
Introduction
Folate is essential for DNA synthesis and cell growth in rapidly dividing cells. In cancer, folate uptake and metabolism are frequently up-regulated to meet the elevated need for nucleotides. Inhibition of folate metabolism is the basis for several widely used cancer drugs, including methotrexate [1-3], which exerts its toxicity by inhibiting dihydrofolate reductase (DHFR) thereby disrupting purine and thymidylate biosynthesis, inhibiting DNA replication and promoting cell death. This folate antagonist has been used as a chemotherapeutic agent to treat a variety of hematological malignancies and solid tumors including acute lymphoblastic leukemia, non-Hodgkin’s lymphoma, osteosarcoma, head and neck cancer, choriocarcinoma, small cell lung cancer, and breast cancer [4].
In the 1970s, clinical trials of methotrexate treatment in children with neuroblastoma showed high levels of toxicity and low response rates [5, 6]. No further clinical trials examining the efficacy of methotrexate treatment have been undertaken in neuroblastoma and methotrexate is currently not considered a treatment option for children with this disease. However, these early clinical trials included only small numbers of patients and were conducted in an era when the prognostic value of MYCN testing was yet to be established. Amplification of the MYCN oncogene is now known to occur in approximately 20% of primary neuroblastomas and is consistently associated with poorer clinical outcome [7, 8]. Similar to other members of the Myc family, N-Myc is an oncogenic transcription factor that dimerizes with partner proteins and binds at E-box sequences (6 nucleotide binding motif) within the promoter of target genes [9]. N-Myc expression has been shown to correlate with growth potential of neuroblastoma cells [10, 11], and may promote an aggressive tumor phenotype through regulation of genes particular to the cell cycle, DNA damage response, differentiation and apoptosis [12]. Relatively little is known about the relationship between folate metabolism and neuroblastoma biology, however folate-mediated one-carbon metabolism has previously been identified as correlating with neuroblastoma aggressiveness in bioinformatics analyses [13], suggesting that inhibition of folate metabolism may be beneficial in high-risk disease.
The SLC19A1 gene encodes the reduced folate carrier (RFC), a high capacity reduced folate transporter that is the most efficient of the cellular folate uptake mechanisms [3]. RFC is also the principle uptake mechanism for the antifolate methotrexate [3] and decreased RFC activity has been shown to be one of the most common mechanisms of methotrexate resistance in osteosarcoma [14-16]. RFC is ubiquitously expressed in rapidly dividing fetal and tumor cells, reflecting its essential role in supplying folate cofactors for DNA synthesis and cell proliferation. As a previous large-scale screen suggested that SLC19A1 can be regulated by Myc [17], and N-Myc and Myc are largely functionally equivalent [18], we hypothesized that SLC19A1 would also be regulated by N-Myc and would be expressed at elevated levels in MYCN-amplified tumors, allowing effective methotrexate uptake.
This study investigated the reliance of MYCN-amplified neuroblastoma cell lines on folates, their sensitivity to the antifolate methotrexate and the contribution of RFC to methotrexate uptake. The relationship between SLC19A1 and MYCN expression was examined in tumors from two independent patient cohorts, in a panel of 13 neuroblastoma cell lines and in cell lines with inducible and silenced MYCN expression. The regulation of SLC19A1 by N-Myc was examined using quantitative chromatin immunoprecipitation (ChIP) and luciferase reporter assays. The association between SLC19A1 gene expression levels and risk of relapse or death was also examined in patient samples.
RESULTS
MYCN-amplified neuroblastoma cells have higher folate requirements than non-amplified cells
To gain insight into the biological pathways upregulated in MYCN-amplified neuroblastoma, Gene Set Enrichment Analysis [19] was performed on pre-existing microarray data from a cohort of tumors from 650 neuroblastoma patients [20]. Using a false discovery rate (FDR) of 0.25 as a cut-off, 20 pathways were positively correlated with MYCN amplification (Supplementary Table S3). The most highly enriched gene sets were those for the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways “Ribosome” (ribosome components), “DNA replication” (DNA replication machinery), “Base excision repair” (base excision repair machinery), “One carbon pool by folate” (folate-mediated one-carbon metabolism) and “RNA polymerase” (RNA polymerase subunits), each of which had a FDR less than 0.05 and an Enrichment Score (ES) greater than 0.65. Folate-mediated one-carbon metabolism (Figure 1A) was of particular interest having previously been identified as correlating with neuroblastoma aggressiveness in bioinformatics analyses [13], and being targetable by existing agents such as methotrexate.
To test whether upregulation of the folate-mediated one-carbon metabolism pathway corresponds to an increased reliance on folate in MYCN-amplified cells, we assessed the colony forming ability of two MYCN-amplified cell lines (CHP-134 and Kelly) and two non-amplified lines (SH-EP and NB69) in folate-free medium supplemented with increasing concentrations of folinic acid. As expected, none of the cell lines formed colonies in the complete absence of folates (Figure 1B). In the presence of 0.5 nM folinic acid, the MYCN-amplified lines formed few colonies (1% of the 10 nM value for CHP-134 and 14% for Kelly; P < 0.001 for each in comparison to 5 nM folinic acid) while the non-amplified lines exhibited partial rescue of colony formation (34%, P < 0.001 for SH-EP and 52%, P < 0.001 for NB69). In the presence of 1 nM folinic acid the MYCN-amplified lines exhibited partial rescue (33% of the 10 nM value, P < 0.001 for CHP-134 and 70%, P = 0.256 for Kelly) while the non-amplified lines exhibited complete rescue (90% of the 10 nM value, P = 0.453 and P = 0.410 for SH-EP and NB69 respectively). MYCN-amplified neuroblastoma cells therefore appear to have a greater requirement for folates than their non-amplified counterparts.
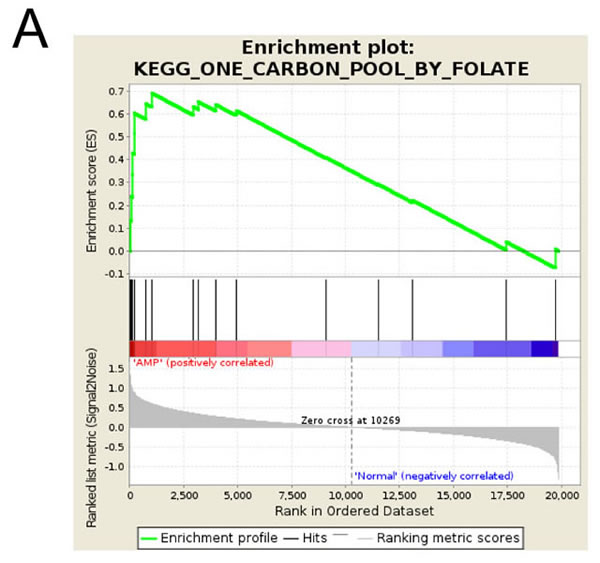
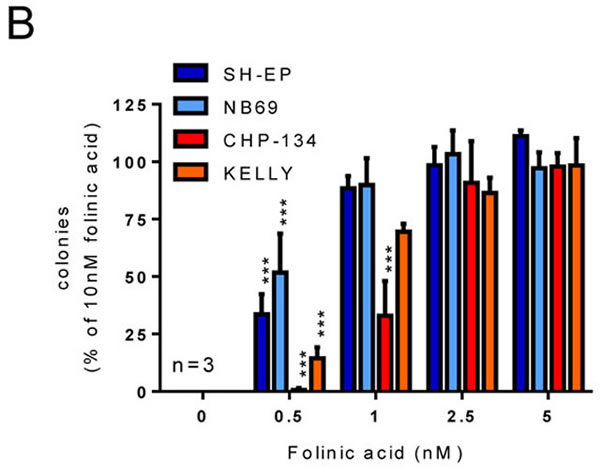
Figure 1: MYCN-amplified cell lines have a higher requirement for folate than those without amplification. (A) GSEA enrichment plot for the KEGG “One carbon by folate” pathway, showing correlation with MYCN amplification. Genes from this pathway are over-represented at the top of the whole gene list when ranked based on their relationship to MYCN amplification status. (B) Colony formation for MYCN single copy (SH-EP, NB69) and MYCN-amplified (CHP-134, Kelly) cell lines in a range of folinic acid concentrations. Colony numbers are normalised to 10 nM folinic acid at 100%, and comparisons made to the 5 nM folinic acid values. Data show means and SD derived from three independent experiments.
MYCN-amplified neuroblastoma cells have enhanced methotrexate sensitivity, with uptake mediated by RFC
Folate-mediated one-carbon metabolism is targetable by the antifolate methotrexate. We therefore asked whether MYCN-amplified cells are more sensitive to methotrexate than those without MYCN amplification by generating dose-response data for a panel of 13 neuroblastoma cell lines, 7 of which were MYCN-amplified and 6 single copy. The IC50 for methotrexate in MYCN-amplified cell lines ranged from 0.05 to 1.18 μM (Figure 2A, Supplementary Table S4). In contrast, the methotrexate IC50 was not reached for any of the 6 single copy cell lines (Figure 2B, Supplementary Table S4) despite testing up to 1 mM methotrexate. To exclude the possibility that this striking difference was simply a consequence of more rapid proliferation in MYCN-amplified cell lines, we determined the doubling times of each cell line. The mean doubling times of MYCN-amplified and non-amplified lines were not significantly different across the cell line panel (Figure 2C, P = 0.233, Supplementary Table S4).
Since methotrexate uptake is mediated principally by the reduced folate carrier (RFC; SLC19A1) [3], we investigated the relevance of RFC expression for methotrexate uptake in neuroblastoma. Following down-regulation of SLC19A1 in the MYCN-amplified cell line BE(2)-C using siRNA, SLC19A1 mRNA expression was reduced 7-fold (paired t-test; P < 0.001; Figure 2D). At protein level, RFC expression was substantially reduced (Figure 2E, full-length Western blots presented in Supplementary Figure S1). As a result of RFC depletion, the rate constant for [3H]methotrexate uptake was reduced from 0.016 min-1 to 0.008 min-1 (Figure 2F, P = 0.005) indicating that RFC is a major contributor to methotrexate uptake in these cells.
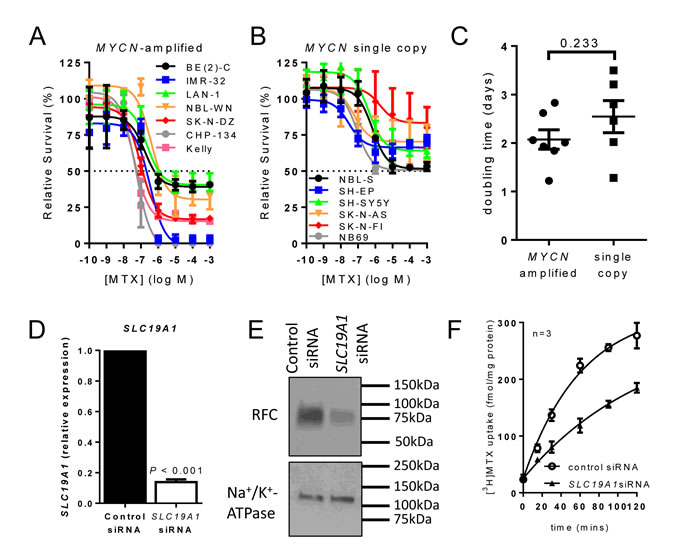
Figure 2: MYCN-amplified cell lines are more sensitive to methotrexate than non-amplified lines, with Reduced Folate Carrier (SLC19A1) a major contributor to methotrexate uptake. (A, B) Methotrexate dose-response curves at 72 h for a panel of 7 MYCN-amplified (A) and 6 MYCN single copy (B) cell lines showing the means and SD derived from three independent experiments. (C) Cell doubling times for MYCN-amplified and MYCN single copy cell lines do not differ significantly (P = 0.233, unpaired t-test). (D) Transfection of MYCN-amplified BE(2)-C cells with SLC19A1 siRNA reduced SLC19A1 expression 87% (7-fold) compared to control siRNA (paired t-test; P < 0.001). (E) Western blot of RFC protein levels in control and SLC19A1 siRNA treated BE(2)-C cells. Representative results from triplicate experiments are shown, and full-length Western blots are shown in Supplementary Figure S1. (F) Knockdown of SLC19A1 in BE(2)-C cells by siRNA halved the rate constant for [3H]methotrexate uptake from 0.016 min-1 to 0.008 min-1 (extra sum-of-square F test; P = 0.005). Data show means and SD derived from three independent experiments.
SLC19A1 and MYCN expression are closely correlated in neuroblastoma tumors and cell lines
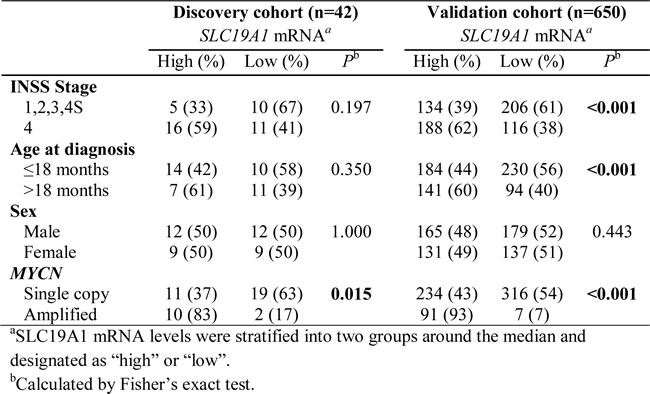
We subsequently investigated the expression of SLC19A1 in primary neuroblastoma tumors using a 42 sample discovery cohort and a 650 sample validation cohort. As seen in Table 1, high levels of SLC19A1 mRNA expression were significantly associated with MYCN amplification in both cohorts (Fisher’s exact, P = 0.015 and P < 0.001 for the 42 and 650 tumor cohorts, respectively). In contrast, SLC19A1 mRNA expression was not associated with sex, age or International Neuroblastoma Staging System (INSS) stage at diagnosis in the 42 tumor discovery cohort, but was associated with age and stage at diagnosis in the 650 sample validation cohort (P < 0.001). High SLC19A1 expression therefore appears to be a common feature of MYCN amplified tumors.
Table 1: SLC19A1 mRNA expression and clinical characteristics for the 42 sample discovery and 650 sample validation cohorts.
Next, we correlated expression of MYCN and SLC19A1 following determination of MYCN mRNA levels in 36 of the 42 discovery cohort samples (insufficient cDNA for 6 samples). MYCN and SLC19A1 expression showed a strong positive association (Figure 3A, Spearman’s ρ = 0.599, P < 0.001). Similarly, a strong positive association was observed in the 650 sample validation cohort (Figure 3B, Spearman’s ρ = 0.445, P < 0.001), and in the panel of 13 neuroblastoma cell lines (Figure 3C, Spearman’s ρ = 0.659, P = 0.017).
The relationship between MYCN and SLC19A1 expression was further investigated in MYCN-inducible Tet21N cells. Following removal of tetracycline, MYCN levels were induced 445-fold (Figure 3D, paired t-test, P < 0.001), accompanied by a 12-fold increase in SLC19A1 expression (paired t-test, P < 0.001). The increase in SLC19A1 was comparable to the 14-fold increase observed for ODC1, a well-established Myc family target gene [21] (paired t-test, P < 0.001). Conversely, down-regulation of MYCN in MYCN-amplified BE(2)-C cells using siRNA resulted in a 96.6% reduction in MYCN mRNA (Figure 3E, paired t-test, P < 0.001) accompanied by an 85.8% reduction in SLC19A1 expression (paired t-test, P = 0.002).
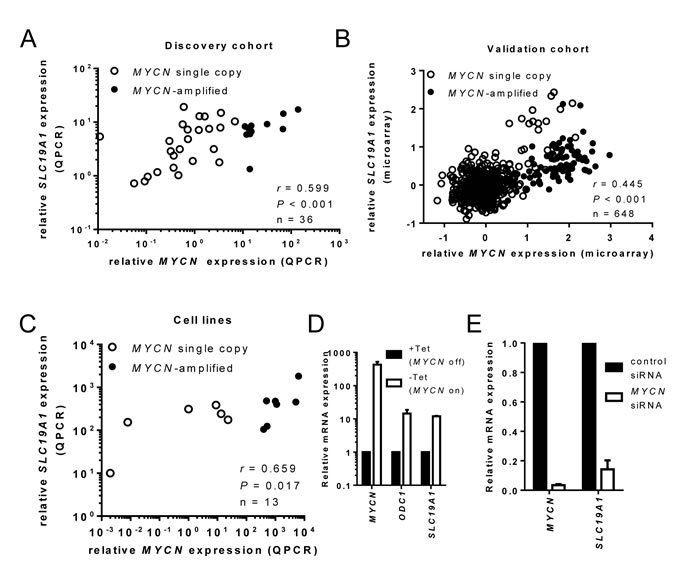
Figure 3: MYCN and SLC19A1 expression are positively correlated in neuroblastoma tumors and cell lines. MYCN and SLC19A1 mRNA expression were positively correlated in (A) a panel of 37 neuroblastoma patients, (B) an independent cohort of 650 neuroblastoma patients and (C) 13 neuroblastoma cell lines. Log-transformed expression levels relative to calibrator are shown in A and C; log-transformed, zero-centred expression levels obtained from microarray dataset are shown in B. (D) MYCN, SLC19A1 and ODC1 expression in SH-EP Tet21N cells with tetracycline treatment (MYCN OFF), or without tetracycline (MYCN ON). MYCN induction (445-fold; paired t-test; p < 0.001) increased SLC19A1 12-fold (paired t-test; P < 0.001) and ODC1 14-fold (paired t-test; P < 0.001). (E) MYCN and SLC19A1 expression in BE(2)-C cells transfected with control siRNA or MYCN siRNA. Silencing of MYCN (96.6% or 29-fold reduction in MYCN expression; paired t-test; P < 0.001) decreased SLC19A1 expression 85.5% or 7-fold (paired t-test; P = 0.002). Bars represent the mean and SD from 3 independent experiments.
N-Myc regulates SLC19A1 expression
To determine whether N-Myc and its binding partner Max associate with E-box sites in the promoter region of the SLC19A1 gene in neuroblastoma cell lines, we employed chromatin immunoprecipitation (ChIP) assays. In both BE(2)-C and SH-EP Tet21N cells, N-Myc and Max bind to a region adjacent to the putative transcriptional start site which contains four non-canonical E-box sequences (Figure 4A–4B, amplicon C). Very weak enrichment was observed at the other two E-box-containing amplicons (amplicons B and D) and no enrichment was observed at the control amplicon (amplicon A) which does not contain an E-box. The fold enrichment for SLC19A1 at amplicon C was comparable to that of the positive control APEX1 in both BE(2)-C and MYCN-induced SH-EP Tet21N cells and no signal was detected for the negative control ABCA10.
The SLC19A1 gene promoter was then cloned upstream of the luciferase reporter gene to determine whether N-Myc regulates SLC19A1 transcription. Repression of N-Myc expression in SH-EP Tet21N cells reduced luciferase activity of the SLC19A1 promoter/luciferase reporter construct with repression comparable to that observed for the established N-Myc target gene ABCC1 [22] (Figure 4C). Collectively, these results indicate that SLC19A1 is a direct transcriptional target of N-Myc in neuroblastoma cells.
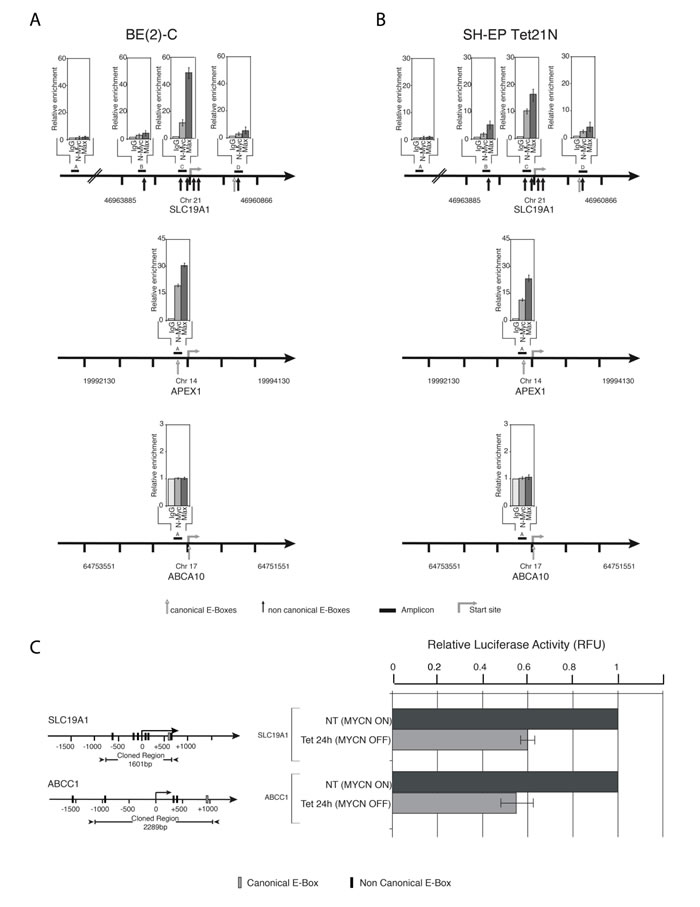
Figure 4: N-Myc is a direct transcriptional regulator of SLC19A1. Quantitative ChIP was applied to (A) BE(2)-C cells and (B) SH-EP Tet21N cells following MYCN induction. Fold enrichment is relative to the pre-immune serum and results represent the mean and SD from three independent ChIP experiments in which each region was measured for expression using QPCR in duplicate. Both positive (APEX1) and negative (ABCA10) controls are shown. (C) N-Myc directly activates SLC19A1 transcription. Luciferase activity was determined following transient transfection of SLC19A1 reporter construct into SH-EP Tet21N cells in the absence (MYCN ON) and presence (MYCN OFF) of tetracycline for 12 hours (unpaired t-test, P = 0.002). ABCC1 is included as a positive control.
High SLC19A1 expression is associated with an increased risk of relapse or death
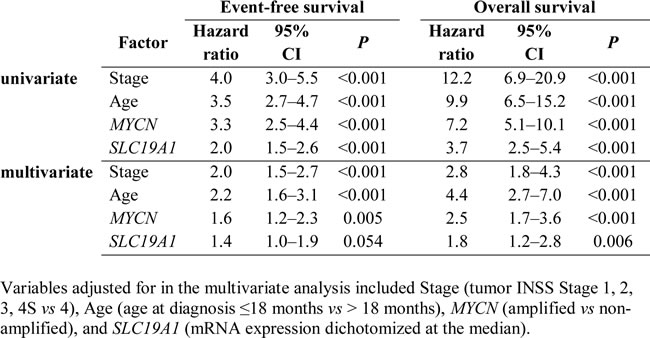
Finally, we investigated the association between SLC19A1 expression, established prognostic factors, and the risk of relapse and/or death in the cohort of 650 neuroblastoma patients. As expected, an increased risk of relapse and/or death was associated with MYCN amplification (P < 0.001), INSS Stage 4 (P < 0.001) and age >18 months at diagnosis (P < 0.001) (Table 2). When levels of SLC19A1 mRNA expression were dichotomized around the median, higher levels of SLC19A1 expression were strongly associated with reduced event-free survival (EFS) (HR = 1.98, 95%CI: 1.51–2.61; P < 0.001) with a 5-year survival rate of 72% ± 3% and 54% ± 3% in patients with low or high expression of SLC19A1, respectively. Similarly, high expression of SLC19A1 was associated with poorer overall survival (OS) (HR = 3.66; 95%CI: 2.50–5.35; P < 0.001) with a 5-year OS rate of 85% ± 3% and 58% ± 4% in patients with low or high SLC19A1 expression, respectively. Both EFS and OS were significantly lower in patients with higher SLC19A1 expression in the entire cohort (Figure 5A, P < 0.001 and Figure 5D, P < 0.001) and in the subset of patients without MYCN amplification (Figure 5B, P = 0.020 and Figure 5E, P < 0.001). SLC19A1 expression was not significantly associated with EFS or OS in patients with MYCN-amplified tumors (Figure 5C, P = 0.202 and Figure 5F, P = 0.324), most likely as the number of patients in the “low” category was very small. In multivariate analysis conducted on the entire cohort, high expression of SLC19A1 was found to be independently associated with reduced EFS or OS after adjusting for MYCN amplification status, INSS Stage and age at diagnosis (EFS: HR = 1.36; 95%CI: 0.99–1.85; P = 0.054; OS: HR = 1.84; 95%CI: 1.19–2.83; P = 0.006; Table 2). High SLC19A1 expression therefore appears to be a common feature of poor-outcome neuroblastoma, particularly in tumors with MYCN amplification.
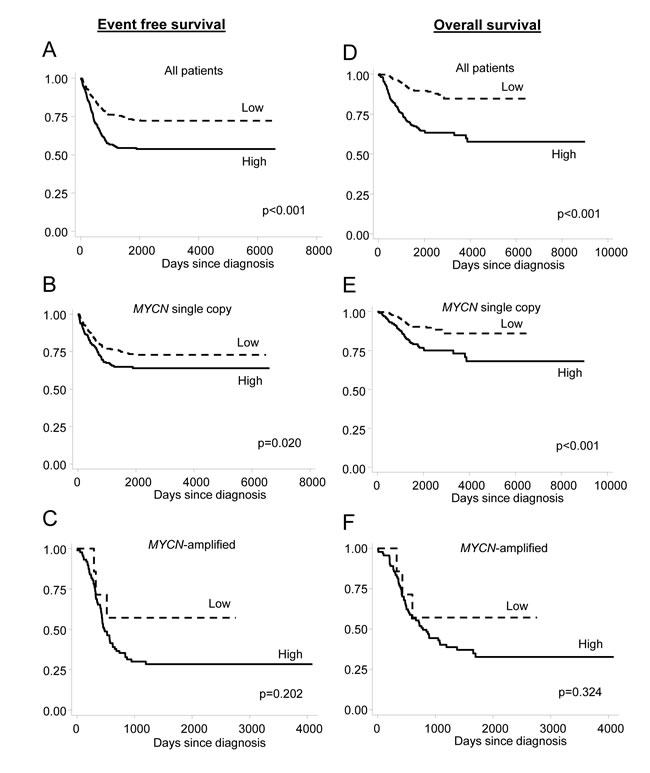
Figure 5: High SLC19A1 expression is associated with an increased risk of relapse or death in neuroblastoma. Kaplan-Meier curves showing the probability of EFS and OS for the entire 650 patient cohort (A, D), patients with tumors lacking MYCN amplification (B, E), and patients with MYCN amplification (C, F). Tumors were dichotomized into “high” and “low” SLC19A1 expression groups around the median. Survival curves were analyzed by log-rank test.
Table 2: Univariate and multivariate analysis of prognostic factors and SLC19A1 gene expression for event-free survival and overall survival (validation cohort).
DISCUSSION
Altered metabolism is a defining feature of tumor cells [23, 24]. Activation of the Myc oncogene in particular reprograms metabolic pathways, providing increased biosynthetic and bioenergetic capacity [25] through upregulation of protein synthesis [26], glycolysis [27], glutaminolysis [28], nucleotide synthesis [29] and polyamine synthesis [21], all of which contribute to rapid proliferation. Genes of the folate-mediated one-carbon metabolic pathway have previously been shown to be upregulated with c-Myc expression [30] and to be correlated with neuroblastoma aggressiveness in bioinformatics analyses [13]. Furthermore, tumors with upregulated folate metabolism have been proposed to be sensitive to folate antagonists [13, 31]. The current study extends these observations to demonstrate experimentally that MYCN-amplified neuroblastoma cell lines have increased folate requirements and are more sensitive to methotrexate treatment than their non-amplified counterparts. Interestingly, enhanced folate requirements and methotrexate sensitivity were not simply a consequence of more rapid proliferation in MYCN-amplified cell lines, as the proliferation rates of MYCN-amplified and non-amplified neuroblastoma lines were not significantly different across the cell line panel despite the colony assays being conducted over a timeframe well in excess of cell doubling times.
While there is a clear increase in methotrexate sensitivity with MYCN amplification in cell lines, a key question is whether metabolic changes associated with MYCN amplification can be targeted clinically. Despite the widespread use of methotrexate as a chemotherapeutic agent to treat a variety of haematological malignancies and solid tumors [4], methotrexate is not routinely used to treat neuroblastoma, largely due to high rates of toxicity and low response rates observed in clinical trials conducted in the 1970s. Of these studies, one reported toxicity in three neuroblastoma patients treated with methotrexate and observed no apparent therapeutic responses [6]. The second study investigated the use of methotrexate in 16 patients with previously treated metastatic neuroblastoma and reported no objective responses, despite a majority of patients achieving plasma methotrexate concentrations exceeding 5 µM [5]. No subsequent clinical trials have examined the efficacy of methotrexate in children diagnosed with neuroblastoma. While these studies were performed prior to the prognostic use of MYCN amplification and therefore did not consider MYCN amplification status, MYCN amplification is now known to occur in approximately 20% of primary neuroblastomas [7, 8], and is likely to be over-represented in a trial enrolling patients with metastatic disease [32]. Knowledge of MYCN amplification status alone may not be sufficient to determine those patients most likely to respond to antifolate therapy, and assessment of any potential clinical application of these findings will also have to consider the limitations of methotrexate toxicity and methotrexate availability in the tumor.
To this end, quantification of SLC19A1 expression may be of additional benefit. RFC, encoded by SLC19A1, is the principle cellular uptake mechanism for methotrexate [3] and SLC19A1 mutations or altered RFC expression impact on methotrexate uptake and sensitivity [14-16, 33]. We found that down-regulation of SLC19A1 substantially decreases the rate of methotrexate uptake in neuroblastoma cells, indicating that RFC is also a major methotrexate uptake mechanism in neuroblastoma. Furthermore, we found that SLC19A1 expression is upregulated by N-Myc, and that high SLC19A1 expression is correlated with neuroblastoma outcome, raising the possibility that a subset of high-risk patients with MYCN amplification and high SLC19A1 expression might be appropriate candidates for methotrexate therapy.
These findings may also extend beyond MYCN-amplified neuroblastoma, since MYC family genes, including MYCN, MYC and MYCL1 appear to be dysregulated in some high-risk neuroblastomas that lack MYCN amplification. Recent studies correlating microarray signals from MYC target genes with the clinical outcome of neuroblastoma found that MYC transcriptional signatures are predictive of disease outcome independent of MYCN amplification status [34, 35]. As SLC19A1 has previously been identified as a Myc target gene [17], there may also be a subset of non-amplified tumours with highly efficient methotrexate uptake.
The findings presented in this study, although preliminary, provide new evidence to suggest that MYCN amplification leads to antifolate sensitivity, and that methotrexate may be beneficial in a subset of patients with MYCN-amplified neuroblastoma and high SLC19A1 expression. These observations warrant further investigation in pre-clinical animal models and may also have relevance for other tumor types where MYC or MYCN amplification is observed, such as breast cancer, small-cell lung cancer, retinoblastoma, medulloblastoma, rhabdomyosarcoma, and astrocytoma.
MaterialS and Methods
Patient samples
A discovery set of 42 primary neuroblastoma samples was provided by the Tumour Bank at Children’s Cancer Institute Australia and has been described elsewhere [36]. Clinical data, including age, sex, INSS stage, dates of diagnosis, relapse and death from disease and MYCN status were obtained from medical records. All children were treated using standard protocols according to their tumor stage as previously described [36]. Gene expression and clinical data were also obtained for an independent validation cohort of 650 neuroblastoma patients as previously published [20]. A summary of clinical characteristics for each cohort is presented in Supplementary Table S1.
Gene Set Enrichment Analysis
Gene Set Enrichment Analysis was performed on the 650 neuroblastoma patient dataset [20] with the GSEA application (Broad Institute at MIT, Cambridge, MA) [19] using the KEGG pathway gene sets (186 gene sets). Gene sets showing a false discovery rate of 0.25 or less were considered to be significantly enriched between classes.
Cell lines and culture conditions
BE(2)-C, LAN-1, SH-EP, and SH-SY5Y were provided by Dr June Biedler, Memorial Sloan-Kettering Cancer Centre, New York, NY and NBL-S and NBL-WN by Dr Susan Cohn, University of Chicago, IL. IMR-32 was purchased from American Type Culture Collection (Manassas, VA) and SK-N-AS, SK-N-DZ, SK-N-FI, CHP-134, Kelly, NB69 from European Collection of Cell Cultures. Cell lines were cultured in Roswell Park Memorial Institute (RPMI) 1640 with 10% FCS (Life Technologies, Carlsbad, CA) and 2mM L-glutamine (CHP-134, Kelly, NB69), Dulbecco’s modified Eagle’s medium (DMEM) with either 20% FCS (NBL-WN), 15% FCS (NBL-S) or 10% FCS (remaining cell lines). The identity of each cell line was verified by short tandem repeat genetic profiling (CellBank Australia, Sydney, Australia).
Colony formation assays
Cells were depleted of endogenous folate cofactors by culturing for 10 days in folate-free depletion medium consisting of folic acid free RPMI (Life Technologies) with 10% dialysed FCS (Life Technologies) and 2mM L-glutamine, supplemented with 10 µM thymidine (Sigma-Aldrich, St Louis, MO), and 100 µM adenosine (Sigma-Aldrich) as previously described [37]. For experiments, cells were trypsinized then washed in folate-free assay medium consisting of folic acid free RPMI with 10% dialysed FCS and 2mM L-glutamine. Cells were diluted in folate-free assay medium and plated in 6-well plates at the following cell numbers: CHP-134: 600 cells/well; Kelly: 400 cells/well; NB69: 400 cells/well; SHEP: 150 cells/well supplemented with folinic acid (Sigma-Aldrich) at 0, 0.5nM, 1nM, 2.5nM, 5nM and 10nM. After 9 days (CHP-134, NB69 and SH-EP) or 12 days (Kelly), colonies were stained with 0.5% crystal violet in 50% methanol. Plates were scanned, and colonies counted. Colony numbers are normalised to 10 nM folinic acid at 100%, and comparisons made to the 5 nM folinic acid values. Results are from triplicate experiments.
Methotrexate sensitivity assays
For cell viability assays, cells were plated in 96-well plates and treated the following day with methotrexate. Cell growth was determined after 72 h of continuous exposure to 0.1nM–1mM methotrexate using a resazurin-based assay (Sigma-Aldrich), with fluorescence measured using a Benchmark Plus plate reader (Bio-Rad). Cell doubling times were determined by plating cells in 96-well plates and measuring fluorescence as above every 24 hours for 4 days.
Gene induction and suppression experiments
A SH-EP neuroblastoma cell line harboring an inducible MYCN construct (Tet21N) was provided by Dr Manfred Schwab, German Cancer Research Center, Heidelberg, Germany [10] and cultured as described previously [38]. MYCN expression was suppressed using 2ug/mL tetracycline, with MYCN induction following tetracycline removal. MYCN-induced and control cells were harvested 72 h after tetracycline removal. Silencing of MYCN and SLC19A1 was achieved using Dharmacon ON-TARGET plus SMARTpool siRNAs (Thermo Fisher Scientific, Lafayette, CO) at 10nM with a non-targeting siRNA pool as a negative control using Lipofectamine transfection reagent (Invitrogen, CA). Cells were harvested at 72 hours post-transfection to assess gene expression.
Protein expression analysis
For analysis of RFC protein expression, whole membrane extracts were prepared using a Mem-PER Eukaryotic Membrane Protein Extraction Reagent Kit (Thermo Fisher Scientific, Lafayette, CO). Protein was quantitated by BCA assay (Pierce, Rockford, IL) and 10 μg of each sample was electrophoresed on a 4–15% SDS-PAGE gel and transferred to a nitrocellulose membrane (Biorad, Gladesville, NSW, Australia). Membranes were blocked overnight with 5% skim milk powder in Tris-buffered saline. Membranes were incubated with rabbit polyclonal anti-RFC [39] or rabbit polyclonal Na+/K+-ATPase (H-300, sc-28800; Santa Cruz Biotechnology, Dallas, TX; 1:1000), followed by HRP-conjugated secondary antibodies. Membranes were developed using Supersignal reagent (Progen Biosciences, Brisbane, QLD, Australia), and protein expression was visualised on film.
Methotrexate uptake assay
BE(2)-C cells were transfected with control or SLC19A1 siRNA as described above. At 24 h post-transfection, cells were replated at 3.5x105 cells/well into a 6-well plate for the [3H]methotrexate uptake assay, with the remainder cultured to confirm RFC protein knockdown (both at 72 h post-transfection). For the [3H]methotrexate uptake assay, the cell monolayer was washed once with serum-free DMEM before addition of 0.8 µCi [3', 5', 7'-3H]methotrexate sodium salt (ARC, Bioscientific; specific activity = 40Ci/mmol; final methotrexate concentration = 20nM) to each well in 1mL of serum-free DMEM. Following incubation at 37°C for the specified time, medium was removed and cell monolayers were washed 3 times with ice-cold PBS. 1mL of 0.5M NaOH was added to each well, and cells were lysed by incubation at 70°C for 1 h. 1mL of 0.5M HCl was added to each well and 60uL was removed from each well and kept aside for BCA assay with the remainder transferred to scintillation vials containing 4mL of Ultima Gold™ scintillant. DPM was counted and [3H]methotrexate uptake calculated as fmoles of [3H]methotrexate/mg protein.
RNA isolation and mRNA expression analysis
Total RNA was extracted from primary neuroblastoma tissues and cell lines using guanidinium thiocyanate (GTC) as previously described [40]. Reverse transcription was performed with MMLV reverse transcriptase according to manufacturer’s protocol (Invitrogen). Quantitative PCR (QPCR) was performed using TaqMan gene expression assay (Applied Biosystems, CA). For the discovery set, expression levels of MYCN (primers and probe listed in Supplementary Table S2) and SLC19A1 (inventoried TaqMan assay Hs00953342_m1) were quantified using the comparative threshold cycle (Ct) method and normalized to TaqMan endogenous control assay for β2-microglobulin (B2M; B2M Endogenous control, VIC/MGB probe, primer limited), and expressed relative to a calibrator [41]. Gene expression data from the 650 patient validation cohort was previously described [20] and was derived from single-color gene expression profiles using 44K oligonucleotide microarrays as described previously [42]. Data were log2 transformed and zero centred.
ChIP and luciferase reporter gene assays
Putative N-Myc binding sites, including the canonical E-box sequence CACGTG and other non-canonical sequences (CATGTG and CACGCG) within the promoter region of SLC19A1 were identified using bioinformatics techniques as previously described [43]. Binding sites located within 2000 base pairs either side of the transcription start site of the SLC19A1 gene (as annotated in publically available databases) were chosen for further analysis, as well as a site outside of this region. To assess binding of N-Myc to SLC19A1, quantitative ChIP was performed from SH-EP Tet21N cells cultured with or without tetracycline, as well as from BE(2)-C cells, as previously described [43, 44]. APEX-1 [17] and ABCA10 [22] served as positive and negative controls. Specific primers used for ChIP are listed in Supplementary Table S2. A fragment of DNA containing each of the putative binding sites within 2000 base pairs either side of the SLC19A1 transcription start site was cloned upstream of a luciferase reporter gene. Transcription activity was tested in SH-EP Tet21N cells grown with or without tetracycline, with ABCC1 used as a positive control [22].
Statistical analyses
Statistical analyses utilising patient data were performed using STATA version 10 (StataCorp, College Station, TX). Expression levels of SLC19A1 in tumor samples were stratified into two groups around the median and designated as “high” or “low”. The frequencies of INSS stage, age at diagnosis, sex and MYCN status in these two groups were examined using Fisher’s exact test. Correlation between MYCN and SLC19A1 mRNA levels in tumor samples and cell lines was evaluated using Spearman’s rank correlation coefficient. EFS was defined as the time to relapse or death within 5 years from initial diagnosis, while OS was defined as the time to death within 5 years from initial diagnosis. The cumulative EFS and OS were computed by the Kaplan-Meier method and compared between subgroups using the log-rank test. A Cox proportional hazards model was used to determine whether the expression of SLC19A1 as well as established prognostic factors (MYCN amplification, neuroblastoma INSS stage, and age at diagnosis using an age cut-off of 18 months) were predictive of EFS or OS. Probabilities of survival and hazard ratios are presented with 95% confidence intervals (CIs).
All other statistical analyses were conducted using Prism version 6 (GraphPad Software, La Jolla, CA). Cell doubling times were calculated using an exponential growth equation. [3H]methotrexate uptake was analysed using a one-phase association equation with P values for determining significant differences in the rate constant (K) determined using the extra sum-of-squares F test.
Gene expression in MYCN-induced or silenced cells was compared using paired t-tests with two-sided P values. Dose-response curves were fitted with a three parameter logistic model by non-linear regression in order to calculate methotrexate IC50 and EC50 (half-maximal inhibition) values. P values for determining significant differences in IC50 values were determined using the extra sum-of-squares F test. Colony assay P values were determined using one-way ANOVA, with individual comparisons assessed using Dunnett’s multiple comparison test.
Acknowledgements
This work was supported by funds from the National Health and Medical Research Council [Program Grant APP1016699 to MDN, GMM and MH and Public Health Postgraduate Research Scholarship to DTL]; Cancer Institute New South Wales [Translational Program Grant 10/TPG/1-03 to MDN, GMM and MH and Career Development Fellowship 09CDF217 to JIF]. The Children’s Cancer Institute Australia is affiliated with University of New South Wales (UNSW) and Sydney Children’s Hospital Network, Randwick, Australia.
ConflictS of Interest
There is no conflict of interest that I should disclose.
References
1. Assaraf YG. Molecular basis of antifolate resistance. Cancer Metastasis Rev. 2007; 26:153–181.
2. Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat Rev Cancer. 2013; 13:572–583.
3. Matherly LH, Hou Z and Deng Y. Human reduced folate carrier: translation of basic biology to cancer etiology and therapy. Cancer Metastasis Rev. 2007; 26:111–128.
4. Bertino JR. Karnofsky memorial lecture. Ode to methotrexate. J Clin Oncol. 1993; 11:5–14.
5. Ablin AR, Bleyer WA, Finklestein JZ, Hartmann JR, Leikin S and Hammond GD. Failure of moderate-dose prolonged-infusion methotrexate and citrovorum factor rescue in patients with previously treated metastatic neuroblastoma--a phase II study. Cancer treatment reports. 1978; 62:1097–1099.
6. Fujimoto T, Sasaki K and Goya N. High-dose methotrexat therapy of childhood cancer--observations relating to clinical toxicity. Nihon Gan Chiryo Gakkai shi. 1978; 13:118–125.
7. Brodeur GM, Seeger RC, Schwab M, Varmus HE and Bishop JM. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science. 1984; 224:1121–1124.
8. Seeger RC, Brodeur GM, Sather H, Dalton A, Siegel SE, Wong KY and Hammond D. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N Engl J Med. 1985; 313:1111–1116.
9. Blackwell TK, Kretzner L, Blackwood EM, Eisenman RN and Weintraub H. Sequence-specific DNA binding by the c-Myc protein. Science. 1990; 250:1149–1151.
10. Lutz W, Stohr M, Schurmann J, Wenzel A, Lohr A and Schwab M. Conditional expression of N-myc in human neuroblastoma cells increases expression of alpha-prothymosin and ornithine decarboxylase and accelerates progression into S-phase early after mitogenic stimulation of quiescent cells. Oncogene. 1996; 13:803–812.
11. Schmidt ML, Salwen HR, Manohar CF, Ikegaki N and Cohn SL. The biological effects of antisense N-myc expression in human neuroblastoma. Cell Growth and Differentiation. 1994; 5:171–178.
12. Bell E, Chen L, Liu T, Marshall GM, Lunec J and Tweddle DA. MYCN oncoprotein targets and their therapeutic potential. Cancer Lett. 2010; 293:144–157.
13. Schramm G, Wiesberg S, Diessl N, Kranz AL, Sagulenko V, Oswald M, Reinelt G, Westermann F, Eils R and Konig R. PathWave: discovering patterns of differentially regulated enzymes in metabolic pathways. Bioinformatics (Oxford, England). 2010; 26:1225–1231.
14. Guo W, Healey JH, Meyers PA, Ladanyi M, Huvos AG, Bertino JR and Gorlick R. Mechanisms of methotrexate resistance in osteosarcoma. Clin Cancer Res. 1999; 5:621–627.
15. Serra M, Reverter-Branchat G, Maurici D, Benini S, Shen JN, Chano T, Hattinger CM, Manara MC, Pasello M, Scotlandi K and Picci P. Analysis of dihydrofolate reductase and reduced folate carrier gene status in relation to methotrexate resistance in osteosarcoma cells. Ann Oncol. 2004; 15:151–160.
16. Sowers R, Wenzel BD, Richardson C, Meyers PA, Healey JH, Levy AS and Gorlick R. Impairment of methotrexate transport is common in osteosarcoma tumor samples. Sarcoma. 2011; 2011:834170.
17. Fernandez PC, Frank SR, Wang L, Schroeder M, Liu S, Greene J, Cocito A and Amati B. Genomic targets of the human c-Myc protein. Genes Dev. 2003; 17:1115–1129.
18. Malynn BA, de Alboran IM, O’Hagan RC, Bronson R, Davidson L, DePinho RA and Alt FW. N-myc can functionally replace c-myc in murine development, cellular growth, and differentiation. Genes Dev. 2000; 14:1390–1399.
19. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES and Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005; 102:15545–15550.
20. Kocak H, Ackermann S, Hero B, Kahlert Y, Oberthuer A, Juraeva D, Roels F, Theissen J, Westermann F, Deubzer H, Ehemann V, Brors B, Odenthal M, Berthold F and Fischer M. Hox-C9 activates the intrinsic pathway of apoptosis and is associated with spontaneous regression in neuroblastoma. Cell death & disease. 2013; 4:e586.
21. Bello-Fernandez C, Packham G and Cleveland JL. The ornithine decarboxylase gene is a transcriptional target of c-Myc. Proc Natl Acad Sci U S A. 1993; 90:7804–7808.
22. Porro A, Haber M, Diolaiti D, Iraci N, Henderson M, Gherardi S, Valli E, Munoz MA, Xue C, Flemming C, Schwab M, Wong JH, Marshall GM, Della Valle G, Norris MD and Perini G. Direct and coordinate regulation of ATP-binding cassette transporter genes by Myc factors generates specific transcription signatures that significantly affect the chemoresistance phenotype of cancer cells. J Biol Chem. 2010; 285:19532–19543.
23. DeBerardinis RJ, Lum JJ, Hatzivassiliou G and Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell metabolism. 2008; 7:11–20.
24. Hanahan D and Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–674.
25. Dang CV. Enigmatic MYC Conducts an Unfolding Systems Biology Symphony. Genes & cancer. 2010; 1:526–531.
26. Gordan JD, Thompson CB and Simon MC. HIF and c-Myc: sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell. 2007; 12:108–113.
27. Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, Xu Y, Wonsey D, Lee LA and Dang CV. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem. 2000; 275:21797–21800.
28. Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB and Thompson CB. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A. 2008; 105:18782–18787.
29. Liu YC, Li F, Handler J, Huang CR, Xiang Y, Neretti N, Sedivy JM, Zeller KI and Dang CV. Global regulation of nucleotide biosynthetic genes by c-Myc. PLoS One. 2008; 3:e2722.
30. Morrish F, Neretti N, Sedivy JM and Hockenbery DM. The oncogene c-Myc coordinates regulation of metabolic networks to enable rapid cell cycle entry. Cell Cycle. 2008; 7:1054–1066.
31. Vazquez A, Tedeschi PM and Bertino JR. Overexpression of the mitochondrial folate and glycine-serine pathway: a new determinant of methotrexate selectivity in tumors. Cancer Res. 2013; 73:478–482.
32. DuBois SG, Kalika Y, Lukens JN, Brodeur GM, Seeger RC, Atkinson JB, Haase GM, Black CT, Perez C, Shimada H, Gerbing R, Stram DO and Matthay KK. Metastatic sites in stage IV and IVS neuroblastoma correlate with age, tumor biology, and survival. J Pediatr Hematol Oncol. 1999; 21:181–189.
33. Charasson V, Hillaire-Buys D, Solassol I, Laurand-Quancard A, Pinguet F, Le Morvan V and Robert J. Involvement of gene polymorphisms of the folate pathway enzymes in gene expression and anticancer drug sensitivity using the NCI-60 panel as a model. Eur J Cancer. 2009; 45:2391–2401.
34. Fredlund E, Ringner M, Maris JM and Pahlman S. High Myc pathway activity and low stage of neuronal differentiation associate with poor outcome in neuroblastoma. Proc Natl Acad Sci U S A. 2008; 105:14094–14099.
35. Valentijn LJ, Koster J, Haneveld F, Aissa RA, van Sluis P, Broekmans ME, Molenaar JJ, van Nes J and Versteeg R. Functional MYCN signature predicts outcome of neuroblastoma irrespective of MYCN amplification. Proc Natl Acad Sci U S A. 2012; 109:19190–19195.
36. Ashton LJ, Murray JE, Haber M, Marshall GM, Ashley DM and Norris MD. Polymorphisms in genes encoding drug metabolizing enzymes and their influence on the outcome of children with neuroblastoma. Pharmacogenet Genomics. 2007; 17:709–717.
37. Hou Z, Orr S and Matherly LH. Post-transcriptional regulation of the human reduced folate carrier as a novel adaptive mechanism in response to folate excess or deficiency. Bioscience reports. 2014; 34(4).
38. Manohar CF, Bray JA, Salwen HR, Madafiglio J, Cheng A, Flemming C, Marshall GM, Norris MD, Haber M and Cohn SLC. MYCN Mediated Regulation of the MRP1 Promoter in Human Neuroblastoma. Oncogene. 2004; 23:753–762.
39. Wong SC, Zhang L, Proefke SA and Matherly LH. Effects of the loss of capacity for N-glycosylation on the transport activity and cellular localization of the human reduced folate carrier. Biochimica et biophysica acta. 1998; 1375:6–12.
40. Chomczynski P and Sacchi N. The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: twenty-something years on. Nat Protoc. 2006; 1:581–585.
41. Winer J, Jung CKS, Shackel I and Williams PM. Development and Validation of Real-Time Quantitative Reverse Transcriptase-Polymerase Chain Reaction for Monitoring Gene Expression in Cardiac Myocytes in Vitro. Analytical Biochemistry. 1999; 270:41–49.
42. Oberthuer A, Juraeva D, Li L, Kahlert Y, Westermann F, Eils R, Berthold F, Shi L, Wolfinger RD, Fischer M and Brors B. Comparison of performance of one-color and two-color gene-expression analyses in predicting clinical endpoints of neuroblastoma patients. The pharmacogenomics journal. 2010; 10:258–266.
43. Weinmann AS and Farnham PJ. Identification of unknown target genes of human transcription factors using chromatin immunoprecipitation. Methods. 2002; 26:37–47.
44. Fletcher JI, Gherardi S, Murray J, Burkhart CA, Russell A, Valli E, Smith J, Oberthuer A, Ashton LJ, London WB, Marshall GM, Norris MD, Perini G and Haber M. N-Myc regulates expression of the detoxifying enzyme glutathione transferase GSTP1, a marker of poor outcome in neuroblastoma. Cancer Res. 2012; 72:845–853.