicated. CA stands for competition assay; these samples were treated with 5 mM DTT prior to incubation with BIAM. Cell extracts were labeled with 10 μM BIAM, subjected to SDS-PAGE followed by western blotting with the antibodies indicated. (D) 0.65 μM of human recombinant ANXA2 or ANXA2C8S protein was incubated with the indicated concentrations of H2O2 for 30 minutes, after what 10 μg/ml of catalase was added. Samples were labeled with 20 µM BIAM and subjected to SDS-PAGE followed by western blotting with a streptavidin probe and ANXA2 antibody. Protein band quantification was done using the Licor Odyssey software.
In order to determine the relative abundance of ANXA2 compared with other redox sensitive proteins, we labeled the cell extracts of ANXA2 depleted and control TIME cells with BIAM, followed by SDS-PAGE and western blotting with a streptavidin probe (Figure 5C). We did not detect a BIAM labeled band around 36 KDa (ANXA2 molecular weight) in the ANXA2 depleted TIME cells, and ANXA2 was one of the major proteins that reacted with BIAM in the TIME control cells, indicating the abundance of ANXA2 and the high reactivity of its Cys8 redox sensitive cysteine in these cells. Furthermore, we observed that oxidative stress resulted in a more dramatic and rapid oxidation of cellular proteins in the ANXA2 depleted TIME cells compared to control cells (Figure 5C), further suggesting that ANXA2 plays a role in redox regulation. Pre-incubation of cells with NAC prevented the oxidation of ANXA2 (Figure 5C, lane 11). As an additional control we observed that addition of 5 mM DTT to the cell extracts prior to incubation with BIAM abolished BIAM labeling of ANXA2. Since DTT competes with ANXA2 for the BIAM reagent, this result establishes that ANXA2 biotinylation is due to BIAM labeling of its redox sensitive cysteine and that ANXA2 does not exist as a biotinylated protein in the cells (Figure 5C, lane 12).
The reagent 3-(N-Maleimidylpropionyl)biocytin (NEM-biotin), although distinct in chemical reactivity from BIAM is also commonly used to identify redox sensitive cysteines in proteins [27]. We observed in experiments in which cell extracts were labeled with NEM-biotin that ANXA2 with C8S and C8A substitutions completely failed to react with this probe, while ANXA2 with C132S and C132A substitutions reacted with this probe similarly to ANXA2 WT protein (Figure S4B). This result further supported that Cys8 residue is the only redox sensitive cysteine of ANXA2.
H2O2 is a key ROS that also functions as a second messenger molecule in cells. To investigate whether the redox sensitive cysteine of ANXA2, Cys8, is a direct target for H2O2 oxidation, we incubated human recombinant ANXA2 with increasing amounts of H2O2, quenched the reaction with catalase and labeled the protein with BIAM followed by SDS-PAGE and western blotting with a streptavidin probe. These results showed that ANXA2 reacted with BIAM and that the BIAM labeling of ANXA2 decreased with increasing concentrations of H2O2 establishing that ANXA2 was oxidized by H2O2 in a dose dependent manner (Figure 5D). However, the ANXA2C8S mutant protein did not react with the BIAM reagent as expected, confirming again that Cys8 residue is the redox sensitive cysteine of ANXA2 (Figure 5D).
The NADPH-dependent thioredoxin and glutathione redox systems are the best characterized antioxidant systems that are utilized by the cells to reduce/restore oxidized proteins. We observed that incubation of a cellular lysate with H2O2 caused the oxidation of ANXA2 and that the subsequent addition of NADPH to this cellular lysate reversed ANXA2 oxidation (Figure 6A). This result shows that ANXA2 can be reversibly oxidized by H2O2 and reduced by an NADPH-dependent antioxidant system in the cells. We next investigated if the Trx antioxidant system was involved in the restoration of oxidized ANXA2 to its reduced form. Human recombinant ANXA2 was incubated in the presence or absence of H2O2 and the sample was labeled with BIAM. As expected, incubation of ANXA2 with H2O2 resulted in the oxidation of ANXA2. Interestingly, if the oxidized ANXA2 was then incubated with thioredoxin, both ANXA2 and a 48 KDa complex of ANXA2 and thioredoxin were observed by SDS-PAGE and western blotting under non-reducing conditions. Furthermore, the formation of the ANXA2-thioredoxin complex resulted in the reduction of H2O2-oxidized ANXA2 (Figure 6B). As a control we repeated this experiment with ANXA1, an annexin closely related to ANXA2. ANXA1 does not possess a redox sensitive cysteine and the BIAM reagent did not label this protein. Since ANXA1 contains cysteine residues this result further supports the specific labeling of reactive thiols by BIAM (Figure 6B). We also observed that when human recombinant ANXA2 was incubated with 100 μM H2O2 and NADPH (control), the ANXA2 was rapidly oxidized by 50% within one minute and by 90% after 30 minutes of incubation with H2O2. However, the inclusion of Trx reductase, Trx and NADPH (the Trx antioxidant system) resulted in the reduction of H2O2-oxidized ANXA2 (Figure 6C). ANXA2 reduced form was similar to non treated protein until 5 minutes after addition of H2O2, at 10 minutes post-incubation with H2O2 we observed a mild oxidation of ANXA2, around 30%, which increased by 15 minutes post-incubation to 60%, but ANXA2 reduced levels were almost completely restored by 30 minutes after incubation with 100 μM H2O2 in the presence of the Trx redox system. Together these results indicate that H2O2-oxidized ANXA2 is reversibly reduced by the Trx system.
In order to determine if ANXA2 interacts with Trx in vivo we performed co-immunoprecipitation studies. Cells were incubated in the absence or presence of H2O2 and the cell lysates were immunoprecipitated with antibodies against ANXA2, Trx or JunD (control). These results confirmed that Trx and ANXA2 formed a complex whose levels increased 1.5 fold after stimulation with H2O2 (Figure 6D, E).
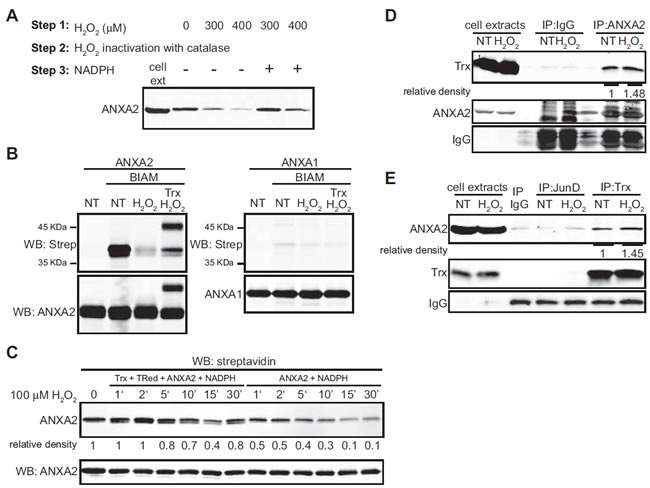
Figure 6: ANXA2 is a substrate of the Trx system. (A) TIME cell extracts were treated with the indicated amounts of H2O2, incubated with catalase after what mock or 500 μM of NADPH was added, as indicated. Lysates were incubated with 10 μM BIAM and labeled proteins were purified with streptavidin beads. Cell extracts of non treated cells (lane 1) and streptavidin purified samples were subjected to SDS-PAGE followed by western blotting for ANXA2. (B) 2.5 μM ANXA2 or ANXA1 were incubated in the absence or presence of 100 μM H2O2 with or without 20 μM of Trx as indicated. Proteins were incubated in the absence or presence of 20 μM BIAM, subjected to SDS-PAGE under non-reducing conditions, followed by western blot analysis with the antibodies indicated. (C) 2 μM of human recombinant ANXA2 were incubated with 0.15 μM Trx reductase, 0.5 μM Trx, 500 μM NADPH (left panel) or with NADPH alone (right panel). 250 μM of H2O2 was added and samples were collected at the times indicated, after what 10 μg/ml of catalase was added. Samples were labeled with 20 μM BIAM and subjected to SDS-PAGE followed by western blotting with a streptavidin probe. (D) A549 cells were either non-treated (NT) or treated with 1 mM H2O2 for 1 hour. Cells were lysed and immunoprecipitated with IgG, anti-JunD or anti-Trx antibodies as indicated; (E) A549 cells were either non-treated (NT) or treated with 1 mM H2O2 for 1 hour. Cells were lysed and immunoprecipitated with either IgG or anti-ANXA2 antibodies as indicated. (D-E) Immunoprecipitates and 20 µg of each cell extract were subjected to SDS-PAGE followed by western blotting with the antibodies indicated. Protein band quantification was done using the Licor Odyssey software.
ANXA2-null mice show increased protein oxidation in the liver and lungs
We used ANXA2-null mice to study the role of ANXA2 in cellular redox regulation in vivo [28]. We reasoned that if ANXA2 played a role in cellular redox regulation its loss would result in enhanced oxidative stress which could be visualized by increased tissue protein oxidation. In order to investigate this possibility, we removed and homogenized organs from age-matched WT and ANXA2-null mice, labeled the cell extracts with BIAM, followed by SDS-PAGE and western blotting with a streptavidin probe. We observed a 50% decrease in the BIAM labeling of liver proteins in ANXA2-null mice compared to WT mice (Figure 7A) and a 30% decrease in BIAM labeling of lung proteins in ANXA2-null mice (Figure 7B). These data established that the liver and lung tissues of ANXA2-null mice were significantly more oxidized compared to WT mice. Interestingly, the oxidation status of spleen proteins was similar in the ANXA2-null and WT mice (Figure 7C).
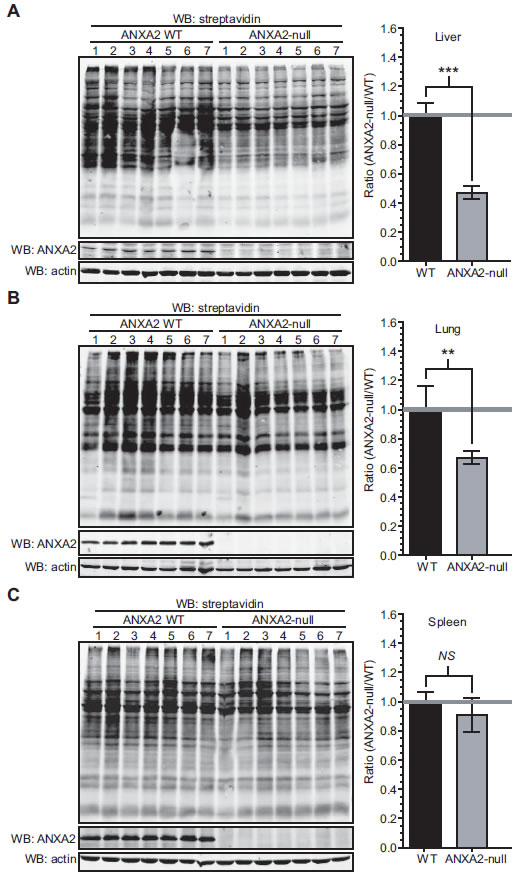
Figure 7: ANXA2-null mice show increased protein oxidation in the liver and lungs. ANXA2 WT or ANXA2-null mice age matched were sacrificed, (A) livers, (B) lungs and (C) spleens were removed, homogenized and labeled with 20 µM BIAM. Samples were subjected to SDS-PAGE followed by western blotting with a streptavidin probe (left panels). The right panels show the quantification of the results on the left panels using the Licor Odyssey software. Data was analyzed using the two tailed Student’s t test and represented as ± StDev (N=7).
ANXA2 redox regulatory function plays a role in tumorigenesis
Tumors are typically under higher oxidative stress compared to neighboring normal cells. We reasoned that if ANXA2 plays a role in cellular redox regulation its depletion will render cancer cells more susceptible to ROS induced damage/death during tumorigenesis. In order to investigate this hypothesis, we established ANXA2 depleted HT1080 and A549 cells and respective control cells and analyzed the early passaged cells for protein oxidation. As assessed by BIAM labeling, we observed no difference in the oxidation of cellular proteins in ANXA2 depleted versus control cell lines while cultured in optimal tissue culture conditions (Figure S5A, F). This was expected as these cells were not under oxidative stress. The ANXA2 depleted and control cells were injected subcutaneously into the right flank of NOD-SCID mice and tumor growth was monitored by caliper measurement. We observed a significant impairment of tumor growth by ANXA2 depleted HT1080 and A549 cancer cells compared to control cells (Figure 8A, B). At the experimental end point, the volume of tumors grown from ANXA2 depleted HT1080 cells was approximately 85% smaller compared to tumors grown from control HT1080 cells. Furthermore, the volume of tumors grown from ANXA2 depleted A549 cancer cells was 70% smaller compared to A549 control tumors. The growth impairment of ANXA2 depleted A549 and HT1080 tumors was not due to decreased cell proliferation since we observed the same growth rate for ANXA2 depleted and control cell lines in culture (Figure S5E, H). In order to investigate if the growth impairment observed for the ANXA2 depleted tumors was due to ROS overload, we repeated the tumor growth experiments using ANXA2 depleted HT1080 and control cells and administered the antioxidant agent, NAC intraperitoneally every two days. These data showed that the growth rate of the ANXA2 depleted HT1080 tumors in mice receiving NAC injections was only slightly lower than the HT1080 control tumors (Figure 8C). At the end point of the experiment the ANXA2 depleted HT1080 tumor volume was only 8% smaller than the HT1080 control tumors. To investigate the redox status of the cellular proteins in the ANXA2 depleted HT1080 or control tumors in the absence or presence of NAC injections, we removed HT1080 tumors from untreated or NAC treated mice and labeled the cellular extracts with BIAM. The tumors grown from ANXA2 depleted HT1080 cells showed a 45% decrease in protein labeling (increased oxidation) compared to tumors grown from control HT1080 cells (Figure 8D, E). However, in mice receiving NAC injections, the labeling of tumor extracts by BIAM suggested similar levels of protein oxidation between tumors grown from ANXA2 depleted or control HT1080 cells (Figure 8F, G). These results indicate that NAC prevented the intracellular protein oxidation observed in tumors grown from ANXA2 depleted HT1080 cells. We examined the redox status of protein extracts obtained from ANXA2 depleted HT1080 and control cells that were grown in culture for the time course of the animal experiments. Interestingly, we observed no difference in the levels of BIAM labeling between ANXA2 depleted HT1080 and control cells that were grown in tissue culture conditions, under no oxidative stress, for three weeks (Figure S5B). This suggested that the oxidative stress that is known to occur during tumor growth, had a more severe effect on the ANXA2 depleted HT1080 cells than the control HT1080 cells. Collectively, these results support a role for ANXA2 in cancer cell redox regulation during tumorigenesis.
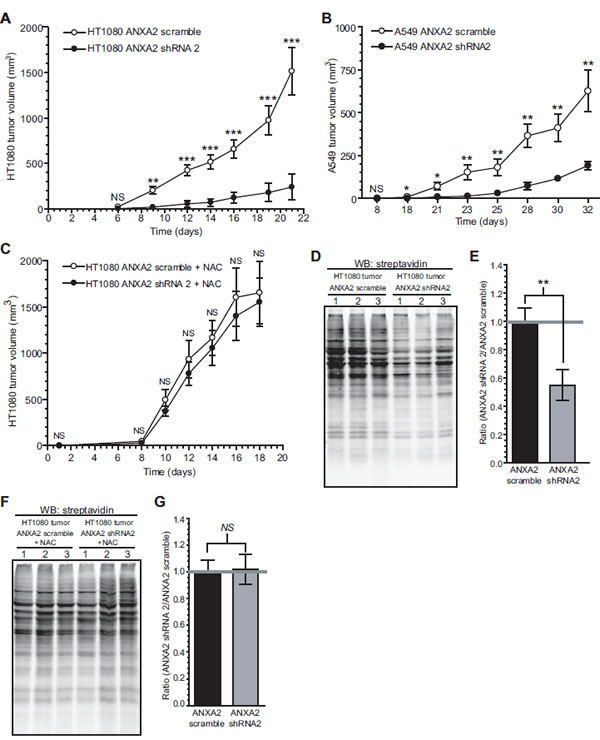
Figure 8: ANXA2 redox regulatory function plays a role in tumorigenesis. (A) 2 X 106 HT1080 ANXA2 shRNA2 or HT1080 ANXA2 scramble cells (N=10 per group) (B) 2 X 106 A549 ANXA2 shRNA2 or A549 ANXA2 scramble cells (N=8 per group) were injected subcutaneously into the right flank of NOD-SCID mice. (C) 2 X 106 HT1080 ANXA2 shRNA2 or HT1080 ANXA2 scramble cells were injected subcutaneously into the right flank of NOD-SCID mice treated with intraperitoneal injections of 150 mg/kg of NAC every two days (N=8 per group). (A-C) Tumor volume was measured for the time indicated using a caliper. Data was analyzed using the 2-way Analysis of Variance (ANOVA) test and represented as ± SEM. (D-G) Three HT1080 ANXA2 shRNA2 and three HT1080 ANXA2 scramble tumors from mice untreated (D) or treated (F) with NAC were homogenized, labeled with 20 μM BIAM, subjected to SDS-PAGE and analyzed by western blotting with a streptavidin probe. (E) and (G) Quantification of the results obtained in (D) and (F) using the Licor Odyssey software, respectively.
ANXA2 redox status in human tumors
We also investigated the redox status of ANXA2 in colon and gastric tumors compared to adjacent normal tissue. We examined six different human colon sample sets and one gastric set, including: normal tissue, outer tumor and core tumor. Cell extracts were labeled with BIAM and purified with streptavidin beads as described previously. We observed that the total levels of ANXA2 were unchanged in all colon samples and moderately elevated in the gastric tumor. However, the levels of the reduced form of ANXA2 were up-regulated in the gastric tumor and in four out of the six colon tumors compared to normal tissues (Figure 9A). Furthermore, in the two sets of colon samples where this did not occur, we observed significant oxidation of total proteins in the tumors (Figure 9 B- colon 1, 4). Colon 1 showed significantly elevated oxidation of total proteins in the outer and core tumors consistent with the high level of ANXA2 oxidation in these samples, whereas in colon 4 where the levels of reduced ANXA2 were similar in normal and tumor samples we only observed a significant oxidation of total proteins in the core/hypoxic tumor, which is under higher oxidative stress due to lack of blood supply.
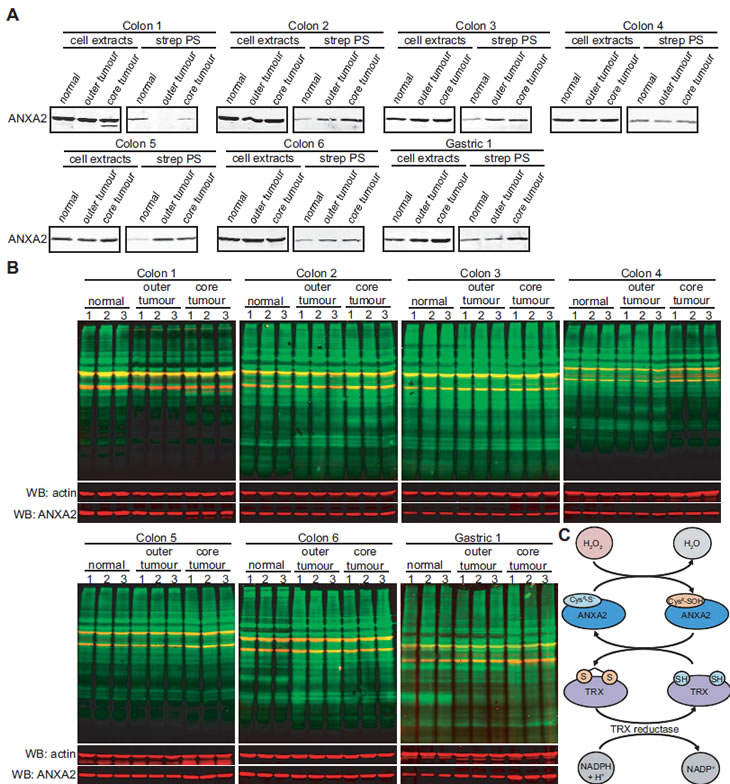
Figure 9: ANXA2 redox status in human normal and tumor tissues. (A,B) Six different sets of human colon tissue extracts and one set of gastric tissue extracts, including normal tissue, outer tumor and core tumor were labeled with 20 μM BIAM; (A) Labeled extracts were purified by incubation with streptavidin beads. Cell extracts and streptavidin purified samples (strep PS) were subjected to SDS-PAGE followed by western blot analysis with the antibodies indicated; (B) 20 µg of each labeled cell extract were subjected to SDS-PAGE, followed by western blotting with a streptavidin probe and the antibodies indicated. (C) H2O2 oxidizes Cys8 of ANXA2 resulting in the conversion of H2O2 to H2O. Oxidized ANXA2 is then reduced by the Trx redox system and can participate in further redox cycles. Thus a single molecule of ANXA2 can degrade several molecules of H2O2.
DISCUSSION
In the current study we identify ANXA2 as a novel cellular redox regulatory protein that plays a significant role in cells undergoing oxidative stress and in particular during tumorigenesis.
With this work we show for the first time that Cys8 residue of ANXA2 is a redox sensitive cysteine that is reversibly oxidized by H2O2 and reduced by the thioredoxin system. Our results allow us to propose the following mechanistic model: H2O2 reacts with the Cys8 of ANXA2 resulting in the oxidation of this residue and the conversion of H2O2 to H2O. Oxidized ANXA2 is then reduced by the Trx redox system and can participate in further redox cycles (Figure 9C). Thus a single molecule of ANXA2 can degrade several molecules of H2O2.
Considering the abundance and wide distribution of ANXA2 in cells and the fact that ANXA2 contains a highly reactive cysteine residue that is a direct target of H2O2, we hypothesized that ANXA2 might play an important role in the cellular response to oxidative stress, particularly H2O2-mediated stress. As mentioned before, H2O2 is a main second messenger in cell signaling transduction and is induced by a number of signaling pathways that regulate cell proliferation, survival and apoptosis. A tight control in the levels of cellular H2O2 is then very important for cell signaling regulation and in order to avoid protein, lipid and DNA oxidative damage. Here we show that ANXA2 depleted cells exhibit significantly enhanced levels of ROS and increased oxidation of redox sensitive proteins upon H2O2 treatment compared to control cells. Although we did not observe differences in the basal levels of ROS in untreated ANXA2 depleted versus control cells using the DCF-DA reagent, it is possible that the DCF-DA reagent is not sensitive enough to detect small differences in the basal concentrations of cellular H2O2 using a fluorometer plate reader. Alternatively, it is possible that ANXA2 only plays a significant redox regulatory role under oxidative stress conditions, while under physiological conditions other antioxidant proteins are able to compensate for the loss of ANXA2.
The enhanced ROS levels observed in the ANXA2 depleted cells upon oxidative stress resulted in increased sensitivity of these cells to ROS induced cell death compared to control cells. Chemotherapeutic agents that induce oxidative stress are thought to selectively kill cancer cells because the elevation of ROS caused by these agents is less toxic to normal cells that have lower levels of endogenous ROS. However, we surprisingly found that our non cancer cell model was more sensitive to oxidative stress compared to cancer cells. Nevertheless, these are different cell types and for this reason it is not appropriate to directly compare their sensitivity to ROS induced death. Furthermore, redox adaptation by cancer cells can in some cases make these cells more resistant to ROS induced death. For instance it has been shown that the MCF10 normal breast cells are more sensitive to H2O2 induced cell death compared to MCF7 breast cancer cells, because MCF7 cells up-regulate the peroxidases, peroxiredoxin I and II in order to counteract the elevation of ROS and prevent cellular damage/death [29]. Interestingly, we observed that ANXA2 protein levels increased by two-fold after treatment of 293T and MCF7 cells with 100 μM H2O2 for two weeks. This result suggests that cells might respond to elevations in ROS levels/ oxidative stress by inducing the expression of the redox regulatory protein, ANXA2.
The observed increased sensitivity of ANXA2 depleted cells to death induced by chemotherapeutics that up-regulate ROS led us to postulate that blocking the expression of ANXA2 in cancer cells might constitute a potential therapeutic strategy. Several reports have shown that up-regulation of ANXA2 in tumors correlates with chemoresistance, but they were unable to explain the mechanism(s) by which this occurs. Our results provide an explanation for this observation, since ANXA2 antioxidant function will protect the cancer cells from oxidative damage/death induced by the chemotherapeutics.
We also analyzed the activation of H2O2 responsive signaling pathways in the ANXA2 depleted versus control cells. The major intracellular sensor of oxidative stress induced by H2O2 is the apoptosis signal-regulated kinase-1 (ASK-1). ASK-1 directly activates MKK3/MKK6 and MKK4/MKK7 kinases which phosphorylate the pro-apoptotic kinases, p38 and JNK [30, 31, 32]. Therefore, the phosphorylation status of p38 and JNK provides a sensitive readout of the redox status of the cell. Elevated levels of H2O2 also lead to activation of the PI3K pathway kinase, Akt through inactivation of PTEN phosphatase [33]. Although Akt signaling is usually associated with cell survival, recently it has been shown that under oxidative stress conditions Akt further increases the intracellular levels of ROS through an increase in oxygen consumption and inhibition of FoxO transcription factors sensitizing cells to ROS-mediated apoptosis [26]. We observed enhanced activation of the pro-apoptotic kinases p38, JNK and Akt in ANXA2 depleted cancer cells treated with H2O2 compared to control cells. These results provide a molecular mechanism that explains the increased sensitivity of these cells to ROS induced death.
If ANXA2 functions as a redox regulatory protein then it is reasonable to expect that the ANXA2-null mice would exhibit a phenotype. However, the cellular redox system is complex with significant cross talk between various proteins and often several proteins compensate for each others function in a relatively efficient way under non pathological conditions. Considering the importance of cellular redox regulation it is not surprising that cells possess redox proteins with overlapping functions. For instance, homozygous catalase-null mice, which are completely deficient in catalase expression, the major cellular enzyme that decomposes H2O2, develop normally and show no gross abnormalities under physiological conditions. In fact, mice deficient in catalase were not more vulnerable to hyperoxia-induced lung injury; nor did their eye lenses showed any increased susceptibility to oxidative stress generated by a photochemical reaction,suggesting that the antioxidant function of catalase in these two models of oxidative injury is either negligible, which is unlikely, or is compensated by other cellular antioxidant protein(s) [34]. The only phenotype observed in these knockout mice was a retarded rate of breakdown of H2O2 by liver, lung and lens tissue slices compared to the wild-type mice.
Our animal studies showed that ANXA2-null mice have enhanced liver and lung protein oxidation compared to WT mice. Liver cells have a high metabolic rate since this organ is involved in protein, lipid and carbohydrate metabolism, synthesis of urea and the manufacture of bile, resulting in increased ROS production by the mitochondria. The lung is constantly exposed to oxidative stress due to gas exchange between the alveoli and the red blood cells. It is not surprising that depletion of the redox regulatory protein, ANXA2 in these organs will lead to increased oxidative stress. We did not observe significant differences in spleen protein oxidation between ANXA2-null and WT mice. This could be due to the fact that most splenocytes show a rapid turnover and are unlikely to accumulate as much ROS induced protein damage as the long-lived liver or lung cells.
During tumorigenesis, cancer cells typically exhibit increased ROS generation. In order to balance the proliferative advantages of ROS up-regulation versus its potential risks due to protein, lipid and DNA damage, cancer cells induce the over-expression and/or activation of antioxidant/redox regulatory proteins. Our in vivo tumor studies showed that tumor growth in animals injected with either ANXA2 depleted HT1080 or ANXA2 depleted A549 cancer cells was severely impaired compared to tumor growth in animals injected with control HT1080 and A549 cancer cells, respectively. In order to investigate if the growth impairment of ANXA2 depleted tumors was specifically due to ANXA2 redox regulatory function we treated mice possessing ANXA2 depleted HT1080 tumors, with the antioxidant NAC. We observed that in the presence of NAC tumor growth in animals injected with ANXA2-depleted cancer cells was identical to tumor growth in animals injected with control cancer cells. These results show that replacement of ANXA2 by another antioxidant reagent, such as NAC, reversed the growth impairment phenotype observed for the ANXA2 depleted tumors. It is then reasonable to propose that ANXA2 depletion from the cancer cells resulted in a loss of cellular redox regulatory capability which became critical as these cancer cells grew into tumors and were subjected to oxidative stress in the tumor site. The loss of ANXA2-dependent redox regulation during tumor growth was restored by the antioxidant activity of NAC. Consistent with this rationale was the observation that in the absence of NAC, the HT1080 ANXA2 depleted tumors showed significantly enhanced protein oxidation (sensor for cellular oxidative damage) compared to control tumors, while there was no enhanced protein oxidation in the ANXA2 depleted tumors grown in mice that received the antioxidant NAC. Taking these data together we therefore propose that ANXA2 plays a major role in tumorigenesis by functioning as a redox regulatory protein.
It was interesting to observe that NAC by itself stimulated tumor growth of the control cancer cells. This result is consistent with the reports that compared to normal cells, cancer cells are under significant oxidative stress and that agents that increase cellular ROS levels can selectively kill cancer cells [35, 36]. In this situation NAC can act by lowering the levels of H2O2 in the HT1080 tumors, thus increasing cell proliferation or decreasing cell death. This result also indicates that oxidative stress plays a major role and is a limiting step in tumor growth under our experimental conditions.
Although controversial, ANXA2 has also been implicated in other cellular processes, in particular plasmin activation, which could also contribute to cancer progression [37]. Several studies have shown that plasmin activation does not play a major role in the subcutaneous growth of primary tumors. It has been shown that sub-cutaneous inoculation of plasminogen negative tumour cells (LLC) in plasminogen-null mice produced tumors only 10-35% smaller compared to control mice [38], while we observed a 70-85% decrease in tumor volume in the A549 and HT1080 ANXA2 depleted tumors compared to control tumors. Another study showed that both WT and plasminogen-null mice carrying the Polyoma middle T antigen under the control of the mouse mammary tumor virus long terminal repeat uniformly developed multiple bilateral mammary tumors that were indistinguishable [39]. More importantly, plasmin generation by ANXA2 or other plasminogen receptors is not regulated by NAC, but is dependent on the presentation of a C-terminal lysine residue by the plasminogen receptor [40, 17, 37]. Consequently, if the growth deficit observed in the ANXA2 depleted tumors was due to decreased plasmin activation, the addition of NAC would not reverse the phenotype and restore the growth of ANXA2 depleted tumors as observed.
Finally, our ex-vivo experiments using human colon and gastric samples showed that five out of the seven sets of samples analyzed exhibited up-regulation of the reduced form of ANXA2 in the tumors compared to normal tissue. Whereas the two tumor samples where we did not observe up-regulation of reduced ANXA2 showed increased total protein oxidation. These results show that ANXA2 antioxidant function is important in maintaining redox equilibrium in the human tumors, since in general tumors show elevated levels of reduced ANXA2 and loss of reduced ANXA2 in tumors led to increased protein oxidation/oxidative stress.
In this study, we show that although ANXA2-null mice do not have an overt phenotype under non-pathological conditions, ANXA2 plays an important role during disease, namely supporting cancer progression by acting as a redox regulatory protein. Our observations that liver and lung tissue proteins from ANXA2-null mice are more oxidized compared to WT mice and that the proteins extracted from tumors produced by ANXA2-depleted cancer cells are more oxidized than proteins extracted from tumors produced by control cancer cells are also consistent with our proposed function for ANXA2 as an antioxidant.
Several reports have shown that up-regulation of ANXA2 levels is positively associated with cancer progression and chemoresistance, nevertheless the function of ANXA2 during these processes remained unknown. This is the first report that elucidates a molecular mechanism by which ANXA2 contributes to tumorigenesis and resistance to chemotherapy, by acting as a redox regulatory protein. Development of strategies that are aimed at preferentially killing cancer cells through mechanisms that cause additional ROS overload are currently being used. Agents that block ANXA2 expression could therefore be useful to preferentially kill malignant cells.
MATERIALS AND METHODS
Cell Culture, transfections and cell lines
MCF-7, A549, HT1080, LLC and 293T cell lines were obtained from ATCC and maintained in Dulbecco’s modified Eagle’s medium (Invitrogen) supplemented with 10% fetal bovine serum (FBS) and 100 U/ml of penicillin/streptomycin, in a humidified incubator in an atmosphere of 5% CO2 at 37ºC. TIME endothelial cells were obtained from Dr McMahon [41] and maintained in EGM2 medium (Lonza) supplemented with 10% FBS and 100U/ml of penicillin/streptomycin, in a humidified incubator in an atmosphere of 5% CO2 at 37ºC. ANXA2 depleted cell lines were obtained by transfection of Phoenix packaging cells with 4 µg of the pSUPER-retro plasmids described below using 12 µl of the lipofectamine 2000 transfection reagent according to the manufacturers’ instructions. 48 hours after transfection the target cells were infected with Phoenix supernatants and selected with 2 µg/ml of puromicin. 293T cells in 6 well plates (60-70% confluency) were transiently transfected for 48 hours with 1 μg of either pcDNA3 (empty vector control), pcDNA3-ANXA2, pcDNA3-ANXA2-Cys-8-Ala, pcDNA3-ANXA2-Cys-8-Ser, pcDNA3-ANXA2-Cys-132-Ala or pcDNA3-ANXA2-Cys-132-Ser plasmids described below using 3 µl of the lipofectamine 2000 transfection reagent according to the manufacturers’ instructions.
Plasmids
pSUPER-retro-ANXA2 shRNA1 was constructed by cloning the dsDNA oligo 5’-GAT CCC CCC TGG TTC AGT GCA TTC AGT TCA AGA GAC TGA ATG CAC TGA ACC AGG TTT TTA-3’ and 5’-AGC TTA AAA ACC TGG TTC AGT GCA TTC AGT CTC TTG AAC TGA ATG CAC TGA ACC AGG GGG-3’ into pSUPER.retro.puro (OligoEngine), pSUPER-retro-ANXA2 shRNA2 was constructed by cloning the dsDNA oligo 5’-GAT CCC CGT GCA TAT GGG TCT GTC AAT TCA AGA GAT TGA CAG ACC CAT ATG CAC TTT TTA-3’ and 5’-AGC TTA AAA AGT GCA TAT GGG TCT GTC AAT CTC TTG AAT TGA CAG ACC CAT ATG CAC GGG-3’ into pSUPER.retro.puro (OligoEngine) and the pSUPER-retro-ANXA2 scramble was constructed by cloning the dsDNA oligo 5’-GAT CCC CGT GCA TAT GGG TCT GTC CAT TAG AGA GAT TGA CAG ACC CAT ATG CAC TTT TTA-3’ and 5’-AGC TTA AAA AGT GCA TAT GGG TCT GTC AAT CTC TCT AAT GGA CAG ACC CAT ATG CAC GGG-3’ into pSUPER.retro.puro (OligoEngine). pSUPER-retro-ANXA2 shRNA3 (Murine) was constructed by cloning the dsDNA oligo into pSUPER.retro.puro (OligoEngine) and pSUPER-retro-ANXA2 shRNA2 (Human specific) was used as ANXA2 scramble (Murine). pcDNA3-ANXA2 plasmid was constructed by PCR amplification of ANXA2 cDNA and cloning it into the pcDNA3 vector. The primers used for PCR amplification were the following: 5’-AAG CTT GGA TCC GCC GCC ACC ATG TCT ACT-3’ and 5’-GAT CGC GGC CGC TCA GTC ATC TCC ACC AC-3’. pcDNA3-ANXA2-Cys-8-Ala was constructed by point mutation of the ANXA2 gene in the pcDNA3-ANXA2 vector using the QuikChange II Site-directed mutagenesis kit (Agilent Technologies). The primers used to introduce the point mutation in the ANXA2 gene were the following: 5’-CT GTT CAC GAA ATC CTG GCA AAG CTC AGC TTG GAG GG-3’ and 5’-CC CTC CAA GCT GAG CTT TGC CAG GAT TTC GTG AAC AG-3’. pcDNA3-ANXA2-Cys-8-Ser was constructed by point mutation of the ANXA2 gene in the pcDNA3-ANXA2 vector using the QuikChange II Site-directed mutagenesis kit (Agilent Technologies). The primers used to introduce the point mutation in the ANXA2 gene were the following: 5’-GTT CAC GAA ATC CTG AGC AAG CTC AGC TTG GAG GG-3’ and 5’-CC CTC CAA GCT GAG CTT GCT CAG GAT TTC GTG AAC-3’. pcDNA3-ANXA2-Cys-132-Ala was constructed by point mutation of the ANXA2 gene in the pcDNA3-ANXA2 vector using the QuikChange II Site-directed mutagenesis kit (Agilent Technologies). The primers used to introduce the point mutation in the ANXA2 gene were the following: 5’-CTC ATT GAG ATC ATC GCT TCC AGA ACC AAC CAG GAG CTG-3’ and 5’- CAG CTC CTG GTT GGT TCT GGA AGC GAT GAT CTC AAT GAG-3’. pcDNA3-ANXA2-Cys-132-Ser was constructed by point mutation of the ANXA2 gene in the pcDNA3-ANXA2 vector using the QuikChange II Site-directed mutagenesis kit (Agilent Technologies). The primers used to introduce the point mutation in the ANXA2 gene were the following: 5’-CTC ATT GAG ATC ATC AGC TCC AGA ACC AAC CAG GAG CTG-3’ and 5’- CAG CTC CTG GTT GGT TCT GGA GCT GAT GAT CTC AAT GAG-3’.
Antibodies
The following antibodies were used for western blot analysis: ANXA2 antibody: 610069 (BD Transduction laboratories), S100A10 antibody: 610071 (BD Transduction laboratories), ANXA1 antibody: sc-12740 (Santa Cruz Biotechnology - SCBT), ANXA4 antibody: sc-1930 (SCBT), ANXA5 antibody: sc-8300 (SCBT), ANXA6 antibody: sc-11388 (SCBT), SOD-1 antibody: sc-11407 (SCBT), SOD-2 antibody: sc-30080 (SCBT), PRDX I antibody: sc-7381, PRDX II antibody: sc-33572, PRDX I-IV antibody: sc-33574 (SCBT), catalase antibody: ab1877-10 (AbCam), GPX 1/2 antibody: sc-133160, Trx antibody: sc-20146 (SCBT), phospho-JNK antibody: 9251S (Cell Signaling), JNK antibody: 9252 (Cell Signaling), phospho-p38 antibody: 9211S (Cell Signaling), p38 antibody: 9212 (Cell Signaling), phospho-Akt antibody: 9271S (Cell Signaling), Akt antibody: 9272 (Cell Signaling), actin antibody (AC-40): A3853 (SIGMA), β-tubulin antibody (H-235): sc-9104 (SCBT), JunD antibody: sc-44 (SCBT).
Immunoprecipitation assays
For Trx, JunD and ANXA2 co-immunoprecipitations, cells were washed two times with PBS, lysed with Triton X-100 lysis buffer (50 mM Tris pH 7.4, 0.15% Triton X-100, 150 mM NaCl, 1.5 mM EGTA, protease inhibitors, 1 mM NaVO4, 10 mM NaF- degassed) for 10 minutes on ice. Cell lysates were pre-cleared for 1 h with protein G-Sepharose, incubated with specific antibodies for 1 h and then with 50% slurry of protein G-Sepharose for 1 h. Beads were washed five times with 500 μl of lysis buffer, resuspended with 25 μl 2X SDS-PAGE loading buffer, boiled for 5 minutes, subjected to SDS-PAGE and analysed by western blotting. The following antibodies were used for immunoprecipitation studies: ANXA2: D1/274.5 mouse monoclonal (made in house), JunD: sc-44 (SCBT) and Trx: sc-20146 (SCBT).
Western blot analysis
For western blot analysis 20 μg of cell lysates, unless noted, were subjected to SDS-PAGE, transferred onto a nitrocellulose membrane, incubated with appropriate antibodies and visualized using a Licor Odyssey scanner (Li-cor Biosciences). Quantification of protein bands was done using the Licor Odyssey scanner software.
Determination of redox sensitive cysteine oxidation
Redox sensitive cysteine oxidation was determined using the thiol-specific biotinylation reagent, BIAM (SIGMA). For experiments that utilized cultured cells, after mock, EGF or H2O2 treatment, cells were washed two times with PBS and scraped with BIAM lysis buffer (10 µM BIAM, 50 mM Tris pH 7.4, 0.2% Triton X-100, 200 mM NaCl, 2 mM EGTA, 1 mM EDTA, 5 μg/ml catalase, protease inhibitors, 1 mM NaVO4, 10 mM NaF-degassed). The resultant lysates were incubated for 20 minutes at 37oC, after which 2 mM iodoacetamide (IA) was added to block any further protein labeling. In other experiments, lysates were prepared by homogenization of tissues or tumor samples with lysis buffer (50 mM Tris pH 7.4, 0.2% Triton X-100, 200 mM NaCl, 2 mM EGTA, 1 mM EDTA, protease inhibitors, 1 mM NaVO4, 10 mM NaF-degassed) followed by incubation at 4ºC for 15 minutes and centrifugation at 20000 g for 20 minutes at 4ºC. The lysates were incubated with 20 µM BIAM for 20 minutes at 37oC followed by addition of 2 mM IA. In order to isolate BIAM-labeled proteins, cell extracts that had been labeled with BIAM were incubated with 50 μl of streptavidin Dynabeads (Invitrogen) for 1 hour at 4oC with rotation. Streptavidin beads were washed 3 times with 500 μl of lysis buffer, resuspended with 25 μl of 2X SDS-PAGE loading buffer and subjected to SDS-PAGE followed by western blotting. All human samples were obtained with patients’ informed consent and the work was approved by the Capital Health Research Ethics Board.
NADPH assay
250 μg of TIME cell lysates were either non-treated or treated with H2O2 for 15 minutes. Lysates were incubated with 100 μg/ml of catalase for 10 minutes at 37oC, after which 500 μM of NADPH was added to the samples and incubated for 30 minutes at 37oC. Samples were incubated with 10μM BIAM reagent and labeled proteins were purified by incubation with streptavidin beads. Cell extracts and BIAM labeled purified samples were subjected to SDS-PAGE and analyzed by western blotting.
Trx assay
2.5 μM of either ANXA2 or ANXA1 were either non treated or treated with 100 μM of H2O2 in the presence or absence of 20 μM Trx for 15 minutes at 37ºC. Samples were labeled with 20 μM BIAM for 20 minutes at 37oC, subjected to SDS-PAGE under non reducing conditions and analyzed by western blotting.
Intracellular ROS measurement
The fluorescent dye DCF-DA (Invitrogen) was used to measure the levels of ROS in cells. 2 X 104 cells/well were seeded in 96 wells plate and incubated overnight at 37oC. Cell medium was removed and replaced with 1% FBS medium containing 50 µM DCF-DA. Cells were incubated for 30 minutes at 37oC, washed with PBS and complete medium was added. Cells were treated with H2O2 ± NAC, incubated for 1 hour at 37oC and fluorescence was measured (Excitation: 488 nm, Emission: 535 nm) using a fluorometer plate reader.
Cell viability assay
Cell viability was determined by using the CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay (Promega) according to the manufacturer’s instructions.
Tumor development in NOD-SCID mice
NOD-SCID mice were injected subcutaneously into the right flank with 2 X 106 HT1080 ANXA2 shRNA 2, HT1080 ANXA2 scramble, A549 ANXA2 shRNA2 or A549 ANXA2 scramble cells in 100 µl of PBS in the absence or presence of 150 mg/kg of NAC administered intraperitoneally every two days. Tumor volume was measured every two/three days by caliper measurement and volume calculated by the formula (V=0.5 X L X W2). At the experimental endpoint mice were euthanized by CO2 inhalation, the ANXA2 depleted HT1080 and control tumors were removed and protein lysates were prepared. Tumors were homogenized with 1ml lysis buffer (50 mM Tris pH 7.4, 0.2 % Triton-X100, 200 mM NaCl, 1 mM EDTA, 2 mM EGTA, 1 mM NaVO4, 10 mM NaF and protease inhibitors-degassed). Homogenates were centrifuged at 20000 g for 20 minutes at 4ºC and supernatants were stored. All mouse experiments were performed in accordance with protocols approved by the University Committee on Laboratory Animals (UCLA) at Dalhousie University, Halifax, N.S., Canada.
Statistical analysis
The statistical significance of the difference in cytotoxicity, DCF quantification and protein oxidation was evaluated using two tailed Student’s t test. The statistical difference in tumor growth between the ANXA2 depleted HT1080 versus control cells with or without NAC and ANXA2 depleted A549 versus control cells was analyzed using the 2-way Analysis of Variance (ANOVA) test. These data analyses were performed using the Prism software (GraphPad, San Diego, CA). In every case a P value of less than 0.05 (*), less than 0.01(**) and 0.001 (***) was considered statistically significant.
Inventory of Supplemental data
Supplemental data includes supplemental methods, five figures and one table.
ACKNOWLEDGEMENTS
We are grateful to Dr Katherine Hajjar for providing us with ANXA2-null mice.
This work was supported by grants from the Canadian Cancer Society Research Institute (CCSRI) and the Canadian Institutes of Health Research (CIHR). Patricia A Madureira was supported by a trainee award from The Beatrice Hunter Cancer Research Institute with funds provided by The Terry Fox Foundation Strategic Health Research Training Program in Cancer Research at CIHR and by the CCSRI.
REFERENCES
1. Fridovich I. Fundamental aspects of reactive oxygen species, or what’s the matter with oxygen? Ann N Y Acad Sci. 1999; 893, 13-8.
2. Rhee SG. Cell signaling. H2O2, a necessary evil for cell signaling. Science. 2006; 312, 1882-1883.
3. Trachootham D, Lu W, Ogasawara MA, Nilsa RV, Huang P. Redox regulation of cell survival. Antioxid Redox Signal. 2008; 10, 1343-74.
4. Veal EA, Day AM, Morgan BA. Hydrogen peroxide sensing and signaling. Mol Cell. 2007; 26, 1-14.
5. Jackson AL, Loeb LA. The contribution of endogenous sources of DNA damage to the multiple mutations in cancer. Mutat. Res. 2001; 477, 7-21.
6. Rubin R, Farber JL. Mechanisms of the killing of cultured hepatocytes by hydrogen peroxide. Arch. Biochem. Biophys. 1984; 228, 450-459.
7. Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991; 51, 794-8.
8. Trachootham D, Zhou Y, Zhang H, Demizu Y, Chen Z, Pelicano H, Chiao PJ, Achanta G, Arlinghaus RB, Liu J, Huang P. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell. 2006; 10, 241-252.
9. Lukosz M, Jakob S, Büchner N, Zschauer TC, Altschmied J, Haendeler J. Nuclear redox signaling. Antioxid Redox Signal. 2010; 12(6):713-42.
10. Moss SE, Morgan RO. The annexins. Genome Biol. 2004; 5, 219.
11. Gerke V, Creutz C, Moss S. Annexins: linking Ca2+ signaling to membrane dynamics. Nat.Rev.Mol.Cell Biol. 2005; 6, 449-461.
12. Ross DT, Scherf U, Eisen MB, Perou CM, Rees C, Spellman P, Iyer V, Jeffrey SS, Van de Rijn M, Waltham M, Pergamenschikov A, Lee JC, Lashkari D, Shalon D, Myers TG, Weinstein JN, Botstein D, Brown PO. Systematic variation in gene expression patterns in human cancer cell lines. Nat. Genet. 2000; 24, 227-235.
13. Cicek M, Samant RS, Kinter M, Welch DR, Casey G. Identification of metastasis-associated proteins through protein analysis of metastatic MDA-MB-435 and metastasis-suppressed BRMS1 transfected-MDA-MB-435 cells. Clin. Exp. Metastasis. 2004; 21, 149-157.
14. Mai J, Waisman D, Sloane B. Cell surface complex of cathepsin B/annexin II tetramer in malignant progression. Biochim.Biophys.Acta. 2000; 1477, 215-230.
15. Takano S, Togawa A, Yoshitomi H, Shida T, Kimura F, Shimizu H, Yoshidome H, Ohtsuka M, Kato A, Tomonaga T, Nomura F, Miyazaki M. Annexin II overexpression predicts rapid recurrence after surgery in pancreatic cancer patients undergoing gemcitabine-adjuvant chemotherapy. Ann. Surg. Oncol. 2008; 15, 3157-3168.
16. Chuthapisith S, Bean BE, Cowley G, Eremin JM, Samphao S, Layfield R, Kerr ID, Wiseman J, El-Sheemy M, Sreenivasan T, Eremin O. Annexins in human breast cancer: Possible predictors of pathological response to neoadjuvant chemotherapy. Eur. J. Cancer. 2009; 45, 1274-1281.
17. Kwon M, Yoon C, Jeong W, Rhee S, Waisman D. Annexin A2-S100A10 heterotetramer, a novel substrate of thioredoxin. J.Biol.Chem. 2005; 280, 23584-23592.
18. Park HS, Lee SH, Park D, Lee JS, Ryu SH, Lee WJ, Rhee SG, Bae YS. Sequential activation of phosphatidylinositol 3-kinase, beta Pix, Rac1, and Nox1 in growth factor-induced production of H2O2. Mol. Cell. Biol. 2004; 24, 4384-4394.
19. Choi KS, Fitzpatrick SL, Filipenko NR, Fogg DK, Kassam G, Magliocco AM, Waisman DM. Regulation of plasmin-dependent fibrin clot lysis by annexin II heterotetramer. J.Biol.Chem. 2001; 276(27):25212-21.
20. Lee TH, Kim SU, Yu SL, Kim SH, Park DS, Moon HB, Dho SH, Kwon KS, Kwon HJ, Han YH, Jeong S, Kang SW, Shin HS, Lee KK, Rhee SG, Yu DY. Peroxiredoxin II is essential for sustaining life span of erythrocytes in mice. Blood. 2003; 101(12):5033-8.
21. Bedard K, Krause KH. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiological Reviews. 2007; 87(1):245 -313.
22. Formichi P, Radi E, Battisti C, Tarquini E, Leonini A, Di Stefano A, Federico A. Human fibroblasts undergo oxidative stress-induced apoptosis without internucleosomal DNA fragmentation. J. Cell. Physiol. 2006; 208, 289-297.
23. Saito Y, Nishio K, Ogawa Y, Kimata J, Kinumi T, Yoshida Y, Noguchi N, Niki E. Turning point in apoptosis/necrosis induced by hydrogen peroxide. Free Radic. Res. 2006; 40, 619-630.
24. Oh SY, Sohn YW, Park JW, Park HJ, Jeon HM, Kim TK, Lee JS, Jung JE, Jin X, Chung YG, Choi YK, You S, Lee JB, Kim H. Selective cell death of oncogenic Akt-transduced brain cancer cells by etoposide through reactive oxygen species mediated damage. Mol Cancer Ther. 2007; 6(8):2178-87.
25. Dolado I, Swat A, Ajenjo N, De Vita G, Cuadrado A, Nebreda AR. p38α MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell. 2007; 11, 191-205.
26. Nogueira V, Park Y, Chen CC, Xu PZ, Chen ML, Tonic I, Unterman T, Hay N. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell. 2008; 14, 458-70.
27. Hill BG, Reily C, Oh J-Y, Johnson MS, Landar A. Methods for the determination and quantification of the reactive thiol proteome. Free Radic Biol Med. 2009; 47(6):675-683.
28. Ling Q, Jacovina AT, Deora A, Febbraio M, Simantov R, Silverstein RL, Hempstead B, Mark WH, Hajjar KA. Annexin II regulates fibrin homeostasis and neoangiogenesis in vivo. J.Clin.Invest. 2004; 113(1):38-48.
29. Bae JY, Ahn SJ, Han W, Noh DY. Peroxiredoxin I and II inhibit H2O2-induced cell death in MCF-7 cell lines. J Cell Biochem. 2007; 101(4):1038-45.
30. Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T, Takagi M, Matsumoto K, Miyazono K, Gotoh Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997; 275, 90-94.
31. Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998; 17, 2596-2606.
32. Goldman EH, Chen L, and Fu H. Activation of apoptosis signal-regulating kinase 1 by reactive oxygen species through dephosphorylation at serine 967 and 14-3-3 dissociation. J. Biol. Chem. 2004; 279, 10442-10449.
33. Leslie NR, Bennett D, Lindsay YE, Stewart H, Gray A, Downes CP. Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J. 2003; 22, 5501-5510.
34. Ho YS, Xiong Y, Ma W, Spector A, Ho DS. Mice lacking catalase develop normally but show differential sensitivity to oxidant tissue injury. J Biol Chem. 2004;279(31):32804-12.
35. Alexandre J, Batteux F, Nicco C, Chéreau C, Laurent A, Guillevin L, Weill B, Goldwasser F. Accumulation of hydrogen peroxide is an early and crucial step for paclitaxel-induced cancer cell death both in vitro and in vivo. Int J Cancer. 2006; 119(1):41-8.
36. Raj L, Ide T, Gurkar AU, Foley M, Schenone M, Li X, Tolliday NJ, Golub TR, Carr SA, Shamji AF, Stern AM, Mandinova A, Schreiber SL, Lee SW. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature. 2011; 475(7355):231-4.
37. Madureira PA, Surette AP, Phipps KD, Taboski MA, Miller VA, Waisman DM. The role of the annexin A2 heterotetramer (AIIt) in vascular fibrinolysis. Blood. 2011; 118:4789-4797
38. Bugge TH, Kombrinck KW, Xiao Q, Holmbäck K, Daugherty CC, Witte DP, Degen JL. Growth and dissemination of Lewis lung carcinoma in plasminogen-deficient mice. Blood. 1997; 90(11):4522-31.
39. Bugge TH, Lund LR, Kombrinck KK, Nielsen BS, Holmbäck K, Drew AF, Flick MJ, Witte DP, Danø K, Degen JL. Reduced metastasis of Polyoma virus middle T antigen-induced mammary cancer in plasminogen-deficient mice. Oncogene. 1998; 16(24):3097-104.
40. Miles LA, Hawley SB, Baik N, Andronicos NM, Castellino FJ, Parmer RJ. Plasminogen receptors: the sine qua non of cell surface plasminogen activation. Front Biosci. 2005; 10:1754-62.
41. Venetsanakos E, Mirza A, Fanton C, Romanov SR, Tlsty T, McMahon M. Induction of tubulogenesis in telomerase-immortalized human microvascular endothelial cells by glioblastoma cells. Exp Cell Res. 2002; 273(1):21-33.