
Introduction
Randomized clinical trials conducted in non-small-cell lung cancer (NSCLC) carrying activating mutations of the epidermal growth factor receptor (EGFR) have clearly shown that tyrosine kinase inhibitors (TKIs) dramatically contribute to improve prognosis, disease control, symptoms and quality of life when compared to traditional platinum-based chemotherapy [1-5]. A recent meta-analysis confirmed that EGFR mutant NSCLC patients derived a significant progression-free-survival (PFS) advantage from TKIs over platinum-doublet chemotherapy as first-line treatment, although a significant differential benefit may be observed according to smoking status (HR for never-smokers 0.29 versus 0.54 for ever-smokers; p < 0.007) and to the type of EGFR mutation (HR for exon 19 deletion 0.25 versus 0.44 for exon 21 substitution; p < 0.001) [6].
Therefore, the anti-EGFR TKIs gefitinib, erlotinib and afatinib are currently employed for the treatment of patients with advanced lung adenocarcinoma harboring EGFR activating mutations. However, the duration of response is variable and almost 25% of patients rapidly progress during treatment, many at the first disease assessment time-point. The question arises whether there are additional candidate clinical and/or molecular predictive factors permitting a further selection (‘super-selection’) of patients with EGFR mutated cancers to implement prediction of the awaited TKIs efficacy and identify those patients not beneficiating from TKIs despite the presence of the EGFR alteration.
The onset of resistance seems to represent an inevitable consequence of targeted therapies in solid tumors. Classically, the resistance develops after an initial response to therapy (acquired resistance) and may be pharmacological (failure of delivery of the drug to its target) or biological, primarily deriving from the activation of coexisting pathways, bypassing the oncogenic dependency of a given driven alteration [7].
Although the widely validated role of the EGFR T790M mutation as the main mechanism of acquired resistance to erlotinib and gefitinib [8, 9], the reliable impact of this genetic alteration at diagnosis is still debatable, relying on contradictory data regarding its true incidence and clinico-biological role. The rate of pretreatment T790M mutation is strongly dependent on the sensitivity of the detection method (ranging from 2% to 35%) [10, 11]. Recently, Costa et al. detected a high frequency (65%) of coexisting EGFR T790M before treatment, using a highly sensitive method based on laser microdissection and peptide-nucleic acid-clamping PCR [12]. The presence of a pretreatment T790M EGFR mutation seems to be associated with worse clinical outcomes (in term of objective response and PFS) to EGFR TKIs compared with patients with classic EGFR activating alterations without any detected T790M mutation [13, 14].
Other genetic abnormalities and signaling pathways are currently under investigation because of their potential implication in the development of TKIs resistance. TP53 represents the most frequently mutated gene in lung cancer, occurring in over half of adenocarcinoma, 80% of squamous cell carcinoma and 70% of small-cell-lung cancer [15]. Although the prognostic role of TP53 is still debatable, some preclinical data seem to suggest an intriguing predictive influence of TP53 mutation. In this regard, the persistent STAT3 activation has been observed in the residual survivor lung cancer cells both in vitro and in vivo under targeted TKIs, suggesting that early STAT3 phosphorylation may represent an important transcriptional programming event prior to the resurgence of resistant tumor survivors [16]. Moreover, TP53 and PTEN knowkdown synergize to activate pro-inflammatory interleukin-6/STAT3/nuclear factor kB signaling generating highly metastatic epithelial-to-mesenchymal transition-like cancer stem cells. The constitutive activation of this loop leads to the suppression of SOCS3 (suppressor of cytokine signaling 3), a critical negative regulator of pro-inflammatory pathways, suggesting interesting connection between inflammation and carcinogenesis [17].
Several clinical approaches can help to maintain the disease control in the resistance setting, including the use of radiation to treat isolated areas of progression (classically the central nervous system) and switching to cytotoxic chemotherapy. Moreover, novel approaches have already demonstrated a strong signal of activity, such as the development of second-generation and third-generation inhibitors and the combination of some of these inhibitors with antibodies directed against the same target. In this continuously evolving setting, the increased understanding of the spectrum of resistance is mandatory to make progress in clinical research [18].
Recently, many new facets emerged, highlighting the fact that EGFR mutations may be potentially targeted even with drugs without a peculiar EGFR action, such as bisphosphonates [19] and ibrutinib (a Bruton tyrosine kinase and BMX inhibitor) [20], that have demonstrated activity in EGFR-mutant NSCLC cell lines, including erlotinib-resistant tumors. Other promising data suggest the fact that the EGFR mutation may represent a genetic biomarker predicting enhanced sensitivity to topoisomerase II inhibitor (such as etoposide), in response to a methyltransferase EZH2 inhibitor, supporting the biological rationale underlying the possibility of a combined approach with these molecules [21]. With regard to the hypothesis of a multi-targeted approach, another recent preclinical analysis demonstrated that adding chloroquine (acting as an autophagy inhibitor) to EGFR and AKT inhibition might potentially improve tumor responses in EGFR mutant NSCLC cells [22]. Several experimental approaches, such as the establishment of pooled short-hairpin RNAs library screen, are currently under investigation to identify promising drugs and pathways for further study in EGFR mutant NSCLC, including TKIs-resistant NSCLC [23].
Tumor heterogeneity strongly contribute to primary resistance. The demonstration that human cancers are frequently represented by a molecular mosaic of cells, spatially and temporally different, derived from several genomic studies (reviewed in [24]). Next-generation sequencing approaches in renal and pancreatic cancers have demonstrated the existence of a strong heterogeneity among different regions within the same tumor and between the primary tumor and metastasis [25, 26].
The codification of tumor heterogeneity, improving the understanding of the oncogenic mechanism inclusive of the totality of the genomic and epigenomic processes, may help to identify clinically relevant subgroups of patients, leading to a better management of the therapeutic resistance to targeted agents.
In this context, the assessment of molecular heterogeneity with the innovative multigene next generation sequencing (NGS) technology may help to concurrent screen for additional genetic abnormalities potentially deputed to drive cancer predictive testing for therapeutic decisions [27]. The implications for future research are challenging: the EGFR mutated dosage as well as coexisting mutations could become a new predictive tool for lung cancer patients and new technologies, such as NGS, may potentially be introduced in routine practice.
Results
Eighteen patients from two institutions, whose characteristics are reported in Table 1, were studied. Median age was 71 years (range 37-83). Smokers were significantly more represented in the poor group than in the others (66.7% versus 10.0%, p[Fisher]=0.04). Seventeen patients were evaluable for PFS at least at first evaluation. At a median follow-up of 8 months (range: 1-33), 14 events of progression and 7 deaths were recorded. None of the patients had rearrangements of the ALK gene. Six, 3 and 8 patients were grouped as poor, intermediate and good responders, respectively, according to the treatment resistance to gefitinib and PFS (Table 2). The median PFS was 1.7, 6.1 and 17.3 months for poor, intermediate and good responders, respectively (p < 0.0001, Table 2).
Table 1: Patients’ characteristics (overall patients’ sample).
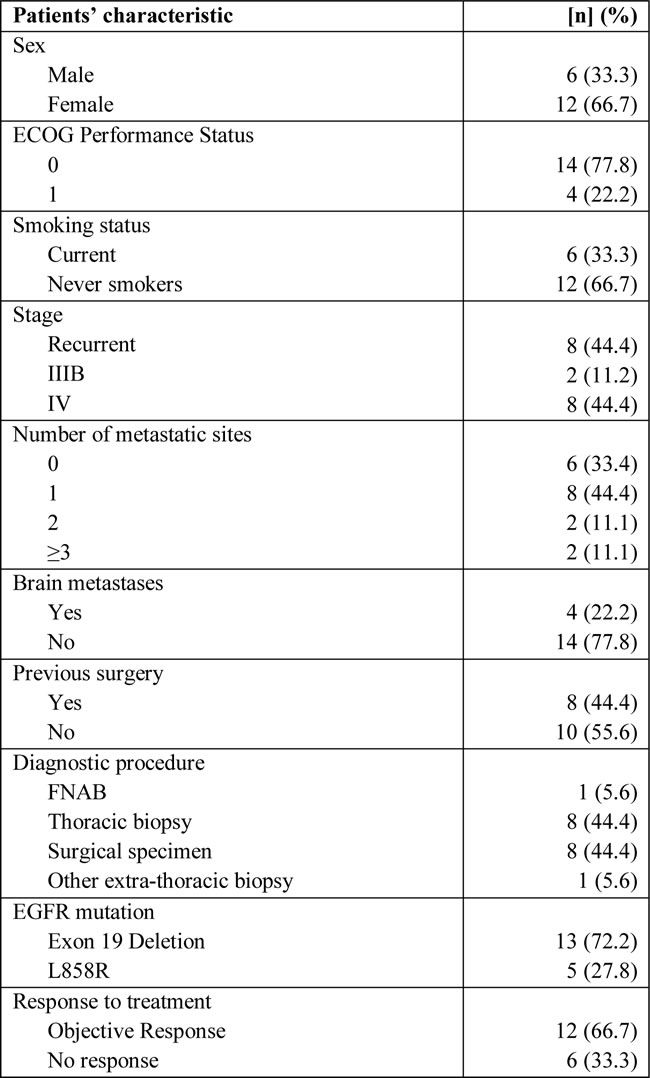
n, number of patients; ECOG, Eastern Cooperative Oncology Group; FNAB: fine needle aspiration biopsy; EGFR: epidermal growth factor receptor.
The proportion of cancer cells carrying mutated EGFR was calculated on the basis of the output of NGS analysis that furnishes the proportion of the mutated alleles (PMA), i.e., the number of mutated alleles over the total alleles analyzed. Thus, as the alleles in a cell are two, the proportion of cancer cells carrying a mutated allele is generally the double of the PMA indicated by NGS analysis, and referred to a cancer cellularity of at least 60% obtained through microdissection. As for EGFR mutations, median PMA was 0.2 (range 0.12-0.36), 0.16 (range 0.15-0.65) and 0.35 (range 0.17-0.61) for poor, intermediate and good responders, respectively. A non-significant trend between the good responder group and the 2 others was found in median PMA (0.35 versus 0.21, p[t-student]=0.08; p-value[Mann-Whitney]=0.16). A rate of 0.36 for PMA was identified at the ROC analysis as the best cut-off to split patients according to PFS.
Table 2: Patients’ groups according to resistance to Gefitinib and Progression-Free-Survival; 17 evaluable patients (Log-Rank p < 0.0001).
Group |
Definition |
Pts (%) |
Median PFS (months, 95%CI) |
Poor |
Progression at 1st assessment |
6 (35.2) |
1.7 (0.1-3.2) |
Intermediate |
Progression within 12 months |
3 (17.7) |
6.1 (3.0-9.2) |
Good |
Progression ≥ 12 months or treatment ongoing |
8 (47.1) |
17.3 (9.0-25.5) |
Pts: patients; PFS: progression-free-survival; CI: confidence intervals.
Thirteen mutations in addition to the one in EGFR were found in the TP53 (n=6), KRAS (n=2), CTNNB1 (n=2), PIK3CA, SMAD4 and MET (n=1 each) gene. All mutations were confirmed by Sanger sequencing. None of the patients had more than 2 concomitant mutations. The association of additional mutations and prognostic groups are reported in Table 3 and Supplementary Table 1. TP53 mutations (median PMA 0.45) were exclusively found among poor and intermediate patients and lacked in good responders (66.7% versus 0%, p[Fisher]=0.009) (Figure 1, Panel A).
Table 3: Relative frequency of gene mutations according to patients’ groups (17 evaluable patients).
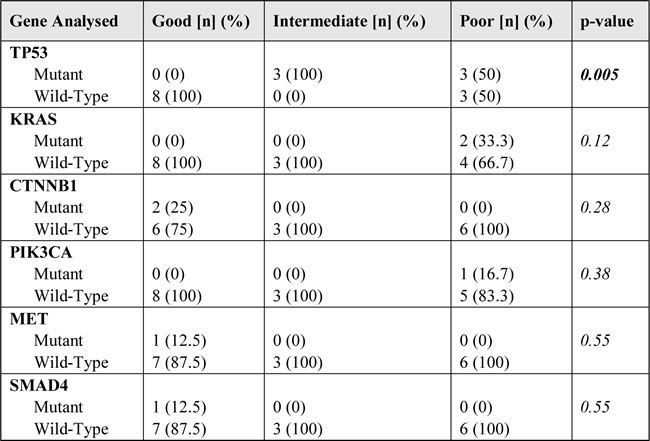
n, number of patients; p-value: chi-square test.
A paradigmatic case of a poor responder patient with a clear molecular heterogeneity is shown in Figure 1 (panels B-C) where an EGFR deletion in exon 19 in an area of the lesion presenting a well-differentiated adenocarcinoma (panels B-2 and C-3) is associated with a concomitant TP53 mutation (R248W) in a more dedifferentiated area (panels B-1 and C-3); both mutations were confirmed by Sanger Sequencing (Figure 1, Panel C-4).
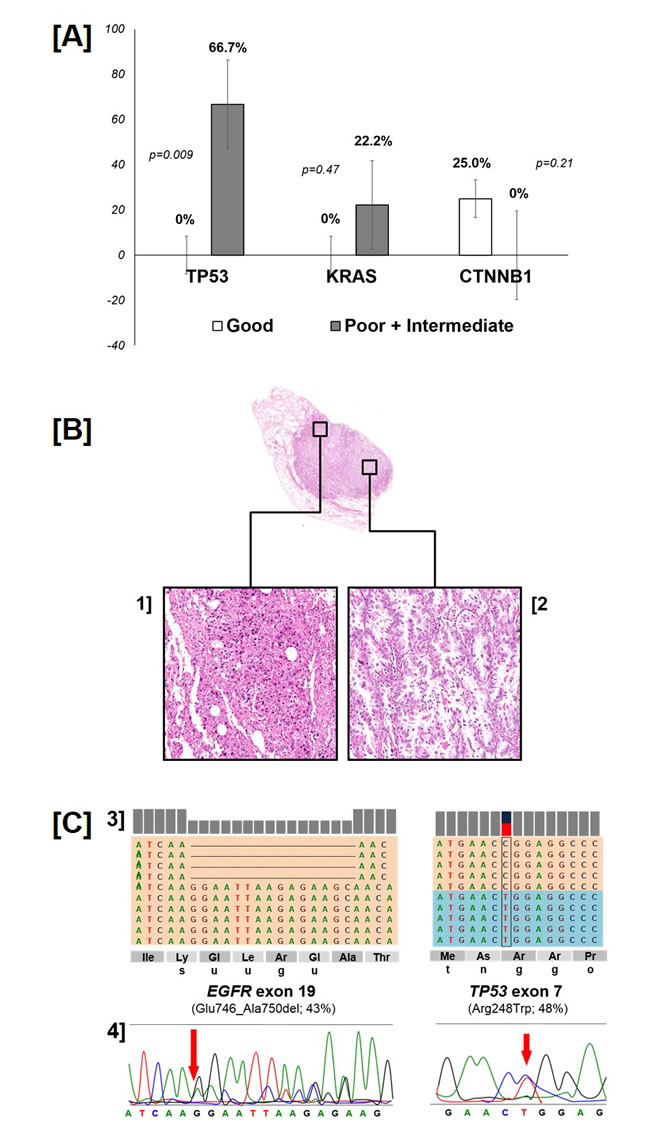
Figure 1: Distribution of TP53, KRAS and CTNNB1 gene mutation according to group (p-value: Fisher’s exact test - panel A). A representative case of intratumor histologic and molecular heterogeneity (panel B-C). The poor responder case presented well-differentiated coexisting with de-differentiated areas within the same specimen (panel B; original magnifications 4x and 20x). Of interest, an EGFR deletion in exon 19 was observed in the well-differentiated adenocarcinoma, that was associated with a concomitant TP53 mutation (R248W) in the more de-differentiated area. The representation of the reads obtained by Ion Torrent sequencing, aligned to the reference genome as provided by the Integrative Genomics Viewer (IGV v.2.1, Broad Institute) software for the mutations in EGFR and TP53 genes, and the corresponding Sanger sequencing are reported. (panel C).
No significant difference according to PMA in term of PFS was found (PMA ≥ 0.36, median PFS 12.0 months, 95% CI 11-13; 1-year PFS 62.0%, 2-year PFS 22.2% versus PMA < 0.36, median PFS 4.0 months, 95% CI 1-9; 1-year PFS 33.0%, 2-year PFS 20.8%; p=0.31), with a HR of 1.53 (95% CI 0.47-5.01, p=0.48) (Figure 2, Panel A). With regard to OS, no significant difference according to PMA was found (PMA ≥ 0.36, median OS 18 months, 95% CI 11-24; 1-year OS 83.5%, 2-year OS 41.7% versus PMA < 0.36, median OS not reached; 1-year OS 56.0%, 2-year OS 55.6%; p=0.59), with a HR of 1.21 (95% CI 0.28-5.16, p=0.79) (Figure 2, Panel B). V
A borderline significant difference in favor of the 8 patients with ACM=0 (median PFS 12.3 months, 95% CI 6.1-18.6; 1-year PFS 61.5%, 2-year PFS 31.3%) in comparison with those 9 with a ACM≥1 (median PFS 3.0 months, 95% CI 0.7-5.3; 1-year PFS 22.0%, 2-year PFS 11.1%) was found (p=0.03), with a HR of 2.88 (95% CI 0.92-9.02; p=0.068) (Figure 2, Panel C). With regard to OS, a significant difference in favor of the 8 patients with ACM=0 (median OS not reached, 1-year OS 88.0%, 2-year OS 70.0%) in comparison with those 9 with a ACM ≥ 1 (median OS 3.6 months, 95% CI 0.0-7.8; 1-year OS 43.5%, 2-year OS 22.2%) was found (p=0.03), with a HR of 5.07 (95% CI 0.99-26.03; p=0.052) (Figure 2, Panel D).
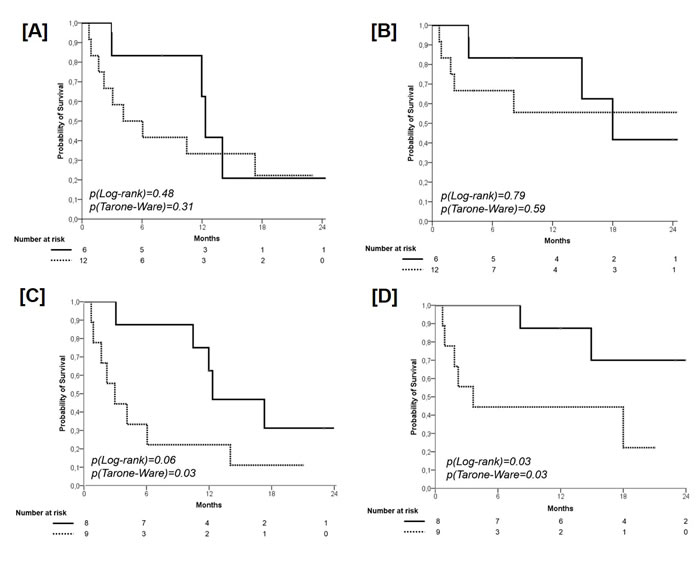
Figure 2: Progression-Free Survival according to PMA (cut-off 0.36). Solid line: patients with PMA ≥ 0.36; dashed line: patients with PMA < 0.36 (panel A). Overall Survival according to PMA (cut-off 0.36). Solid line: patients with PMA ≥ 0.36; dashed line: patients with PMA < 0.36 (panel B). Progression-Free Survival according to the presence of additional coexisting mutations (ACM). Solid line: patients with ACM = 0; dashed line: patients with ACM ≥ 1 (panel C). Overall Survival according to the presence of additional coexisting mutations (ACM). Solid line: patients with ACM = 0; dashed line: patients with ACM ≥ 1 (panel D).
A significant difference in favor of the 11 patients with wild type TP53 (median PFS 14 months, 95% CI 11.0-17.0; 1-year PFS 62.5%, 2-year PFS 31.8%) in comparison with those 7 with a mutant TP53 (median PFS 4.0 months, 95% CI 1.0-7.0; 1-year PFS 0%) was found (p=0.02), with a HR of 4.66 (95% CI 1.12-19.37; p=0.03) (Figure 3, Panel A). With regard to OS, no significant difference according the presence of p53 status was found (wild type TP53, median OS not reached; 1-year OS 71.7%, 2-year OS 52.0% versus mutant TP53, median OS not reached; 1-year OS 58.0%, 2-year OS 57.0%; p=0.31), with a HR of 2.25 (95% CI 0.43-5.16, p=0.34) (Figure 3, Panel B).
A significant difference in favor of the 7 patients with an EGFR PMA ≥ 0.36 and ACM=0 (median PFS 17.3 months, 95% CI 11.5-23.1; 1-year PFS 70.5%, 2-year PFS 35.7%) in comparison with those 10 with an EGFR PMA < 0.36 and ACM≥1 (median PFS 3.0 months, 95% CI 1.0-6.0; 1-year PFS 19.4%, 2-year PFS 10%) was found (p=0.03), with a HR of 3.26 (95% CI 0.78-10.85, p=0.054) (Figure 3, Panel C). With regard to OS, a trend towards significance in favor of patients with an PMA ≥ 0.36 and ACM=0 (median OS not reached; 1-year OS 86.0%, 2-year OS 68.6%) in comparison with patients with an EGFR PMA < 0.36 and ACM ≥ 1 (median OS 4.0 months, 95% CI 1-18; 1-year OS 50.5%, 2-year OS 25.0%) was found (p0.06), with a HR of 4.01 (95% CI 0.78-20.59, p=0.10) (Figure 3, Panel D).
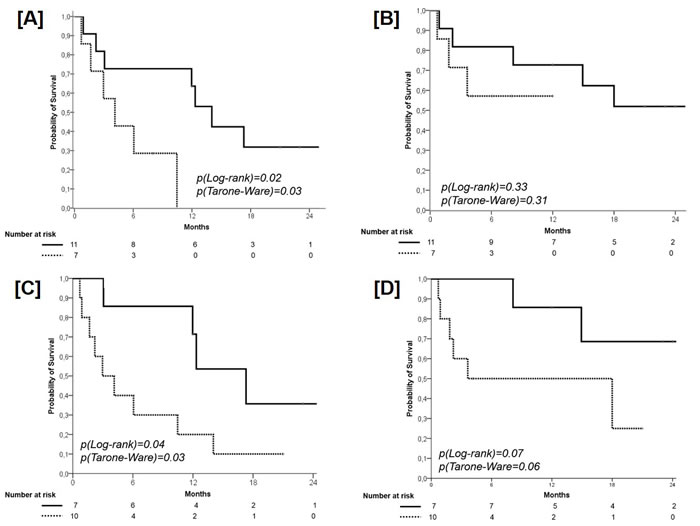
Figure 3: Progression-Free Survival according to the TP53 Mutation. Solid line: patients with wild type TP53; dashed line: patients with mutant TP53 (panel A). Overall Survival according to the TP53 Mutation. Solid line: patients with wild type TP53; dashed line: patients with mutant TP53 (panel B). Progression-Free Survival according to the combination of PMA (cut-off 0.36) and the presence of additional coexisting mutations (ACM). Solid line: patients with PMA ≥ 0.36 and ACM = 0; dashed line: patients with PMA < 0.36 and ACM ≥ 1 (panel C). Overall Survival according to the combination of PMA (cut-off 0.36) and the presence of additional coexisting mutations (ACM). Solid line: patients with PMA ≥ 0.36 and ACM = 0; dashed line: patients with PMA < 0.36 and ACM ≥ 1 (panel D).
Discussion
Despite exploratory and unpowered for conclusive interpretations, the results reported herein indicate that the application of NGS technology may furnish a baseline genetic portrait of advanced NSCLC that gives information on the presence of actionable and additional mutations that may impair the efficacy of targeted therapies. A prospective validation in a large set of patients is required to corroborate our results.
Our study on a series of EGFR mutant advanced lung cancers receiving 1st line Gefitinib suggests that the presence of ACM significantly decreases the expected benefit of TKIs.
Among the identified ACMs, TP53 mutations were exclusively documented among poor/intermediate responders than in good responders (66.7% versus 0%, p=0.009), suggesting the potential implication of this gene in influencing the chance of response to the anti-EGFR agents. In lung cancer, TP53 represents the most frequently mutated gene, occurring in over half of adenocarcinoma, 80% of squamous cell carcinoma and 70% of small-cell-lung cancer [15], and, despite debated [28], it has been suggested as an independent marker for shorter survival in advanced NSCLC. A recent evidence demonstrated that not all TP53 mutations are equal: non-disruptive TP53 mutations (those preserving some functional properties of the protein) represents an independent prognostic factor of shorter survival in advanced NSCLC (13.3 versus 24.6 months; HR=2.08; p < 0.001) [29].
In contrast to EGFR-activating mutations, KRAS mutations are usually detected in smokers, and associated with poor prognosis and no benefit from TKIs and adjuvant chemotherapy. However, some evidences are available regarding the coexistence of KRAS and EGFR mutations, raising questions about the relative values of these genetic abnormalities as predictors of outcome in NSCLC [30]. Marchetti et al. reported that patients carrying both mutations were resistant to TKIs and showed a shorter survival compared with those patients with only EGFR mutations, suggesting that the biological power determined by the presence of KRAS mutation, even if only in minor cellular clones, may potentially overcome the dependence to EGFR [31].
EGFR mutations are very uncommon among smokers and former smokers, enforcing the rationale that coexisting mutations may feature the disease. From a purely speculative point-of-view, we observed that 4 out of 6 TP53 mutations, KRAS mutations (n=2) and the MET mutation (n=1) have been detected in the smaller group of EGFR mutant patients that were smoker, suggesting a potential correlation between smoking status and the presence of ACMs.
Our data may contribute to generate hypotheses with regard to the biological intrinsic difference between primary and acquired resistance, being the latest event supported by a series of data indicating the EGFR T790M mutation as the more common mechanism of acquired resistance to erlotinib and gefitinib [8]. Indeed, approximately 50-80% of rebiopses of EGFR-mutant patients progressing under the selective pressure of EGFR TKIs display this kind of mutation [8, 9]. Globally considered, our results support the hypothesis that TP53 (and perhaps other tumor suppressor genes) may affect the efficacy of the traditional target therapy in molecularly addicted patients, thus triggering cell proliferation stimulus, and bypassing the oncogenic power of the EGFR pathway.
In this population carrying ‘bad’ genetic alterations, concurrently with the classical EGFR-activating mutations, the employment of a combination strategy of chemotherapy and target agents may preserve a clinico-biological rationale. Although no benefit was demonstrated for adding TKIs to chemotherapy (INTACT-1 and 2 [32, 33], TRIBUTE [34] and TALENT trials [35]), intercalated erlotinib to cisplatin or carboplatin demonstrated a significant benefit in the context of an EGFR mutation-positive subgroup (FASTACT-1 and 2) [36, 37], although these evidences warrant to be replicate in the context of non-asian patients’ populations.
In addition to this ‘qualitative’ analysis exploring the number of ACM, we tried to determine if the ‘quantitative’ analysis of the EGFR mutation (PMA) might have a role in influencing the efficacy of TKIs. These data allow speculating upon the exploration of those key processes critical to the tumor development and progression, as the tumor heterogeneity.
With regard to the ‘quantitative’ analysis of the EGFR mutation, Zhou et al. firstly hypothesized that the quantification of EGFR mutations might predict the extent of EGFR TKIs benefit, demonstrating an advantage in term of PFS for those patients with high abundance of EGFR mutations (detected concurrently with two methods with a different sensitivity) [38]. Our results support this hypothesis, although, given the limited number of patients, only a weak trend in favor of those patients with a high PMA (≥ 0.36) was found.
Nevertheless, given the intrinsic differences in the adopted EGFR detection methods, the reliability of the observed evidences and their overall conclusions must be considered purely speculative for further research in this regard. Indeed, as recently demonstrated by Tseng et al., a significant portion of responses to erlotinib in EGFR wild-type patients was related to the limitations of detection methods (both direct sequencing and mutant type-specific sensitive methods) [39].
Although no detailed study on tumor heterogeneity have been published in lung cancer, several data regarding the complexity of the molecular background of lung cancer are available. From the morphological perspective, the most recent clinico-pathological classification of lung adenocarcinoma identifies multiple histologic subtypes, frequently co-existing in the same tumor [40]. From the genomic standpoint, recent data demonstrate the high mutational rate of lung cancer, supporting the hypothesis of a strong heterogeneity [41, 42]. Nevertheless, a series of data suggest that the mutational load of lung adenocarcinoma in non-smoker patients (where the chance of detecting the EGFR mutation is the highest) is significantly lower [43], and that few (but critical) mutations may be enough to switch cells from normal to malignant [44].
While conflicting evidences exist regarding the discordance rate of EGFR mutation status between primary tumors and metastases, a substantial consistency supports the lack of a reliable heterogeneity of EGFR mutations at the intratumor level [24]. Recently, Wright et al. analyzed different areas in the context of a series of lung adenocarcinoma specimens and found that KRAS and BRAF mutations were confined to high-grade morphological domains, while the EGFR mutations were identifiable through all histological subtypes in the tumor according to the driver status of the mutations [45].
Globally considered, these results suggest that the heterogeneity of a driver-gene mutation, such as that of EGFR in lung cancer is a rare molecular event. However, the alterations in the target gene itself (affecting the delivery of the drug to its target) and the concurrent activation or suppression of other signaling pathways (relevant for tumor progression and survival), may justify the heterogeneity in term of the clinical benefit deriving from the employment of target agents.
The data emerging from our analysis support the potential role of the NGS technology in the deeper analysis of the molecular background, also in the context of a recognized oncogene-addicted disease, with the ideal aim to further improve the EGFR mutant patients’ selection. Our group already highlighted the basic contribution of NGS technique in the development of the translational research in the diagnostic field, able to provide, with limited amounts of the biological material (DNA), reliable quantitative and qualitative data, critical for the application of the tailored medicine [46].
Together with other emerging scientific evidences, our analysis may represent a proof-of-principle study supporting the existence of a tumor heterogeneity which, from now on, should be considered clinically relevant and deserving to be deepened for clinical validation [47]. In this new era of precision medicine, our study highlights the potential strong contribution to the understanding of the molecular bases of cancer yielded by NGS technologies, which may allow to provide a ‘qualitative’ (presence of ACM) and ‘quantitative’ (PMA) measure of the tumor heterogeneity [48]. In this regard, the found genetic portrait of cancer cells may mirror the underlying tumor heterogeneity, thus allowing to ultra-stratifying those EGFR mutant NSCLC patients, and estimating a differential clinical benefit of anti-EGFR TKIs in such already-selected subpopulation.
materials and Methods
Patients
A consecutive series of patients carrying a sensitizing mutation of the EGFR gene, receiving first line gefitinib were analyzed for mutations in 22 genes with deep sequencing technology. In all cases, a formalin-fixed and paraffin-embedded (FFPE) tumor excisional/trans-bronchial biopsy was available. Patients were retrospectively grouped according to time to progression during gefitinib treatment in: poor responders (progression at first tumor response assessment), intermediate responders (progression within 12 months), and good responders (progression or treatment still ongoing after 12 months). The principal aim of the study was to correlate the activity of gefitinib (in terms of treatment resistance and Progression-Free Survival, PFS) with: 1) the proportion of cells carrying the EGFR mutation, calculated on the basis of the value of the proportion of mutated alleles (PMA) that is derived from NGS analysis readout [Quantitative analysis], and 2) the presence of Additional Coexisting Mutations [ACM] (other than EGFR mutation) [Qualitative analysis]. All the samples were received anonymously and processed at the Molecular Pathology Unit of the Department of Pathology and Diagnostics of the University of Verona.
Tissue microdissection and DNA preparation
Four 10-µm paraffin sections were manually microdissected to ensure that each tumor sample contained at least 60% of neoplastic cells. DNA was isolated using the QIAmp DNA FFPE tissue kit (Qiagen) and quantified and its quality assessed using NanoDrop® (Invitrogen Life Technologies; Milan, Italy) and Qubit® (Invitrogen Life Technologies) platforms [46, 49].
Deep sequencing of multiplex PCR
Amplicons. Deep sequencing was performed using the Ion Torrent platform (Life Technologies). Briefly, 10 ng of purified genomic DNA were used for library construction with the Ion AmpliSeq Colon and Lung Cancer Panel v2 (Life Technologies) that targets 504 mutational hotspot regions of the following 22 cancer-associated genes, in alphabetical order: AKT1, ALK, BRAF, CTNNB1, DDR2, EGFR, ERBB2, ERBB4, FBXW7, FGFR1, FGFR2, FGFR3, KRAS, MAP2K1, MET, NOTCH1, NRAS, PIK3CA, PTEN, SMAD4, STK11, TP53. Emulsion PCR was performed either manually or with the OneTouch DL system (Life Technologies). The quality of the obtained library was evaluated by the Agilent® 2100 Bioanalyzer on-chip electrophoresis (Agilent Technologies; Santa Clara, CA). Sequencing was run on the Ion Torrent Personal Genome Machine™ (PGM, Life Technologies) loaded with a 316 chip as per manufacturer’s protocol. Data analysis, including alignment to the hg19 human reference genome and variant calling, was done using the Torrent Suite Software v.3.2 (Life Technologies). Filtered variants were annotated using both the Ion Reporter software v1.2 (Life Technologies) and the SnpEff software v.3.0 (alignments visually verified with the Integrative Genomics Viewer; IGV v.2.1, Broad Institute) [46].
DNA sanger sequencing
To validate NGS results, KRAS (exons 2, 3), EGFR (exons 19, 21), TP53 (exons 5, 6, 7, 8) , and CTNNB1, PIK3CA, SMAD4 and MET specific PCR fragments were analyzed by Sanger sequencing. PCR products were purified using Agencourt AMPure XP magnetic beads (Beckman Coulter), labeled with Big Dye terminator v3.1 (Applied Biosystems). Agencourt CleanSEQ magnetic beads (Beckman Coulter) were used for post-labeling purification. Sequence analysis was performed on an Applied Biosystems 3130xl Genetic Analyser.
Statistics
Descriptive statistics was used to summarize pertinent study information. Follow-up was analyzed and reported according to Shuster [50]. The correlation between variables were analyzed according to chi-square, Student’s t, and Mann–Whitney (nonparametric) tests. The PMA value for each patient was normalized taking into account the rate of cellularity of malignant cells for each sample. The receiver operating characteristic (ROC) curve analysis was adopted for dichotomization of continuous variables according to outcome [51]. The hazard ratio (HR) and the 95% confidence intervals (95% CI) was estimated for each variable using the Cox univariate model [52]. Progression-free- and Overall-survival (PFS and OS) were calculated by the Kaplan-Meier product limit method from the date of treatment start until progression or death for any cause. The Log-rank and Tarone-Ware analyses were adopted to assess differences between curves. Significance was defined at the p < 0.05 level. The SPSS® (18.0) licensed statistical program was used for all analyses.
Acknowledgements, Support & Funding
Supported by a grant of the Italian Association for Cancer Research (AIRC My First AIRC Grant n° 14282, Investigator Grant n° 11631, 5X1000 n° 12182 and 12214), and from the Italian Ministry of Education, Universities and Research (MIUR-FIRB RBAP10AHJB).
Conflicts of interest
E.Bria has a consultancy role for Celgene, boards’ participation for Novartis, Astra-Zeneca, Pierre-Fabre, and speaker’s fee from Pfizer; S.Novello declares boards’ participation for Novartis, Astra-Zeneca, MSD, Boehringer Ingelheim, Eli Lilly, Roche and speaker’s fee from Pfizer; M. Milella declares boards’ participation for Celgene, Astra-Zeneca, Boehringer Ingelheim, and speaker’s fee from Pfizer, Novartis, and Celgene; A. Scarpa declares consultancies for Amgen, and boards’ participation for Novartis, Sanofi; G.Tortora has consultancy role for Novartis, Pfizer and GSK. No potential conflicts of interest were disclosed by the other authors.
References
1. Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, Palmero R, Garcia-Gomez R, Pallares C, Sanchez JM, Porta R, Cobo M, Garrido P, Longo F, Moran T, Insa A, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012; 13:239-246.
2. Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J, Seto T, Satouchi M, Tada H, Hirashima T, Asami K, Katakami N, Takada M, Yoshioka H, Shibata K, Kudoh S, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010; 11:121-128.
3. Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I, Fujita Y, Okinaga S, Hirano H, Yoshimori K, Harada T, Ogura T, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010; 362:2380-2388.
4. Sequist LV, Yang JC, Yamamoto N, O’Byrne K, Hirsh V, Mok T, Geater SL, Orlov S, Tsai CM, Boyer M, Su WC, Bennouna J, Kato T, Gorbunova V, Lee KH, Shah R, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013; 31:3327-3334.
5. Pilotto S, Di Maio M, Peretti U, Kinspergher S, Brunelli M, Massari F, Sperduti I, Giannarelli D, De Marinis F, Tortora G and Bria E. Predictors of outcome for patients with lung adenocarcinoma carrying the epidermal growth factor receptor mutation receiving 1st-line tyrosine kinase inhibitors: Sensitivity and meta-regression analysis of randomized trials. Crit Rev Oncol Hematol. 2014; 90:135-145.
6. Hasegawa Y, Ando M, Maemondo M, Yamamoto S, Isa SI, Saka H, Kubo A, Kawaguchi T, Takada M, Rosell R, Kurata T and Ou SH. The Role of Smoking Status on the Progression-Free Survival of Non-Small-Cell Lung Cancer Patients Harboring Activating Epidermal Growth Factor Receptor (EGFR) Mutations Receiving First-Line EGFR Tyrosine Kinase Inhibitor Versus Platinum Double Chemotherapy: A Meta-Analysis of Prospective Randomized Trials. Oncologist. 2015.
7. Lovly CM and Shaw AT. Molecular pathways: resistance to kinase inhibitors and implications for therapeutic strategies. Clin Cancer Res. 2014; 20:2249-2256.
8. Arcila ME, Oxnard GR, Nafa K, Riely GJ, Solomon SB, Zakowski MF, Kris MG, Pao W, Miller VA and Ladanyi M. Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin Cancer Res. 2011; 17:1169-1180.
9. Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, Kris MG, Miller VA, Ladanyi M and Riely GJ. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013; 19:2240-2247.
10. Rosell R, Molina MA, Costa C, Simonetti S, Gimenez-Capitan A, Bertran-Alamillo J, Mayo C, Moran T, Mendez P, Cardenal F, Isla D, Provencio M, Cobo M, Insa A, Garcia-Campelo R, Reguart N, et al. Pretreatment EGFR T790M mutation and BRCA1 mRNA expression in erlotinib-treated advanced non-small-cell lung cancer patients with EGFR mutations. Clin Cancer Res. 2011; 17:1160-1168.
11. Yu HA, Arcila ME, Hellmann MD, Kris MG, Ladanyi M and Riely GJ. Poor response to erlotinib in patients with tumors containing baseline EGFR T790M mutations found by routine clinical molecular testing. Ann Oncol. 2014; 25:423-428.
12. Costa C, Molina MA, Drozdowskyj A, Gimenez-Capitan A, Bertran-Alamillo J, Karachaliou N, Gervais R, Massuti B, Wei J, Moran T, Majem M, Felip E, Carcereny E, Garcia-Campelo R, Viteri S, Taron M, et al. The impact of EGFR T790M mutations and BIM mRNA expression on outcome in patients with EGFR-mutant NSCLC treated with erlotinib or chemotherapy in the randomized phase III EURTAC trial. Clin Cancer Res. 2014; 20:2001-2010.
13. Lee Y, Lee GK, Lee YS, Zhang W, Hwang JA, Nam BH, Kim SH, Kim JH, Yun T, Han JY, Kim HT and Lee JS. Clinical outcome according to the level of preexisting epidermal growth factor receptor T790M mutation in patients with lung cancer harboring sensitive epidermal growth factor receptor mutations. Cancer. 2014; 120:2090-2098.
14. Su KY, Chen HY, Li KC, Kuo ML, Yang JC, Chan WK, Ho BC, Chang GC, Shih JY, Yu SL and Yang PC. Pretreatment epidermal growth factor receptor (EGFR) T790M mutation predicts shorter EGFR tyrosine kinase inhibitor response duration in patients with non-small-cell lung cancer. J Clin Oncol. 2012; 30:433-440.
15. Shtivelman E, Hensing T, Simon GR, Dennis PA, Otterson GA, Bueno R and Salgia R. Molecular pathways and therapeutic targets in lung cancer. Oncotarget. 2014; 5:1392-1433.
16. Fan W, Tang Z, Yin L, Morrison B, Hafez-Khayyata S, Fu P, Huang H, Bagai R, Jiang S, Kresak A, Howell S, Vasanji A, Flask CA, Halmos B, Koon H and Ma PC. MET-independent lung cancer cells evading EGFR kinase inhibitors are therapeutically susceptible to BH3 mimetic agents. Cancer Res. 2011; 71:4494-4505.
17. Kim G, Ouzounova M, Quraishi AA, Davis A, Tawakkol N, Clouthier SG, Malik F, Paulson AK, D’Angelo RC, Korkaya S, Baker TL, Esen ES, Prat A, Liu S, Kleer CG, Thomas DG, et al. SOCS3-mediated regulation of inflammatory cytokines in PTEN and p53 inactivated triple negative breast cancer model. Oncogene. 2015; 34:671-680.
18. Camidge DR, Pao W and Sequist LV. Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nat Rev Clin Oncol. 2014; 11:473-481.
19. Stachnik A, Yuen T, Iqbal J, Sgobba M, Gupta Y, Lu P, Colaianni G, Ji Y, Zhu LL, Kim SM, Li J, Liu P, Izadmehr S, Sangodkar J, Scherer T, Mujtaba S, et al. Repurposing of bisphosphonates for the prevention and therapy of nonsmall cell lung and breast cancer. Proc Natl Acad Sci U S A. 2014; 111:17995-18000.
20. Gao W, Wang M, Wang L, Lu H, Wu S, Dai B, Ou Z, Zhang L, Heymach JV, Gold KA, Minna J, Roth JA, Hofstetter WL, Swisher SG and Fang B. Selective antitumor activity of ibrutinib in EGFR-mutant non-small cell lung cancer cells. J Natl Cancer Inst. 2014; 106.
21. Fillmore CM, Xu C, Desai PT, Berry JM, Rowbotham SP, Lin YJ, Zhang H, Marquez VE, Hammerman PS, Wong KK and Kim CF. EZH2 inhibition sensitizes BRG1 and EGFR mutant lung tumours to TopoII inhibitors. Nature. 2015; 520:239-242.
22. Bokobza SM, Jiang Y, Weber AM, Devery AM and Ryan AJ. Combining AKT inhibition with chloroquine and gefitinib prevents compensatory autophagy and induces cell death in EGFR mutated NSCLC cells. Oncotarget. 2014; 5:4765-4778.
23. Sudo M, Mori S, Madan V, Yang H, Leong G and Koeffler HP. Short-hairpin RNA library: identification of therapeutic partners for gefitinib-resistant non-small cell lung cancer. Oncotarget. 2015; 6:814-824.
24. Mitsudomi T, Suda K and Yatabe Y. Surgery for NSCLC in the era of personalized medicine. Nat Rev Clin Oncol. 2013; 10:235-244.
25. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012; 366:883-892.
26. Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, Kamiyama M, Hruban RH, Eshleman JR, Nowak MA, Velculescu VE, Kinzler KW, Vogelstein B and Iacobuzio-Donahue CA. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010; 467:1114-1117.
27. Liu B, Morrison CD, Johnson CS, Trump DL, Qin M, Conroy JC, Wang J and Liu S. Computational methods for detecting copy number variations in cancer genome using next generation sequencing: principles and challenges. Oncotarget. 2013; 4:1868-1881.
28. Mitsudomi T, Hamajima N, Ogawa M and Takahashi T. Prognostic significance of p53 alterations in patients with non-small cell lung cancer: a meta-analysis. Clin Cancer Res. 2000; 6:4055-4063.
29. Molina-Vila MA, Bertran-Alamillo J, Gasco A, Mayo-de-Las-Casas C, Sanchez-Ronco M, Pujantell-Pastor L, Bonanno L, Favaretto A, Cardona AF, Vergnenegre A, Majem M, Massuti B, Moran T, Carcereny E, Viteri S and Rosell R. Nondisruptive p53 mutations are associated with shorter survival in advanced non-small-cell lung cancer patients. Clin Cancer Res. 2014.
30. Choughule A, Sharma R, Trivedi V, Thavamani A, Noronha V, Joshi A, Desai S, Chandrani P, Sundaram P, Utture S, Jambhekar N, Gupta S, Aich J, Prabhash K and Dutt A. Coexistence of KRAS mutation with mutant but not wild-type EGFR predicts response to tyrosine-kinase inhibitors in human lung cancer. Br J Cancer. 2014; 111:2203-2204.
31. Marchetti A, Milella M, Felicioni L, Cappuzzo F, Irtelli L, Del Grammastro M, Sciarrotta M, Malatesta S, Nuzzo C, Finocchiaro G, Perrucci B, Carlone D, Gelibter AJ, Ceribelli A, Mezzetti A, Iacobelli S, et al. Clinical implications of KRAS mutations in lung cancer patients treated with tyrosine kinase inhibitors: an important role for mutations in minor clones. Neoplasia. 2009; 11:1084-1092.
32. Giaccone G, Herbst RS, Manegold C, Scagliotti G, Rosell R, Miller V, Natale RB, Schiller JH, Von Pawel J, Pluzanska A, Gatzemeier U, Grous J, Ochs JS, Averbuch SD, Wolf MK, Rennie P, et al. Gefitinib in combination with gemcitabine and cisplatin in advanced non-small-cell lung cancer: a phase III trial--INTACT 1. J Clin Oncol. 2004; 22:777-784.
33. Herbst RS, Giaccone G, Schiller JH, Natale RB, Miller V, Manegold C, Scagliotti G, Rosell R, Oliff I, Reeves JA, Wolf MK, Krebs AD, Averbuch SD, Ochs JS, Grous J, Fandi A, et al. Gefitinib in combination with paclitaxel and carboplatin in advanced non-small-cell lung cancer: a phase III trial--INTACT 2. J Clin Oncol. 2004; 22:785-794.
34. Herbst RS, Prager D, Hermann R, Fehrenbacher L, Johnson BE, Sandler A, Kris MG, Tran HT, Klein P, Li X, Ramies D, Johnson DH, Miller VA and Group TI. TRIBUTE: a phase III trial of erlotinib hydrochloride (OSI-774) combined with carboplatin and paclitaxel chemotherapy in advanced non-small-cell lung cancer. J Clin Oncol. 2005; 23:5892-5899.
35. Gatzemeier U, Pluzanska A, Szczesna A, Kaukel E, Roubec J, De Rosa F, Milanowski J, Karnicka-Mlodkowski H, Pesek M, Serwatowski P, Ramlau R, Janaskova T, Vansteenkiste J, Strausz J, Manikhas GM and Von Pawel J. Phase III study of erlotinib in combination with cisplatin and gemcitabine in advanced non-small-cell lung cancer: the Tarceva Lung Cancer Investigation Trial. J Clin Oncol. 2007; 25:1545-1552.
36. Mok TS, Wu YL, Yu CJ, Zhou C, Chen YM, Zhang L, Ignacio J, Liao M, Srimuninnimit V, Boyer MJ, Chua-Tan M, Sriuranpong V, Sudoyo AW, Jin K, Johnston M, Chui W, et al. Randomized, placebo-controlled, phase II study of sequential erlotinib and chemotherapy as first-line treatment for advanced non-small-cell lung cancer. J Clin Oncol. 2009; 27:5080-5087.
37. Wu YL, Lee JS, Thongprasert S, Yu CJ, Zhang L, Ladrera G, Srimuninnimit V, Sriuranpong V, Sandoval-Tan J, Zhu Y, Liao M, Zhou C, Pan H, Lee V, Chen YM, Sun Y, et al. Intercalated combination of chemotherapy and erlotinib for patients with advanced stage non-small-cell lung cancer (FASTACT-2): a randomised, double-blind trial. Lancet Oncol. 2013; 14:777-786.
38. Zhou Q, Zhang XC, Chen ZH, Yin XL, Yang JJ, Xu CR, Yan HH, Chen HJ, Su J, Zhong WZ, Yang XN, An SJ, Wang BC, Huang YS, Wang Z and Wu YL. Relative abundance of EGFR mutations predicts benefit from gefitinib treatment for advanced non-small-cell lung cancer. J Clin Oncol. 2011; 29:3316-3321.
39. Tseng JS, Wang CL, Huang MS, Chen CY, Chang CY, Yang TY, Tsai CR, Chen KC, Hsu KH, Tsai MH, Yu SL, Su KY, Wu CW, Yang CT, Chen YM and Chang GC. Impact of EGFR mutation detection methods on the efficacy of erlotinib in patients with advanced EGFR-wild type lung adenocarcinoma. PLoS One. 2014; 9:e107160.
40. Travis WD, Brambilla E, Noguchi M, Nicholson AG, Geisinger KR, Yatabe Y, Beer DG, Powell CA, Riely GJ, Van Schil PE, Garg K, Austin JH, Asamura H, Rusch VW, Hirsch FR, Scagliotti G, et al. International association for the study of lung cancer/american thoracic society/european respiratory society international multidisciplinary classification of lung adenocarcinoma. J Thorac Oncol. 2011; 6:244-285.
41. Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, Sougnez C, Auclair D, Lawrence MS, Stojanov P, Cibulskis K, Choi K, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012; 150:1107-1120.
42. Cancer Genome Atlas Research N. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012; 489:519-525.
43. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr. and Kinzler KW. Cancer genome landscapes. Science. 2013; 339:1546-1558.
44. Tomasetti C, Marchionni L, Nowak MA, Parmigiani G and Vogelstein B. Only three driver gene mutations are required for the development of lung and colorectal cancers. Proceedings of the National Academy of Sciences of the United States of America. 2015; 112:118-123.
45. Wright GM, Do H, Weiss J, Alam NZ, Rathi V, Walkiewicz M, John T, Russell PA and Dobrovic A. Mapping of actionable mutations to histological subtype domains in lung adenocarcinoma: implications for precision medicine. Oncotarget. 2014; 5:2107-2115.
46. Simbolo M, Gottardi M, Corbo V, Fassan M, Mafficini A, Malpeli G, Lawlor RT and Scarpa A. DNA qualification workflow for next generation sequencing of histopathological samples. PLoS One. 2013; 8:e62692.
47. De Mattos-Arruda L, Weigelt B, Cortes J, Won HH, Ng CK, Nuciforo P, Bidard FC, Aura C, Saura C, Peg V, Piscuoglio S, Oliveira M, Smolders Y, Patel P, Norton L, Tabernero J, et al. Capturing intra-tumor genetic heterogeneity by de novo mutation profiling of circulating cell-free tumor DNA: a proof-of-principle. Ann Oncol. 2014; 25:1729-1735.
48. Mafficini A, Amato E, Fassan M, Simbolo M, Antonello D, Vicentini C, Scardoni M, Bersani S, Gottardi M, Rusev B, Malpeli G, Corbo V, Barbi S, Sikora KO, Lawlor RT, Tortora G, et al. Reporting tumor molecular heterogeneity in histopathological diagnosis. PloS one. 2014; 9:e104979.
49. Scarpa A, Sikora K, Fassan M, Rachiglio AM, Cappellesso R, Antonello D, Amato E, Mafficini A, Lambiase M, Esposito C, Bria E, Simonato F, Scardoni M, Turri G, Chilosi M, Tortora G, et al. Molecular typing of lung adenocarcinoma on cytological samples using a multigene next generation sequencing panel. PLoS One. 2013; 8:e80478.
50. Shuster JJ. Median follow-up in clinical trials. J Clin Oncol. 1991; 9:191-192.
51. Hanley JA and McNeil BJ. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology. 1982; 143:29-36.
52. Hess KR. Graphical methods for assessing violations of the proportional hazards assumption in Cox regression. Stat Med. 1995; 14:1707-1723.