
Introduction
Breast cancer brain metastasis is a growing public health concern as advances in systemic therapy have helped to contain metastatic growth in most organs except the brain [1]. Brain metastasis occurs in 10-15% of patients with metastatic breast cancer [2-4], and is associated with an extremely poor prognosis with a median survival of only 3-6 months [5, 6]. Patients with HER2+ or triple negative breast cancer (TNBC) have a greater propensity to develop brain metastasis [7-13].
Three processes may control brain metastasis. The first may involve a minority of primary tumor cells with unique mutations that impart proclivity for brain metastasis. Recent massively parallel sequencing of primary tumor and a brain metastasis from the same patient suggested this possibility [14]. The second is that mutations and/or epigenetic changes in cancer cells bestow blood-brain-barrier (BBB) permeability and consequently brain metastasis. The third is that every cancer cell has the ability to reach the brain but only a few cells that can acquire neuronal cell function through either additional mutations in cancer cells or brain microenvironment-induced epigenetic changes in cancer cells that are essential for metastatic growth proliferate in the brain. For example, circulating tumor cells that metastasize to the brain overexpress proteins such as heparanase (HPSE) that allow cancer cells to interact with brain vasculature [15]. Brain metastatic cancer cells express SERPINE1, which helps in vascular co-adaptation in the brain [16]. The Biology of Brain Metastasis Workshop organized by the National Cancer Institute (NCI) has set several research priorities with respect to biology of brain metastasis [17]. These include investigations into the pathogenic mechanisms of metastasis to brain, identification of commonalities and uniqueness of brain metastasis versus other sites of metastasis, differentiation of indolent and aggressive lesions by understanding heterogeneity among different brain metastatic lesions, investigation of the relationship between “stem cell” features and brain metastasis, and understanding the mechanisms responsible for tumor cell homing to the brain.
Progress in addressing the above issues is limited largely due to the lack of suitable model system. Most of our current knowledge on brain metastasis is derived from studies using brain-seeking variants developed from HER2 -amplified BT474 cells and triple negative breast cancer/mesenchymal stem cell-like cell line MDA-MB-231 [18, 19]. Analyses of MDA-MB-231 derivatives enabled development of a brain metastasis signature and identification of a set of genes that may be involved in BBB extravasation. Genes identified in these studies include the brain-specific sialyltransferase ST6GALNAC5, COX2, ANGPTL4 and EGFR ligands epiregulin and HBEGF [20]. NF-κB inducible genes MMP-1 and FSCIN-1 are also associated with brain metastasis [21]. In experimental models, brain-seeking metastatic variants but not the variants that metastasize to other organs have the ability to establish a unique pattern of vascularization required for growth [22]. However, gene expression changes in brain metastatic cells as an adaptive response in the brain microenvironment are just beginning to get attention.
To begin to address these complexities, we compared gene expression patterns in cancer cells isolated from a brain metastasis with parental cells in culture, mammary fat pad tumor-derived cells, and cancer cells that have metastasized to lungs, bone, and the adrenal gland. We identified a set of genes that are upregulated only in brain metastatic cells compared with all other cell types. Several of these genes have neuronal function suggesting that these genes are “reactivated” in the metastatic cell to enable them to adapt to growth conditions in the brain and utilize neuronal signaling networks for their advantage. Comparison among cells isolated from different metastatic sites revealed significantly higher transcriptome changes in brain metastatic cancer cells and unique pathway alterations involved in drug detoxification. In general, metastasis, irrespective of organs of metastasis, was associated with gain of gene expression suggesting that hyper-activation of general transcriptional machinery is a contributing factor of metastasis.
Results
Brain metastatic variants of MDA-MB-231 (231-BR) cells expressed a unique set of genes compared with parental cells, mammary fat pad tumor, or variants from other organs of metastasis
We recently reported an organ-specific metastasis model of MDA-MB-231 cells that included establishing cell lines from metastases in the lung, the bone, and the adrenal gland [23]. The same cell line model has been used to develop brain metastasis variants [24]. Using these cell lines, we had demonstrated upregulation of 20 and downregulation of seven microRNAs in metastatic cancer cells compared with mammary fat pad tumor cells [23]. We subjected parental MDA-MB-231 cells from two labs (one from us used for developing tumor and metastatic variants except brain metastasis and the other used for developing brain metastatic cells- these cells are labeled MD-231P), mammary fat pad tumor derived cell line (TMD-231), lung metastasis (LMD-231), bone metastasis (BMD-231), adrenal metastasis (ADMD-231), and brain metastasis (231-BR) to microarray mRNA expression analysis. For the different sets of cell lines, we used PAM [25] to identify signature genes for a specific metastasis site compared with all other sites. PAM classifier is based on the nearest shrunken centroid algorithm and identifies signature genes based on the variability of genes in a group. Using this method, we compared each metastatic site’s gene expression profile to all other metastatic expression profiles, tumor-derived cells, and parental cells to compile a set of genes constituting a signature for that metastatic site only. This stringent analysis generated signatures that were unique to brain and adrenal metastasis (Table S1). However, lung and bone metastasis signatures were not as statistically robust as brain and adrenal signatures and demonstrated a higher error rate (Table S1).
231-BR cells showed upregulation of 396 genes and downregulation of 77 genes compared with all other cell types (p < 0.01) (Table S2). In general, metastatic cells showed a higher number of upregulated genes compared with MDA-MB-231 or TMD-231 cells suggesting that acquiring new gene expression rather than loss of gene expression is associated with metastasis (Table S2, see rows 1 and 2). The top 25-upregulated genes in 231-BR cells are shown in Table 1. Several genes in this table (indicated in bold) are linked to neuronal activity. For example, translation elongation factor eEF1A2 variant is expressed in a restricted pattern compared with ubiquitously expressed eEF1A1, and the expression is dominant in adult brain [26]. TMEM47 and SLITRK2 are linked to neuronal development and/or brain tumors [27, 28]. TMEM47 is also called brain cell membrane protein 1 and is related to claudins [29]. Thus, upregulation of genes linked to neuronal function in 231-BR cells support the hypothesis that cancer cells acquire their expression to adapt to the brain microenvironment. We confirmed specific upregulation of LYPD1, TMEM47, and SLITRK2 in 231-BR cells compared with other cell types, as these genes are not part of any previously described brain metastatic signatures (Figure 1A). ESM1, which is upregulated in all metastatic cell types compared with parental or TMD-231 cells in the microarray assay, also showed higher expression levels in LMD, BMD, ADMD-231 cells and 231-BR cells compared with parental or TMD-231 cells by qRT-PCR (Figure 1A). Results of technical replicates are shown because of wide variation in fold induction between experiments. For example, the level of TMEM47 was higher in 231-BR by 193-, 92-, 153-, 891- and 631-fold compared with MD-231P cells in five experiments. Similarly, LYPD1 levels were higher by 19-, 49-, 6-, 49- and 44-fold in 231-BR cells compared with MD-231P cells in five experiments (see also Figure 5 below for statistical analysis).
Table 1: Genes overexpressed (>2 fold, p < 0.0002) in 231-BR compared with cells metastasized to other organs. Genes shown in bold have neuronal functions.
Genes |
p value |
Fold change 231-BR vs. other metastasis |
TMEM47 |
6.83E-11 |
8.92 |
LYPD1 |
9.89E-12 |
6.18 |
CD96 |
8.2E-06 |
5.89 |
TFAP2C |
3.65E-06 |
5.61 |
SLITRK2 |
4.37E-08 |
5.58 |
LOC10013407 |
2.58E-10 |
3.56 |
EEF1A2 |
4.40E-05 |
3.49 |
SHISA2 |
9.84E-06 |
3.48 |
AKR1C3 |
9.67E-05 |
3.23 |
MYH10 |
2.36E-05 |
2.99 |
HOXB5 |
3.06E-06 |
2.98 |
NINJ2 |
3.70E-05 |
2.95 |
Hs.580229 |
1.52E-08 |
2.82 |
SERPINF1 |
3.07E-07 |
2.80 |
CPE |
8.68E-05 |
2.76 |
MAGEE1 |
4.11E-06 |
2.72 |
GZMA |
0.000157 |
2.53 |
RPPH1 |
8.27E-06 |
2.51 |
C17ORF70 |
5.68E-06 |
2.50 |
C8ORF13 |
0.00013 |
2.31 |
ZNF204 |
0.000183 |
2.28 |
SPIN4 |
2.28E-05 |
2.27 |
CLGN |
9.05E-07 |
2.16 |
NCKAP1L |
1.86E-09 |
2.14 |
TIE1 |
8.02E-06 |
2.08 |
LYPD1, TMEM47, and SLITRK2 are transmembrane proteins likely involved in ligand-dependent signal transduction. SLITRK2 family genes, including SLITRK2, are expressed predominantly in brain [28]. These genes likely play critical roles in cancer progression because cBioPortal analysis revealed amplification and /or mutations of TMEM47, LYPD1 and SLITRK2 in a variety of cancers including breast cancer [30]. Four percent of patient-derived breast cancer xenografts in the cBioPortal show amplification of LYPD1 and mutation in SLITRK2. However, there are limited reports on the function of these proteins. To gain insight into their function, we analyzed the STRING database to identify potential interacting partners [31]. While no proteins interacting with TMEM47 were found, LYPD1 and SLITRK2 appear to be involved in various signaling including cell adhesion and neurotransmitter signaling (Figure 1B). For example, GPR39, a G-protein-coupled receptor expressed mostly in brain, is at the top of the list of LYPD1 interacting partners [32]. Neuronal pentraxin-1 is the major interacting partner of SLITRK2, which mediates synaptic remodeling [33]. Future studies will determine the critical role played by these proteins in adaptation of brain metastatic cells to the brain microenvironment.
We created another brain metastatic variant from BMD-231 cells. A nude mice injected with BMD-231 cells via intra-cardiac route developed brain metastasis and metastatic cells were established in culture. These cells, called 231-BR-2, overexpressed TMEM47 but not other genes tested compared with BMD-231 cells (Figure 1C). We also observed elevated TMEM47 expression in brain metastatic variant of MCF-7 cells overexpressing HER2 (MCF-7HER2-BR) compared with parental MCF-7 cells overexpressing HER2 oncogene (Figure 1D) [34]. Thus, TMEM47 is a new brain metastasis-associated gene. Please note that CT values of SLITRK2 expression in 231-BR-2 and MCF-7HER2-BR cells were above 30 and thus are not reliable.
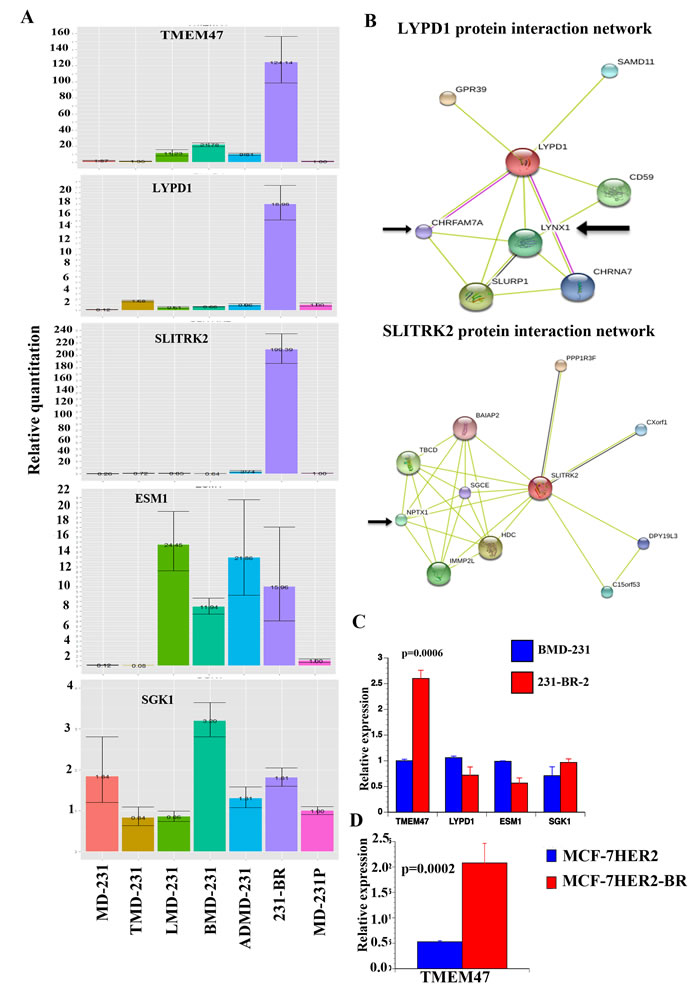
Figure 1: Validation of genes differentially expressed in brain metastatic cells. A) qRT-PCR analysis of select genes in parental, tumor-derived, and organ-specific metastatic cells. β-actin was used as a normalization control. B) Protein-protein interaction network of two genes expressed preferentially in 231-BR cells. Data were generated using STRING network [31]. Arrow indicates proteins involved in neuronal signaling. C) TMEM47 expression is elevated in another brain-metastasis variant of MDA-MB-231 cells. This variant was derived from BMD-231 cells. D) TMEM47 expression in MCF-7HER2 and its brain metastatic variant.
Analysis of a public database [35], which contains gene expression data in on primary tumors but not metastases, for the prognostic value of combined expression of the top 17 genes overexpressed (> 2 fold, p<0.0001, TMEM47, LYPD1, CD96, TFAP2C, EEF1A2, DDX, MYH10, HOXB5, NINJ2, SERPINF1, CPE, MAGEC2, CTLA3, C17orf70, ZNF704, NCKAP1L and TIE1) in 231-BR cells for which data were available showed elevated expression correlating with poor recurrence-free survival of patients with basal or luminal B breast cancer (Figure 2A and 2B). With respect to brain metastasis-free survival, overexpression of TMEM47 was associated with poor brain metastasis-free survival [20] (Figure 2C). TMEM47 displayed prognostic value in estrogen receptor and progesterone negative but not in estrogen receptor and progesterone receptor positive breast cancer (data not shown). LYPD1 and SLITRK2 did not show any significance.
We next determined whether the expression of LYPD1, TMEM47 and/or SLITRK2 is enriched in a specific intrinsic subtype of breast cancer. Three public datasets were analyzed (GSE2607, GSE10886, and GSE19783) [36-38]. LYPD1 but not SLITRK2 showed a trend of elevated expression in basal subtype but differences reached statistical significance only in the GSE19783 dataset (Figure S1 and Figure S2). In this dataset, TMEM47 also showed elevated expression in Basal and HER2+ breast cancers compared with luminal A and B breast cancers. Thus, overexpression of these genes may not be unique to a subtype of breast cancer. Alternatively, the brain microenvironment influences the expression of these genes in metastatic cells irrespective of the subtype.
Since no adrenal metastasis signature has been defined so far, we examined 10 genes overexpressed in ADMD-231 (>1.5 fold, p < 0.01) compared with other metastases for prognostic relevance. Genes were selected based on the availability of data in the public database and included CYB5R2, TAGLN, HAND1, RAB3IL1, TRMT12, TSPAN8, MMP3, STXBP6, AP1S2, and HSPB8 [35]. Overexpression of these genes was associated with poor recurrence-free survival and distant metastasis-free in basal breast cancer (Figure 2D and 2E). Please note that these genes did not show prognostic relevance in other intrinsic subtypes of breast cancer.
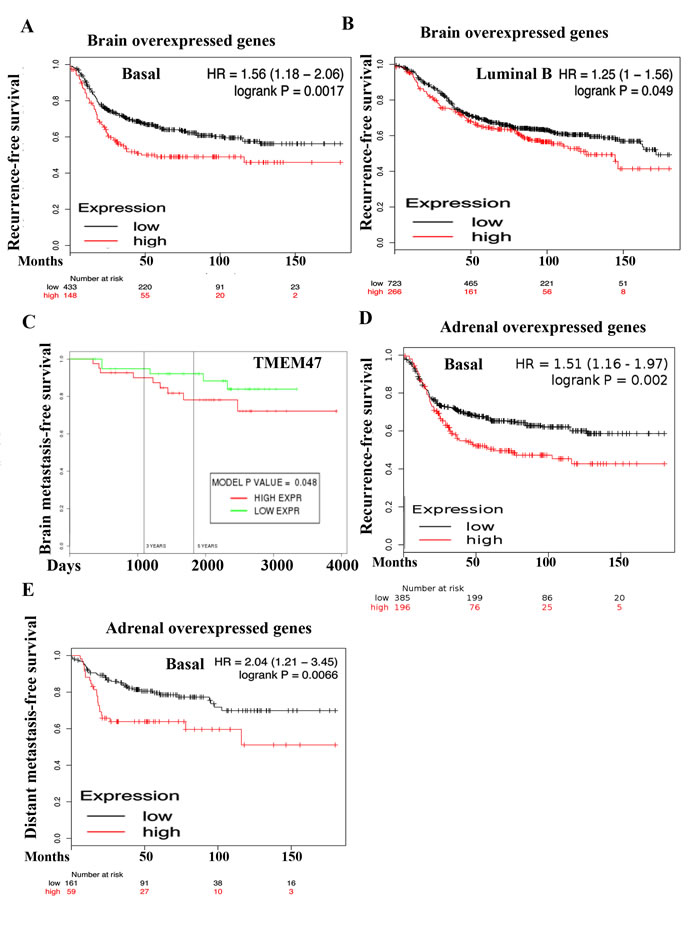
Figure 2: Prognostic value of genes overexpressed in 231-BR and ADMD-231 cells. A) Elevated expression of 231-BR overexpressed genes (TMEM47, LYPD1, CD96, TFAP2C, EEF1A2, DDX, MYH10, HOXB5, NINJ2, SERPINF1, CPE, MAGEC2, CTLA3, C17orf70, ZNF704, NCKAP1L, and TIE1) in primary breast tumor is associated with poor recurrence-free survival among patients with basal breast cancer. Patients were split by median to classify into high or low expressers. B) Elevated expression of 231-BR specific genes in luminal B breast cancer is also associated with poor recurrence-free survival. C) TMEM47 overexpression is associated with poor brain metastasis-free survival. D) Elevated expression of ADMD-231 overexpressed genes (CYB5R2, TAGLN, HAND1, RAB3IL1, TRMT12, TSPAN8, MMP3, STXBP6, AP1S2, and HSPB8) in primary tumor is associated poor recurrence-free survival among patients with basal breast cancer. E) ADMD-231 overexpressed genes are also associated with poor distant metastasis-free survival among patients with basal breast cancer.
Genes differentially expressed in organ-specific metastatic cells were linked to unique and shared signaling networks
To determine the signaling pathways active in cells isolated from different sites of metastasis, we subjected differentially expressed genes to Ingenuity pathway analysis. Glutathione-mediated detoxification, NRF2-mediated oxidative stress response, and Semaphorin signaling in neurons are a few of the signaling pathways in 231-BR cells (Figure 3A). The top two networks in 231-BR cells included SRC-ERK-growth hormone and NF-κB (Figure 3B and 3C). SRC pathway activation has also been noted previously in the BT474 HER2-positive cell brain metastasis model [18]. Notch, LXR/RXR and FXR/RXR pathways are the three major pathways activated in ADMD-231 cells (Figure 4A). Networks in these cells included Notch-ERK-AKT and NF-κB (Figure 4B and 4C).
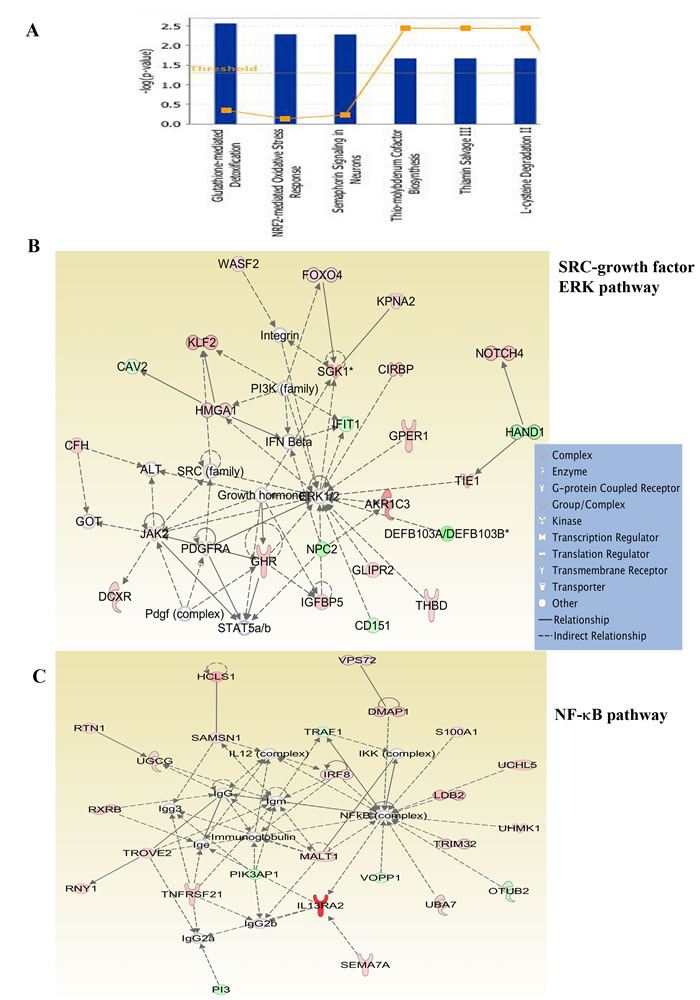
Figure 3: Ingenuity pathway analysis of genes differentially expressed in 231-BR cells. A) Major signaling pathways in 231-BR cells. B) 231-BR cells show activation of SRC-ERK-growth hormone network. C) NF-κB signaling network is active in 231-BR cells. Genes labeled in red are overexpressed, whereas genes in green are expressed at lower levels in 231-BR cells compared with other metastatic cells.
LMD-231 cells showed activation of acute phase response signaling, primary immunodeficiency signaling, and glutamate receptor signaling (Figure S3A). Networks included TNF-CEBPA-p53 and NF-κB (Figure S3B and S3C). Involvement of CEBPA in the network is interesting because of its critical role in lung maturation [39]. BMD-231 cells displayed activation of cytokine signaling, hematopoiesis from pluripotent stem cells and JAK1/JAK3 cytokine signaling (Figure S4A). Signaling networks in these cells included ERK-growth hormone and TNF-p53 (Figure S4B and S4C). Activation of neuronal, orphan nuclear receptor, acute phase response, and cytokine signaling in brain, adrenal, lung, and bone metastatic cells, respectively, further suggests organ-specific adaptive response in metastatic cells.
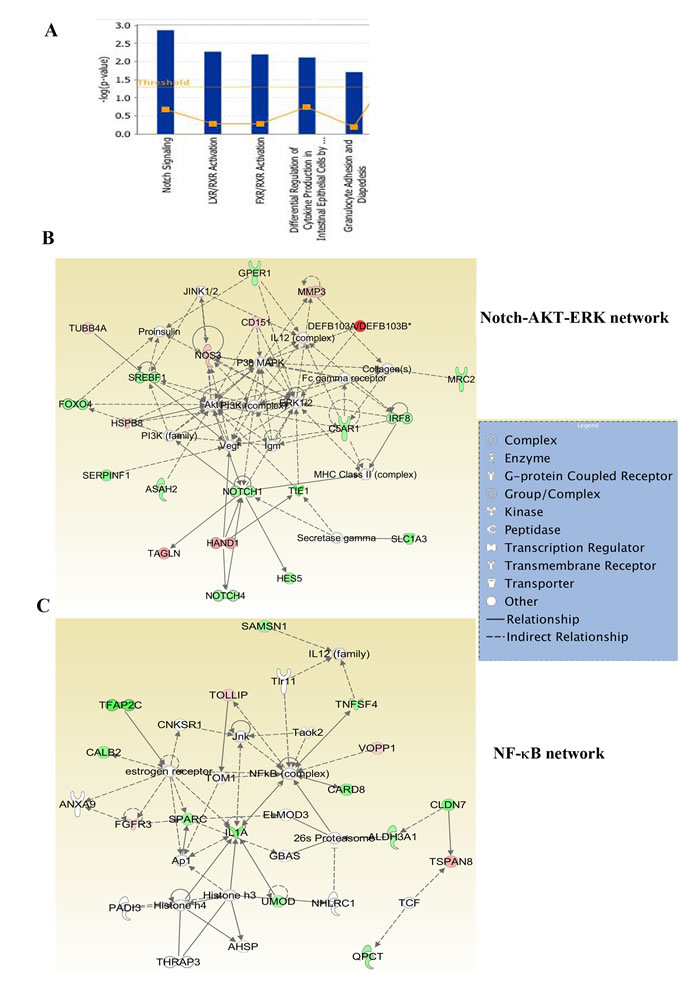
Figure 4: Ingenuity pathway analysis of genes differentially expressed in ADMD-231 cells. A) Notch, FXR/RXR and LXR/RXR networks involved in steroidogenesis similar to adrenal gland are the major pathways in ADMD-231 cells. B) ADMD-231 cells show activation of Notch-ERK-AKT network. C) NF-κB signaling network is active in ADMD-231 cells.
231-BR cells displayed elevated NF-κB DNA binding activity, which was sensitive to DMAPT
To extend the above observation from Ingenuity pathway analysis, we examined NF-κB DNA binding activity in MD-231P and 231-BR cells by electrophoretic mobility shift assays (EMSAs). As we reported previously [40], MD-231P cells displayed constitutive NF-κB DNA binding activity, which was further elevated in 231-BR cells (Figure 5A). NF-κB:DNA complex contained p65 and p50 subunits as per super-shift assay. We next examined the effects of netropsin, which inhibits NF-κB when DNA binding is dependent on HMGA2 [41], and DMAPT, a direct NF-κB inhibitor. DMAPT is a water-soluble parthenolide derivative and has been characterized for anti-tumor activity in vitro and in vivo [42-45]. While netropsin had minimum effect, DMAPT significantly reduced NF-κB DNA binding activity (Figure 5A). 231-BR cells expressed ~65-fold higher levels of CXCL1, an NF-κB inducible chemokine involved in metastasis [46], compared with MD-231P cells, which was reduced by DMAPT (Figure 5B). DMAPT reduced the expression levels of LYPD1 (from 50 –fold to 10 fold) but not TMEM47 suggesting that NF-κB controls the expression of select genes of the brain metastasis signature (Figure 5B). In cell proliferation assays, while both MD-231P and 231-BR cells were sensitive to DMAPT, the concentration of drug required to inhibit 231-BR cells was lower than that for MD-231P cells (p = 0.0001) suggesting that 231-BR cells are dependent on NF-κB for survival (Figure 5C).
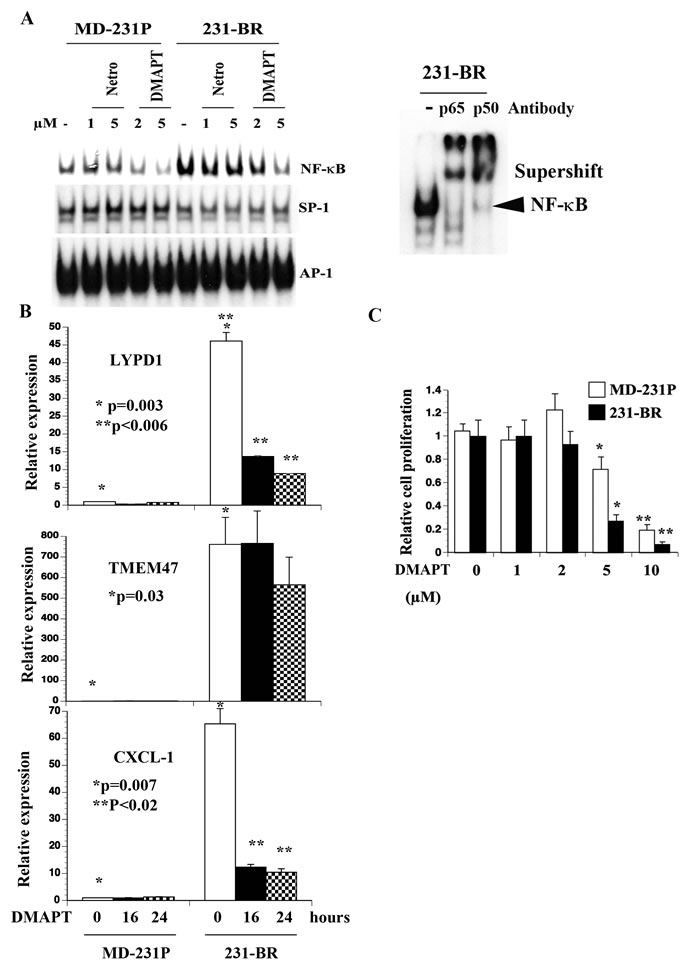
Figure 5: Elevated NF-κB activity in 231-BR cells compared with parental cells. A) DMAPT but not netropsin (netro) inhibited NF-κB DNA binding activity in 231-BR cells. Supershift assays showed p50:p65 NF-κB complex in 231-BR cells. B) DMAPT (10 µM) reduced CXCL1 and LYPD1 but not TMEM47 expression. qRT-PCR was performed to measure mRNA levels. * P values MD-231P versus 231-BR; ** P values untreated 231-BR versus DMAPT-treated 231-BR cells. C) DMAPT inhibited proliferation of 231-BR cells.
4T1-BR cells showed elevated NF-κB compared with 4T1 cells and were sensitive to DMAPT
To determine whether elevated NF-κB activity is observed in additional brain metastasis models, we compared NF-κB in parental 4T1 and a brain-seeking variant of this cell line [45]. 4T1 cells are derived from a spontaneous mammary tumor in BALB/c mice and form highly metastatic tumors upon mammary fat pad injection in syngeneic mice [47]. NF-κB DNA binding activity was elevated in 4T1-BR cells compared with parental 4T1 cells and DMAPT reduced this binding activity (Figure 6A). Note that AP-1 DNA binding activity was lower in 4T1-BR cells compared with 4T1 cells suggesting transcription factor switch with specific upregulation of NF-κB in brain metastatic cells. Unlike 231-BR cells, 4T1-BR cells and parental 4T1 cells were similarly sensitive to DMAPT (Figure 6B). Thus, brain metastatic cells in both model systems show elevated NF-κB activity and can potentially be targeted by NF-κB inhibitors.
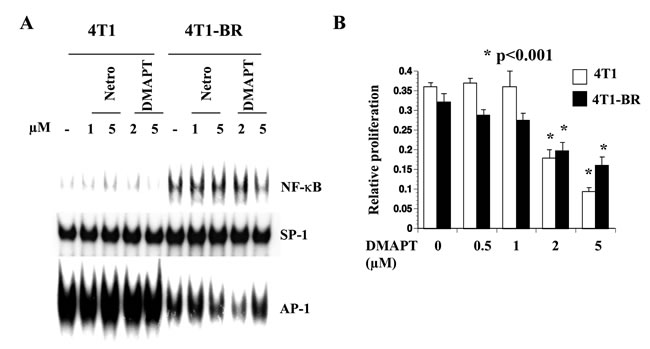
Figure 6: 4T1-BR cells displayed elevated NF-κB activity compared with 4T1 cells. A) DMAPT inhibited NF-κB activity in 4T1-BR cells. Note lower AP-1 DNA binding activity in 4T1-BR cells compared with 4T1 cells. B) DMAPT inhibited 4T1-BR cell proliferation.
Discussion
There have been several attempts to identify genes associated with brain metastasis and to functionally validate these genes for imparting blood brain barrier extravasation, vascular co-adaptation, interaction with brain microenvironment, and cell survival function. Using brain metastatic cell lines derived from four different models, Valiente et al. showed upregulation of seven genes in three out of four models [16]. The authors then focused on SERPIN1 and SERPINB2 and demonstrated their role in establishing vascular adaptation in brain [16]. Although SERPIN1 was not one of the upregulated genes in 231-BR cells (1.22-fold increase but p=0.18), significant upregulation of SERPINB2 and SERPINIF1, a serpine family member without protease inhibitory activity but with neurotropic activity [48], was observed in 213-BR cells compared with other metastatic cells (Table S2). Among the other six genes (CTCF, DUSP1, GALC, HIST1HIC, LEF1, and PCDH7), we found upregulation of DUSP1 and GALC in all metastatic cells compared with parental cells, irrespective of sites of metastasis. Among the recently described DNA repair genes upregulated in brain metastatic cells [49], we noted upregulation of RAD51 (1.1-fold, p = 0.03) and RAD51C (1.4 fold, p = 0.016) but not BARD1 in 231-BR cells compared with other metastatic cells (Table S2). However, 231-BR cells did not show specific changes in the expression levels of the recently described BRCA1 deficient-like gene signature enriched in the brain metastasis of HER2+ breast cancer patients [50, 51]. Nonetheless, four among 13 genes of this signature (NDRG1, CCND1, BOP1, and Myc) were upregulated in metastatic cells irrespective of sites of metastasis compared with parental or TMD-231 cells (Table S2). Similarly, we did not find any overlap between the brain metastasis signatures described in our study and the signature described by Salhia et al. [52]. However, the CRYAB gene, which was downregulated in the brain metastasis in the study described by Salhia et al was also downregulated in all metastatic cells in our analysis. Differences in the types of comparison adapted in different studies may partly be responsible for minimum overlap in genes between signatures. For example, our evaluation involved comparison between parental and metastasis as well as between organ-specific metastatic cells whereas other studies compared brain metastasis with only primary tumor.
Glutathione-mediated detoxification, NRF2-mediated oxidative response, and Semaphorin signaling pathways activated in brain metastatic cells suggest a unique biology of these metastatic cancer cells and potentially explain their relative resistance to standard chemotherapy and possibly challenges the widely held belief that poor BBB permeability of chemotherapeutic drugs is the main reason for treatment failure. Inherent ability to detoxify these drugs may be one of the main reasons for treatment failure. In this respect, a recent study has shown that physical interaction between cancer cells and astrocytes leads to upregulation of glutathione transferase 5A, which contributes to drug resistance [53]. DMAPT, the NF-κB inhibitor tested in this study, has previously been shown to deplete glutathione and cause the death of leukemic cells [54]. Thus, sensitivity of 231-BR cells to DMAPT could be related to their dependency on glutathione and the NF-κB signaling network and the ability of DMAPT to inhibit both pathways. We also noted activation of a signaling network involving SRC kinases in 231-BR cells (Figure 3B), which has recently been suggested to be a therapeutic target for brain metastasis [18].
Although there have been large efforts in defining gene expression signatures for bone and lung metastases [55-57], an adrenal gland metastasis signature is yet to be described. Minn et al. described MDA-MB-231 variants that metastasize to both lungs and adrenal or bone and adrenal but did not define an adrenal-specific gene expression signature [58]. This study, to our knowledge, describes the first adrenal metastatic signature for breast cancer. Ten genes, which are expressed 1.5-fold (p<0.01) higher in ADMD-231 compared with other metastatic cells, displayed prognostic significance in basal breast cancer but not in other subtypes (Figure 2). There is limited literature on the consequences of adrenal metastasis, although a rare case of adrenal failure due to adrenal metastasis in breast cancer has been reported [59]. Transcriptome changes associated with adrenal metastasis were significantly higher compared with bone or lung metastasis (p = 0.0001, Chi-square with Yates correction) suggesting the need for substantial genomic changes to achieve this metastasis. Notch and orphan nuclear receptor signaling were dominant in adrenal metastatic cells. The Notch pathway plays a major role in adrenal gland development, whereas LXR signaling controls steroidogenic pathways in the adrenal gland [60]. Thus, it is likely that cancer cells that have metastasized to the adrenal gland undergo genomic changes that help them to adapt to the adrenal gland or primary tumor cells with these pathways activation metastasize preferentially to the adrenal gland.
The majority of studies on breast cancer metastasis utilized MDA-MB-231 cells. This cell line represents the claudin-low/mesenchymal subtype, which overexpresses stem cell-enriched genes [61] and has a natural tendency to metastasize to brain and lungs. Brain and lung metastatic signatures developed using this cell line have shown clinical utility [62]. Several of the genes that were part of the original bone metastatic signature developed using this cell line, including CXCR4, CTGF, and MMP1, were part of the general metastatic signature irrespective of organ-specificity (Table S1) [55]. Because prior knowledge exists on a lung metastatic signature, we did not perform an extensive analysis. However, identification of acute phase response and glutamate signaling networks in lung metastatic cells suggests an adaptive response of these cells to lung.
In summation, our results indicate that organ-specific metastatic cells acquire the ability to adapt to sites of metastasis, which may involve genomic or epigenomic changes. We observed general upregulation of transcription in metastatic cells compared with parental cells suggesting that genomic aberrations leading to enhanced RNA polymerase II activity are sufficient for metastasis. Alternatively, genomic/epigenomic changes in a few primary tumor cells may activate organ-specific adaptive gene networks prior to their exit from the primary site. In this respect, patients with primary tumors that overexpressed brain or adrenal metastasis signature genes had poor outcome (Figure 2). While inhibitors of NF-κB signaling can be potential chemosensitizers of the majority of metastases as we have demonstrated previously with lung metastasis [44], organ-specific signaling networks identified in this study can be used to develop drugs targeting specific sites of metastasis to limit toxicity. In addition, our study identified TMEM47, a transmembrane protein with little known biology, as a brain metastasis associated gene. TMEM47 could potentially be developed as a biomarker and targeted therapeutically based on its cellular localization at the membrane.
Materials and Methods
Cell lines
MDA-MB-231 and its mammary fat pad tumor and metastatic derivatives have been described [23]. 231-BR and its parental counter part MD-231P, 4T1 and 4T1-BR cells have been described [49, 63]. We also generated a new brain metastatic variant (231-BR-2) from a nude mouse that developed brain metastasis after intra-cardiac injection of bone metastatic variant (BMD-231). Despite the potential for genomic drift during growth within animals and/or during culturing after isolation from sites of metastasis, cell line identification using short tandem repeat profiling (DNA Diagnosis Center, Fairfield, Ohio, USA and Genetica DNA Laboratories, Cincinnati, Ohio, USA) confirmed that all variants are genomically similar to parental MDA-MB-231 cells. MDA-MB-231 and its derivatives were maintained in MEM plus 10% FBS, whereas 4T1 and 4T1-BR cells were maintained in DMEM plus 10% FBS.
RNA preparation, microarray and quantitative reverse transcription polymerase chain reaction (qRT-PCR)
RNA was prepared using RNeasy kit (Qiagen, Valencia, CA, USA) and cDNA from four µgs of RNA was synthesized using the cDNA Synthesis kit (Bio-Rad, Hercules, CA, USA). qRT-PCR was performed using SyberGreen on a TaqMan 7900HT instrument (Applied Biosystems, Carlsbad, CA, USA). Sequences of primers used for qRT-PCR are listed in the supplementary Table S3. Illumina HumanHT-12 V4 expression beadchip was used for microarray analysis of biological triplicates. Genes that showed insignificant signals in a majority of samples were removed and only those probes that showed statistically significant signal in at least half of samples of at least one group were retained. The probe level data were then collapsed to gene level data by retaining only the probes, which showed a maximal coefficient of variation across all samples. The data were imported into Partek genomics suite for differential expression analysis. ANOVA analysis was performed to identify genes differentially expressed between different cell lines. Genes differentially expressed at p value of <0.01 were considered further for pathway analysis using Ingenuity pathway analysis software (© Ingenuity systems, CA, USA). Prognostic relevance of overexpressed genes was determined using a public database as well as a new database created by us [35, 64]. Microarray dataset is available in the Gene Expression Omnibus (GSE66495).
Generation of metastasis signatures
Predictive analysis of Microarray (PAM) [25] was used to identify signature genes capable of discriminating phenotypes with minimal classification error. PAM analysis was performed on a 1 X Rest basis comparing one phenotype with all other phenotypes to identify a group of genes that could differentiate that phenotype from the rest of phenotypes arriving at gene signatures specific for each phenotypic group. Thresholds for PAM analysis were adjusted to identify the smallest possible gene signature with minimal misclassification error rate.
Electrophoretic Mobility Shift Assays (EMSA) and cell proliferation studies
EMSA with whole cell lysates was performed as described previously [65]. Probes for the assay were purchased from Promega (Madison, WI, USA), whereas antibodies for supershift were from EMD Millipore (Billerica, MA, USA). For cell proliferation studies, 1000 cells/well were plated on a 96-well plate and cells were treated with DMAPT (eight wells per drug and highest and lowest numbers were discarded during analysis). Cell proliferation was measured 48 hours after drug treatment using Bromodeoxyuridine-ELISA (Calbiochem/ EMD Millipore). Results are from two or more independent experiments.
Statistical analysis
Graphpad Prism (Graphpad.com) was used for statistical analysis of qRT-PCR and cell proliferation assay. Analysis of variance was used to determine the P-values between mean measurements. A P-value of <0.05 was deemed significant.
AcknowledgmentS
We thank Dr. Patricia Steeg, National Cancer Institute, USA, for MD-231P, 231-BR, 4T1 and 4T1-BR cells. This work was supported by grants from Susan G. Komen for the Cure (SAC110025), Zeta Tau Sorority, 100 Voices of Hope, and IUPUI Signature Center to HN. RMB was supported by the National Institutes of Health training grant CA111198. The IU Medical Scientist Training Program Grant 5T32GM77229-5 supported KEC.
ConflictS of Interest
PC, WPM and HN are co-founders of Leuchemix, Inc, which is developing DMAPT as cancer therapeutics. Other authors have no conflict of interest to declare.
References
1. Lin NU, Amiri-Kordestani L, Palmieri D, Liewehr DJ, Steeg PS: CNS metastases in breast cancer: old challenge, new frontiers. Clin Cancer Res 2013, 19:6404-6418.
2. Tsukada Y, Fouad A, Pickren JW, Lane WW: Central nervous system metastasis from breast carcinoma. Autopsy study. Cancer 1983, 52:2349-2354.
3. Flowers A, Levin VA: Management of brain metastases from breast carcinoma. Oncology (Williston Park) 1993, 7:21-26; discussion 31-24.
4. Steeg PS, Camphausen KA, Smith QR: Brain metastases as preventive and therapeutic targets. Nat Rev Cancer, 11:352-363.
5. Engel J, Eckel R, Aydemir U, Aydemir S, Kerr J, Schlesinger-Raab A, Dirschedl P, Holzel D: Determinants and prognoses of locoregional and distant progression in breast cancer. Int J Radiat Oncol Biol Phys 2003, 55:1186-1195.
6. Mahmoud-Ahmed AS, Suh JH, Lee SY, Crownover RL, Barnett GH: Results of whole brain radiotherapy in patients with brain metastases from breast cancer: a retrospective study. Int J Radiat Oncol Biol Phys 2002, 54:810-817.
7. Pestalozzi BC, Zahrieh D, Price KN, Holmberg SB, Lindtner J, Collins J, Crivellari D, Fey MF, Murray E, Pagani O, et al: Identifying breast cancer patients at risk for Central Nervous System (CNS) metastases in trials of the International Breast Cancer Study Group (IBCSG). Ann Oncol 2006, 17:935-944.
8. Kallioniemi OP, Holli K, Visakorpi T, Koivula T, Helin HH, Isola JJ: Association of c-erbB-2 protein over-expression with high rate of cell proliferation, increased risk of visceral metastasis and poor long-term survival in breast cancer. Int J Cancer 1991, 49:650-655.
9. Weil RJ, Palmieri DC, Bronder JL, Stark AM, Steeg PS: Breast cancer metastasis to the central nervous system. Am J Pathol 2005, 167:913-920.
10. Duchnowska R, Dziadziuszko R, Czartoryska-Arlukowicz B, Radecka B, Szostakiewicz B, Sosinska-Mielcarek K, Karpinska A, Staroslawska E, Kubiatowski T, Szczylik C: Risk factors for brain relapse in HER2-positive metastatic breast cancer patients. Breast Cancer Res Treat 2009, 117:297-303.
11. Miller KD, Weathers T, Haney LG, Timmerman R, Dickler M, Shen J, Sledge GW, Jr.: Occult central nervous system involvement in patients with metastatic breast cancer: prevalence, predictive factors and impact on overall survival. Ann Oncol 2003, 14:1072-1077.
12. Hicks DG, Short SM, Prescott NL, Tarr SM, Coleman KA, Yoder BJ, Crowe JP, Choueiri TK, Dawson AE, Budd GT, et al: Breast cancers with brain metastases are more likely to be estrogen receptor negative, express the basal cytokeratin CK5/6, and overexpress HER2 or EGFR. Am J Surg Pathol 2006, 30:1097-1104.
13. Nam BH, Kim SY, Han HS, Kwon Y, Lee KS, Kim TH, Ro J: Breast cancer subtypes and survival in patients with brain metastases. Breast Cancer Res 2008, 10:R20.
14. Ding L, Ellis MJ, Li S, Larson DE, Chen K, Wallis JW, Harris CC, McLellan MD, Fulton RS, Fulton LL, et al: Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature 2010, 464:999-1005.
15. Zhang L, Ridgway LD, Wetzel MD, Ngo J, Yin W, Kumar D, Goodman JC, Groves MD, Marchetti D: The identification and characterization of breast cancer CTCs competent for brain metastasis. Sci Transl Med 2013, 5:180ra148.
16. Valiente M, Obenauf AC, Jin X, Chen Q, Zhang XH, Lee DJ, Chaft JE, Kris MG, Huse JT, Brogi E, Massague J: Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell 2014, 156:1002-1016.
17. Maher EA, Mietz J, Arteaga CL, DePinho RA, Mohla S: Brain metastasis: opportunities in basic and translational research. Cancer Res 2009, 69:6015-6020.
18. Zhang S, Huang WC, Zhang L, Zhang C, Lowery FJ, Ding Z, Guo H, Wang H, Huang S, Sahin AA, et al: SRC family kinases as novel therapeutic targets to treat breast cancer brain metastases. Cancer Res 2013, 73:5764-5774.
19. Kodack DP, Chung E, Yamashita H, Incio J, Duyverman AM, Song Y, Farrar CT, Huang Y, Ager E, Kamoun W, et al: Combined targeting of HER2 and VEGFR2 for effective treatment of HER2-amplified breast cancer brain metastases. Proc Natl Acad Sci U S A 2012, 109:E3119-3127.
20. Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, Minn AJ, van de Vijver MJ, Gerald WL, Foekens JA, Massague J: Genes that mediate breast cancer metastasis to the brain. Nature 2009, 459:1005-1009.
21. Kim MY, Oskarsson T, Acharyya S, Nguyen DX, Zhang XH, Norton L, Massague J: Tumor self-seeding by circulating cancer cells. Cell 2009, 139:1315-1326.
22. Fidler IJ, Yano S, Zhang RD, Fujimaki T, Bucana CD: The seed and soil hypothesis: vascularisation and brain metastases. Lancet Oncol 2002, 3:53-57.
23. Patel JB, Appaiah HN, Burnett RM, Bhat-Nakshatri P, Wang G, Mehta R, Badve S, Thomson MJ, Hammond S, Steeg P, et al: Control of EVI-1 oncogene expression in metastatic breast cancer cells through microRNA miR-22. Oncogene 2011, 30:1290-1301.
24. Gril B, Palmieri D, Bronder JL, Herring JM, Vega-Valle E, Feigenbaum L, Liewehr DJ, Steinberg SM, Merino MJ, Rubin SD, Steeg PS: Effect of lapatinib on the outgrowth of metastatic breast cancer cells to the brain. J Natl Cancer Inst 2008, 100:1092-1103.
25. Tibshirani R, Hastie T, Narasimhan B, Chu G: Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc Natl Acad Sci U S A 2002, 99:6567-6572.
26. Pan J, Ruest LB, Xu S, Wang E: Immuno-characterization of the switch of peptide elongation factors eEF1A-1/EF-1alpha and eEF1A-2/S1 in the central nervous system during mouse development. Brain Res Dev Brain Res 2004, 149:1-8.
27. Utami KH, Hillmer AM, Aksoy I, Chew EG, Teo AS, Zhang Z, Lee CW, Chen PJ, Seng CC, Ariyaratne PN, et al: Detection of chromosomal breakpoints in patients with developmental delay and speech disorders. PLoS One 2014, 9:e90852.
28. Aruga J, Yokota N, Mikoshiba K: Human SLITRK family genes: genomic organization and expression profiling in normal brain and brain tumor tissue. Gene 2003, 315:87-94.
29. Christophe-Hobertus C, Szpirer C, Guyon R, Christophe D: Identification of the gene encoding Brain Cell Membrane Protein 1 (BCMP1), a putative four-transmembrane protein distantly related to the Peripheral Myelin Protein 22 / Epithelial Membrane Proteins and the Claudins. BMC Genomics 2001, 2:3.
30. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al: Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013, 6:pl1.
31. Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C, Jensen LJ: STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res 2013, 41:D808-815.
32. Popovics P, Stewart AJ: GPR39: a Zn(2+)-activated G protein-coupled receptor that regulates pancreatic, gastrointestinal and neuronal functions. Cell Mol Life Sci 2011, 68:85-95.
33. Sia GM, Beique JC, Rumbaugh G, Cho R, Worley PF, Huganir RL: Interaction of the N-terminal domain of the AMPA receptor GluR4 subunit with the neuronal pentraxin NP1 mediates GluR4 synaptic recruitment. Neuron 2007, 55:87-102.
34. Gril B, Palmieri D, Qian Y, Smart D, Ileva L, Liewehr DJ, Steinberg SM, Steeg PS: Pazopanib reveals a role for tumor cell B-Raf in the prevention of HER2+ breast cancer brain metastasis. Clin Cancer Res 2011, 17:142-153.
35. Gyorffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, Szallasi Z: An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat 2010, 123:725-731.
36. Perreard L, Fan C, Quackenbush JF, Mullins M, Gauthier NP, Nelson E, Mone M, Hansen H, Buys SS, Rasmussen K, et al: Classification and risk stratification of invasive breast carcinomas using a real-time quantitative RT-PCR assay. Breast Cancer Res 2006, 8:R23.
37. Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, Davies S, Fauron C, He X, Hu Z, et al: Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 2009, 27:1160-1167.
38. Enerly E, Steinfeld I, Kleivi K, Leivonen SK, Aure MR, Russnes HG, Ronneberg JA, Johnsen H, Navon R, Rodland E, et al: miRNA-mRNA integrated analysis reveals roles for miRNAs in primary breast tumors. PLoS One 2011, 6:e16915.
39. Martis PC, Whitsett JA, Xu Y, Perl AK, Wan H, Ikegami M: C/EBPalpha is required for lung maturation at birth. Development 2006, 133:1155-1164.
40. Nakshatri H, Bhat-Nakshatri P, Martin DA, Goulet RJ, Jr., Sledge GW, Jr.: Constitutive activation of NF-kappaB during progression of breast cancer to hormone-independent growth. Mol Cell Biol 1997, 17:3629-3639.
41. Miao Y, Cui T, Leng F, Wilson WD: Inhibition of high-mobility-group A2 protein binding to DNA by netropsin: a biosensor-surface plasmon resonance assay. Anal Biochem 2008, 374:7-15.
42. Shanmugam R, Kusumanchi P, Appaiah H, Cheng L, Crooks P, Neelakantan S, Peat T, Klaunig J, Matthews W, Nakshatri H, Sweeney CJ: A water soluble parthenolide analog suppresses in vivo tumor growth of two tobacco-associated cancers, lung and bladder cancer, by targeting NF-kappaB and generating reactive oxygen species. Int J Cancer 2011, 128:2481-2494.
43. Shanmugam R, Kusumanchi P, Cheng L, Crooks P, Neelakantan S, Matthews W, Nakshatri H, Sweeney CJ: A water-soluble parthenolide analogue suppresses in vivo prostate cancer growth by targeting NFkappaB and generating reactive oxygen species. Prostate 2010, 70:1074-1086.
44. Sweeney CJ, Mehrotra S, Sadaria MR, Kumar S, Shortle NH, Roman Y, Sheridan C, Campbell RA, Murry DJ, Badve S, Nakshatri H: The sesquiterpene lactone parthenolide in combination with docetaxel reduces metastasis and improves survival in a xenograft model of breast cancer. Mol Cancer Ther 2005, 4:1004-1012.
45. Lockman PR, Mittapalli RK, Taskar KS, Rudraraju V, Gril B, Bohn KA, Adkins CE, Roberts A, Thorsheim HR, Gaasch JA, et al: Heterogeneous blood-tumor barrier permeability determines drug efficacy in experimental brain metastases of breast cancer. Clin Cancer Res 2010, 16:5664-5678.
46. Acharyya S, Oskarsson T, Vanharanta S, Malladi S, Kim J, Morris PG, Manova-Todorova K, Leversha M, Hogg N, Seshan VE, et al: A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell 2012, 150:165-178.
47. Aslakson CJ, Miller FR: Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res 1992, 52:1399-1405.
48. Lopez-Coviella I, Follettie MT, Mellott TJ, Kovacheva VP, Slack BE, Diesl V, Berse B, Thies RS, Blusztajn JK: Bone morphogenetic protein 9 induces the transcriptome of basal forebrain cholinergic neurons. Proc Natl Acad Sci U S A 2005, 102:6984-6989.
49. Woditschka S, Evans L, Duchnowska R, Reed LT, Palmieri D, Qian Y, Badve S, Sledge G, Jr., Gril B, Aladjem MI, et al: DNA double-strand break repair genes and oxidative damage in brain metastasis of breast cancer. J Natl Cancer Inst 2014, 106 (7). pii: dju145. doi: 10.1093/jnci/dju145.
50. McMullin RP, Wittner BS, Yang C, Denton-Schneider BR, Hicks D, Singavarapu R, Moulis S, Lee J, Akbari MR, Narod SA, et al: A BRCA1 deficient-like signature is enriched in breast cancer brain metastases and predicts DNA damage-induced poly (ADP-ribose) polymerase inhibitor sensitivity. Breast Cancer Res 2014, 16:R25.
51. Welcsh PL, Lee MK, Gonzalez-Hernandez RM, Black DJ, Mahadevappa M, Swisher EM, Warrington JA, King MC: BRCA1 transcriptionally regulates genes involved in breast tumorigenesis. Proc Natl Acad Sci U S A 2002, 99:7560-7565.
52. Salhia B, Kiefer J, Ross JT, Metapally R, Martinez RA, Johnson KN, DiPerna DM, Paquette KM, Jung S, Nasser S, et al: Integrated genomic and epigenomic analysis of breast cancer brain metastasis. PLoS One 2014, 9:e85448.
53. Kim SJ, Kim JS, Park ES, Lee JS, Lin Q, Langley RR, Maya M, He J, Kim SW, Weihua Z, et al: Astrocytes upregulate survival genes in tumor cells and induce protection from chemotherapy. Neoplasia 2011, 13:286-298.
54. Pei S, Minhajuddin M, Callahan KP, Balys M, Ashton JM, Neering SJ, Lagadinou ED, Corbett C, Ye H, Liesveld JL, et al: Targeting aberrant glutathione metabolism to eradicate human acute myelogenous leukemia cells. J Biol Chem 2013, 288:33542-33558.
55. Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C, Guise TA, Massague J: A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3:537-549.
56. Gupta GP, Perk J, Acharyya S, de Candia P, Mittal V, Todorova-Manova K, Gerald WL, Brogi E, Benezra R, Massague J: ID genes mediate tumor reinitiation during breast cancer lung metastasis. Proc Natl Acad Sci U S A 2007, 104:19506-19511.
57. Gupta GP, Nguyen DX, Chiang AC, Bos PD, Kim JY, Nadal C, Gomis RR, Manova-Todorova K, Massague J: Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature 2007, 446:765-770.
58. Minn AJ, Kang Y, Serganova I, Gupta GP, Giri DD, Doubrovin M, Ponomarev V, Gerald WL, Blasberg R, Massague J: Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J Clin Invest 2005, 115:44-55.
59. Davi MV, Francia G, Brazzarola P, Olivieri M, Petrozziello A, Orsolini A, Petronio R, Sussi PL: An unusual case of adrenal failure due to isolated metastases of breast cancer. J Endocrinol Invest 1996, 19:488-489.
60. Gallo-Payet N, Battista MC: Steroidogenesis-adrenal cell signal transduction. Compr Physiol 2014, 4:889-964.
61. Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA: Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest 2011, 121:2750-2767.
62. Harrell JC, Prat A, Parker JS, Fan C, He X, Carey L, Anders C, Ewend M, Perou CM: Genomic analysis identifies unique signatures predictive of brain, lung, and liver relapse. Breast Cancer Res Treat 2012, 132:523-535.
63. Palmieri D, Bronder JL, Herring JM, Yoneda T, Weil RJ, Stark AM, Kurek R, Vega-Valle E, Feigenbaum L, Halverson D, et al: Her-2 overexpression increases the metastatic outgrowth of breast cancer cells in the brain. Cancer Res 2007, 67:4190-4198.
64. Goswami CP, Nakshatri H: PROGgene: gene expression based survival analysis web application for multiple cancers. J Clin Bioinforma 2013, 3:22.
65. Nakshatri H, Rice SE, Bhat-Nakshatri P: Antitumor agent parthenolide reverses resistance of breast cancer cells to tumor necrosis factor-related apoptosis-inducing ligand through sustained activation of c-Jun N-terminal kinase. Oncogene 2004, 23:7330-7344.