
Introduction
Extracellular vesicles (EVs) that originate from cancer cells are gradually and consistently emerging as important regulators of tumor progression [1]. Nano-sized particles called exosomes (~100 nm diameter) are generally considered to originate from multivesicular bodies (MVB) of the late endocytic pathway [2]. In addition to exosomes, tumor cells also produce larger EVs (500 to >1000 nm diameter), referred to in general terms as microvesicles (MVs) [3-5] or ectosomes. MVs seem to be generated by budding of the plasma membrane. Exosomes and other EVs play important functions dictated by their cell of origin and their content [6]. After years of research aimed to identify unequivocal exosome markers, it is now clear that “universal exosome markers” are difficult to find, and recent proteomic studies suggest that exosome populations are diverse and can be enriched with distinct proteins that impart specific functions to the particles [7]. Similarly, it is not known whether specific proteins can be expressed in exosomes versus plasma membrane-derived EVs, such as ectosomes/MVs, and vice-versa.
We recently demonstrated the existence of an atypically large class of EVs (1-10 µm in diameter), termed large oncosomes (LO), which can be byproducts of non-apoptotic plasma membrane blebbing induced by silencing of the cytoskeletal regulator Diaphanous-related formin-3 (DIAPH3), by overexpression of oncoproteins such as MyrAkt1, HB-EGF, and caveolin-1 (Cav-1), or by activation of the EGFR [8-11]. In comparison to exosomes and MVs, LO are still a poorly characterized EV type [12]. Using expression of Cav-1, a circulating marker of metastatic prostate cancer [13] to quantify LO, we demonstrated a significant increase of LO enumeration in the circulation of both TRAMP mice, which harbor rapidly progressing prostate tumors, and patients with metastatic disease [9]. However, because Cav-1 is also detected in exosomes, and our earlier method for LO discrimination was simply based on particle size being >1 µm, we went on and analyzed the protein content of LO and nano-sized EVs using mass spectrometry to identify specific cargo.
Here we report a quantitative duplex stable isotope labeling of amino acids in cell culture (SILAC) analysis of large and nano-sized EVs isolated by differential centrifugation from DU145 cells silenced for DIAPH3. We identified uniquely expressed and differentially expressed proteins in the two preparations. Metabolism emerged as a biological process enriched in large versus nano-sized EVs, and this result was functionally validated. In order to validate the mass spectrometry data, to separate EVs from particulate material, and to determine the density of large EVs, we applied equilibrium centrifugation in OptiPrepTM gradients using an upward displacement modality [14-17]. Among the proteins enriched in large EVs, cytokeratin 18 (CK18) emerged as a potential marker for tumor-derived EVs in tissues and in plasma. Our findings suggest that LO, compared to exosomes, contain different protein cargo, are functionally distinct, and harbor proteins that might be employed in their functional characterization and used as tissue and circulating markers of disease progression.
Results
Silencing of DIAPH3 results in increased shedding of large EVs
Consistent with published results, silencing of the cytoskeletal regulator DIAPH3 (shDIAPH3) in DU145 cells resulted in the formation of large plasma membrane blebs (1-10 µm in diameter) (Figure 1A, Supplementary Movie 1) [8, 10], which were shed into the medium as large EVs (Figure 1B). We observed a higher amount of total protein in large EVs from shDIAPH3 than control cells, collected from the same number of donor cells by differential centrifugation (10,000 x g) [3, 10, 18] (Supplementary Figure 1A). Flow-cytometry (FACS) analysis with specific size beads (1-10 µm), previously employed by us to quantify EVs in the size range of LO [10], revealed a significantly higher number of EVs > 1 µm in media from DIAPH3 silenced versus control cells (Figure 1C, Supplementary Figure 1B). The result was even more robust when we gated for events larger than 2 µm (Figure 1D), consistent with our previous observation that the modal distribution of LO shed from prostate cancer cells is 2-3 µm [10].
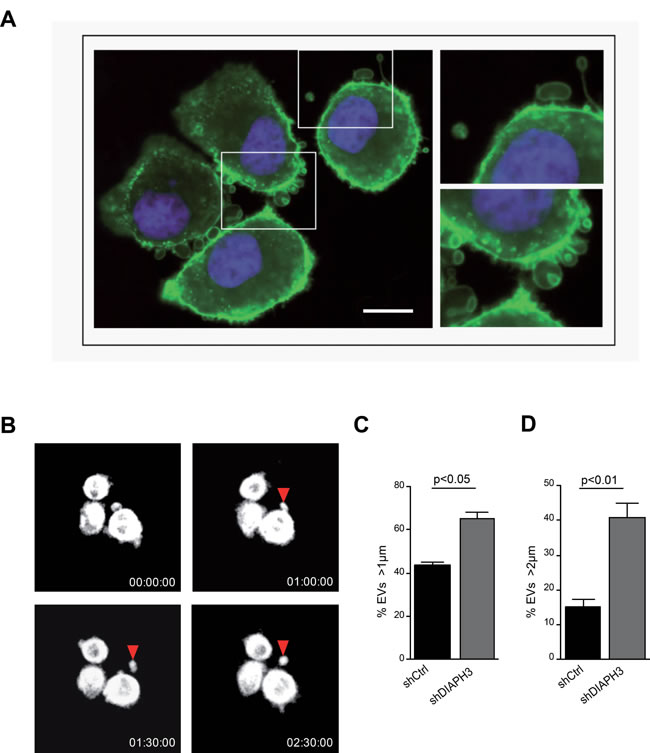
Figure 1: Silencing of DIAPH3 results in increased shedding of large EVs. (A) Membrane blebbing and shedding of EVs of variable size (insets) from DU145 cells stably expressing DIAPH3 shRNA, stained with CTxB-FITC (X 63). Scale bar, 20 µm. (B) 30 min-1h interval frames (from Supplementary Movie 1), acquired by real-time confocal microscopy of DIAPH3-silenced DU145 over-expressing RFP-tubulin. The arrow points to a membrane bleb that is released as a large oncosome. (C, D) EVs from DIAPH3-silenced or parental DU145 cells were analyzed by flow cytometry.
Mass spectrometry identifies differentially expressed and unique proteins in large and nano-sized EVs
Nanoparticle Tracking Analysis (NTA) showed that shDIAPH3 DU145 cells also produce higher numbers of nano-sized EVs [11]; consequently, we used this system as a model to quantitatively compare the protein composition of large and nano-sized EVs, isolated by differential centrifugation, by SILAC. To minimize false-positive hits, two independent SILAC label-swap experiments were conducted. Differentially expressed proteins were identified by an integrative statistical hypothesis testing method with log2-scaled and quantile normalized SILAC intensities [19-21].
A total of 407 proteins were identified, among which 103 (approximately 25% of the total) were differentially represented in large and nano-sized EVs, with a false discovery rate (FDR) < 0.05, and fold change ≥ 2 (Figure 2A, and Supplementary Figure 2A, B). A volcano plot of the differentially expressed proteins showed a noticeable enrichment in nano-sized EVs for tetraspanin family members CD81 and CD9 (Figure 2A, Supplementary Figure 2B), proteins known to be over-represented in exosomes [22-24]. Additional top-ranked differentially expressed proteins (>4 fold) in nano-sized EVs included cell adhesion molecules, such as integrin-α3 (ITGA3), integrin-αV (ITGAV), intercellular adhesion molecule (ICAM) and CD44. Transforming growth factor-β1 (TGFβ1), which has been linked to cancer progression for its role in the tumor microenvironment [25, 26], and which participates in exosome-mediated biological functions [27, 28], was 6-fold more abundant in nano-sized versus large EVs (Figure 2A). Cathepsin proteases (CTSD and CTSA), usually associated with the plasma membrane or secreted into the extracellular environment [29], were also enriched in nano-sized EVs.
In contrast, proteins enriched in large EVs included glyceraldehyde 3-phosphate dehydrogenase (GAPDH), glucose phosphate isomerase (GPI), lactate dehydrogenase B (LDHB), heat shock 70kDa protein 5 (HSPA5), malate dehydrogenase (MDH) and aspartate transaminase (GOT) family members (Figure 2A, Supplementary Figure 2A). These proteins are all involved in metabolic processes relevant to cancer [30, 31].
We also identified “unique proteins” in both large and nano-sized EVs using specific selection criteria (see Methods) (Figure 2B). Proteins uniquely identified in nano-sized EVs, similarly to the differentially expressed, were mainly involved in adhesion, motility, and response to hypoxia, functions frequently attributed to exosome proteins [32], whereas in large EVs metabolic enzymes were also predominant among the unique proteins. Notably, glutaminase (GLS), a cytoplasmic enzyme that converts glutamine to glutamate, was the most abundant protein uniquely identified in large EVs (Figure 2B). Further analysis demonstrated that several proteins significantly enriched (>4 fold) in large EVs and identified in the EV data repository EVpedia [32] were functionally categorized as playing a role in cell migration, resistance to docetaxel, angiogenesis, prostate cancer progression and bone metastasis, and other processes associated with tumor progression (Figure 3). These observations indicate that the two EV populations contain different cargo, and this might be a reflection of differences in function.
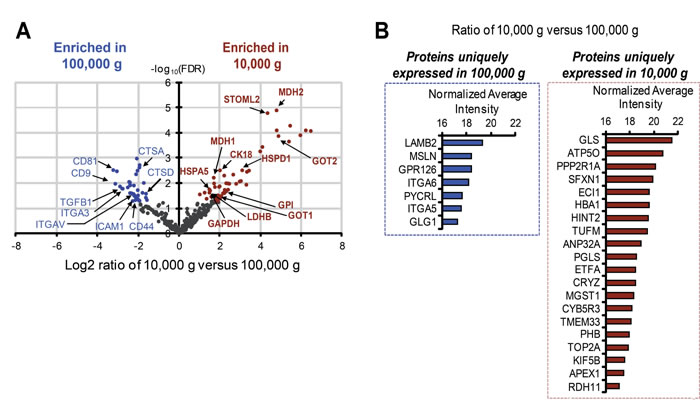
Figure 2: Identification of unique and differentially expressed proteins in large and nano-sized EVs. (A) Volcano plots of the log2 ratio of the averaged, normalized SILAC intensities against the FDR of the differential expression between large and nano-sized EVs. Red and blue dots correspond to proteins enriched in large EVs and nano-sized EVs, respectively. (B) Bar plots show the abundance of unique proteins quantified in large (right) and nano-sized EVs (left). The horizontal axis represents the normalized average log2 ion intensities of uniquely identified proteins.
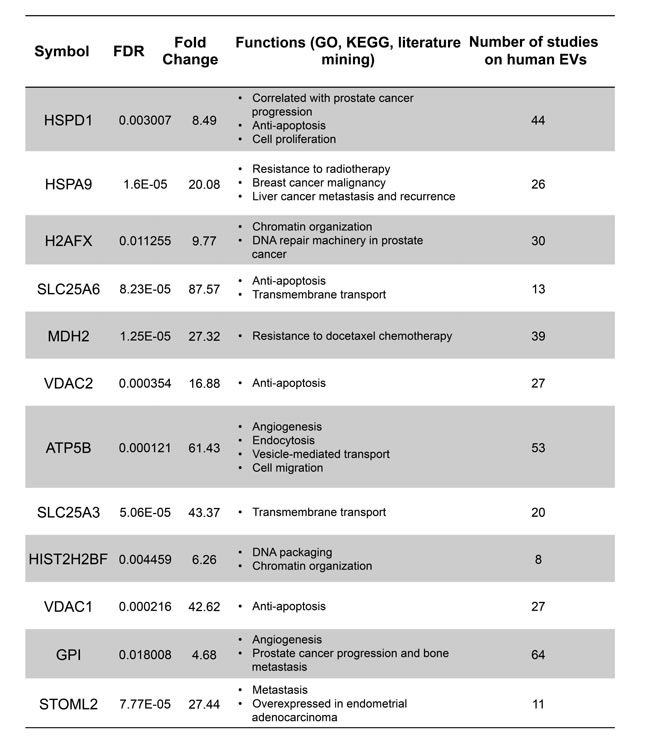
Figure 3: Proteins highly abundant in large EVs are associated with cancer progression. Proteins with >4 fold enrichment in large EVs were functionally annotated by using the GO, KEGG and iHOP literature mining softwares to identify the association with cancer progression. The column on the right indicates the number of studies, obtained from the EVpedia database, in which these proteins were detected.
Functional analysis of the proteins enriched in large EVs suggests an influence on cancer cell glutamine metabolism
Functional enrichment analysis using FunRich software (http://www.funrich.org) further indicated that proteins abundant in large EVs are involved in metabolism of carbohydrates (15.9%), glucose and glutamine metabolism (13.6%), aspartate degradation II (9.1%), and gluconeogenesis (13.6%), metabolic processes relevant to cancer [33] (Figure 4A). This result provided a rationale to determine whether large EVs were capable of inducing alterations in glutamine metabolism in recipient cells, a role that, to the best of our knowledge, has never been tested for any type of EV. Western blotting validated the enrichment of metabolic enzymes GOT1 and GAPDH in large EVs, whereas the tetraspanin CD63 was enriched in nano-sized EVs (Figure 4B). Increased expression of GOT1 was also observed in cancer cells exposed to large EVs as little as 2 h following exposure, suggesting that the protein might be transferred to recipient cells specifically via large EVs. GOT1 transcript levels were not altered by large EV treatment, thereby supporting protein, and not mRNA transfer to recipient cells (Figure 4C).
Because GOT1 catalyzes the reversible conversion from aspartate and α-ketoglutarate to glutamate, we used a functional assay that measures the production of glutamate per unit time as an indication of GOT activity in cancer cells exposed to large and nano-sized EVs [34]. Our results indicate a robust stimulation of the enzyme induced by large EVs, ultra-purified by buoyant density gradient as described in the following paragraph (Figure 4D). We also observed a robust increased activity of GOT in cells treated with large EVs but not vehicle, and cultured in 5% but not in 1% glutamine (Supplementary Figure 3A), suggesting that EVs stimulate tumor metabolic activities in the presence of non-rate limiting amounts of substrate. In line with this result, parallel experiments demonstrated an increased percentage of S-phase in cells treated with large EVs and cultured in 5% but not 1% glutamine (Figure 4E, Supplementary Figure 3B). These results indicate that large EVs can affect specific metabolic functions of cancer cells.
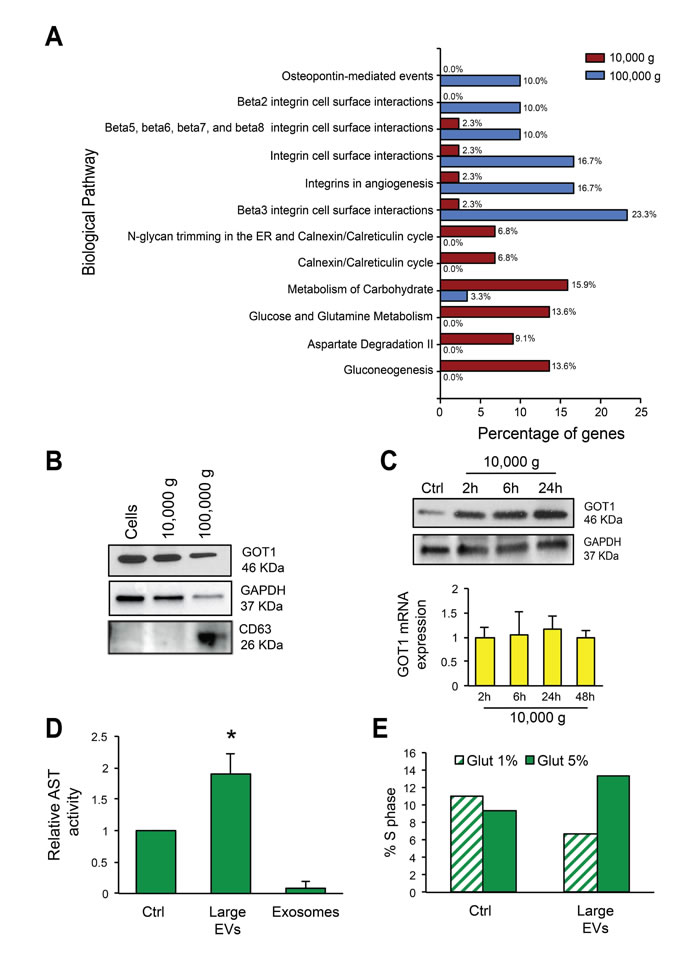
Figure 4: Large EVs are enriched in vesicular markers and alter glutamine metabolism in recipient cells. (A) Functional analysis using FunRich software indicates the biological pathways overrepresented either in large (10,000 g) or nano-sized EVs (100,000 g). (B) Protein lysates from cells, large and nano-sized EVs were blotted with the indicated antibodies. CD63 was expressed specifically in nano-sized EVs, and GOT1 in large EVs. (C) Protein lysates from DU145 cells untreated or treated with large EVs for the indicated times, were blotted with GOT1 antibody. GAPDH was used as a loading control (top panel). GOT1 mRNA expression levels in DU145 cells untreated or treated with large EVs for the indicated times do not exhibit significant changes. The result is displayed as levels of GOT1 transcript after normalization to the housekeeping gene GAPDH in treated versus untreated cells at 2-48h (bottom panel). (D) GOT1 activity was measured in DU145 cells in 5% glutamine with or without treatment with large oncosomes (1.15 g/ml OptiPrepTM density fractions) or exosomes (1.10 g/ml) (20µg/ml of protein lysate). The results from 3 experiments are displayed as relative AST activity in treated cells in comparison with the baseline activity of the enzyme (p=0.024). (E) Cell-cycle was analyzed by flow cytometry in DU145 cells treated with large oncosomes or vehicle in the presence of 1% or 5% glutamine for 24 hours.
Large EVs float to 1.10-1.15 g/ml in iodixanol centrifugation gradients
From the mass spectrometry analysis we found that CK18, a membrane-localized protein we previously used to decorate LO-like features in situ in human prostate cancer tissues [10], was highly abundant in large EVs (top 5th percentile; Figure 5A). In contrast, CD9 and CD81 were expressed at negligible levels in large EVs (Figure 5A, inset; Supplementary Figure 2A). To further validate the SILAC findings, we performed immunoblotting of CK18, which was confirmed to be enriched in large EVs (10,000 x g) in comparison with nano-sized EVs (100,000 x g). In contrast, CD81 was over-represented in nano-sized EVs (Figure 5B).
To determine if the proteins identified using SILAC were associated with EVs, instead of protein clots or debris, and in order to determine the buoyant density of large and nano-sized EVs, we used iodixanol (OptiPrepTM), a medium that is less viscous than sucrose and therefore more likely to enhance the separation of EV populations with differing densities [15]. Large and nano-sized EV pellets, normalized to the same number of cells were separated by flotation in discontinuous 5-60% OptiPrepTM density gradients following deposition of the EV material at the bottom of the tubes (fractionation by upward displacement). Western blot analysis of 10 µg of protein lysate obtained from the gradient fractions derived from the 100,000 x g pellets revealed a population of EVs expressing typical exosome markers, such as CD81 and tumor susceptibility gene 101 (Tsg101), which were detected at a buoyant density of 1.10 g/ml (Figure 5C). Transmission electron microscopy (TEM) of this fraction revealed a homogeneous population of round, cup-shaped vesicles with sizes ranging from 50 to 100 nm, consistent with exosome morphology [35] (Figure 5D). Western blot analysis of gradient fractions derived from the 10,000 x g pellets demonstrated that CK18, GAPDH and HSPA5, identified as potential large EV markers by mass spectrometry, floated at buoyant densities of 1.10 and 1.15 g/ml (Figure 5E). Levels of CD81 and Tsg101 in these fractions were negligible or undetectable. Microscopy of the 1.15 g/ml fraction, labeled with a fluorescent DiO lipophilic dye, revealed the presence of intact EVs, variable in size but larger than 1 µm, consistent with LO morphology as previously described [8, 10, 18] (Figure 5F).
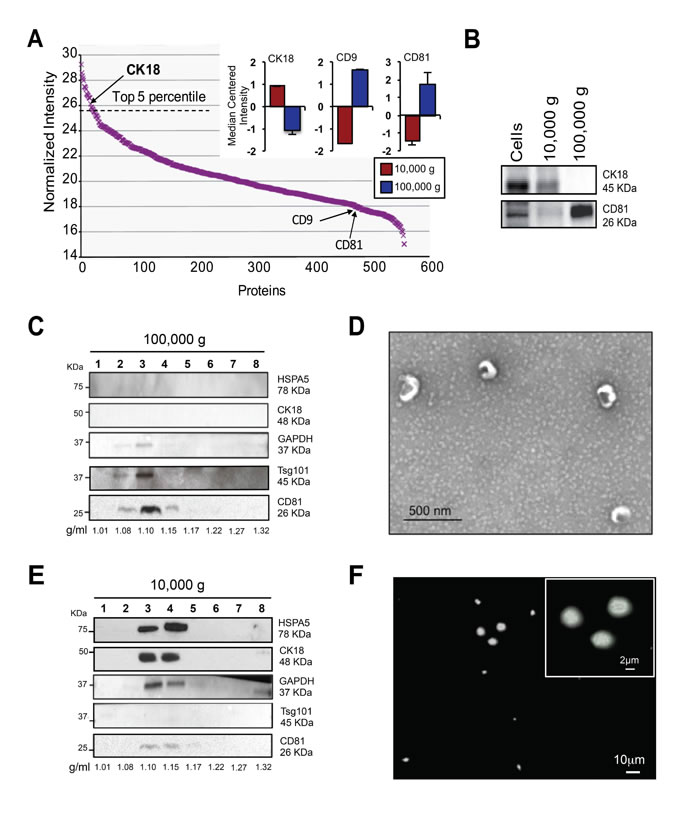
Figure 5: SILAC validation by OptiPrepTM gradient, EM and IF. (A) Rank plot of normalized ion intensities of all proteins identified in large EVs. CK18 is indicated as high abundant and CD9 and CD81 are indicated as low abundant proteins in large EVs. (B) Protein lysates from cells, large EVs and nano-sized EVs were blotted with the indicated antibodies. CD81 was expressed specifically in nano-sized EVs, and CK18 in large EVs. (C) Equal amounts of proteins (10 µg) from OptiPrepTM fractions (1-8) of nano-sized EVs were blotted with the indicated antibodies. Exosome markers CD81 and Tsg101 were identified in fraction 3, corresponding to the buoyant density of 1.10 g/ml. (D) Magnified TEM detail of negative stained EVs corresponding to the buoyant density of 1.10 g/ml, showing cup-shaped vesicles. Scale bars, 500nm. (E) Equal amounts of proteins (10 µg) from OptiPrepTM fractions (1-8) of large EVs were blotted with the indicated antibodies. Large EV enriched proteins (SILAC) such as CK18, GAPDH and HSPA5 were identified in fractions 3 and 4, corresponding to the buoyant density of 1.10-1.15 g/ml. (F) The 1.15 g/ml density fraction, labeled with DiO lipophilic dye, was imaged by IF. Scale bars, 10 µm and 2 µm (inset).
CK18 is a marker of large oncosomes and can be identified in the circulation and in tissues
Having validated enrichment of CK18 in large EVs and specifically in LO by western blotting (Figure 5B, E), we attempted to quantify LO shedding from shDIAPH3 cells by measuring the number of CK18 positive LO by FACS, using differentially sized beads (1-10 µm) to set the gates [10, 18]. We observed a 17-fold increase of events in the PE-positive channel when the EVs were stained with CK18 antibody in comparison with unstained vesicles (Supplementary Figure 4).
We then took an analogous approach to quantitatively analyze circulating CK18 positive EVs >1 µm. We used plasma from a previously described mouse model in which shDIAPH3 DU145 cells, injected into the tail vein, formed a larger number of lung metastatic foci in comparison to control cells [10]. We observed a significant increase in the mean fluorescent intensity (MFI) of the CK18 signal in the plasma EVs of mice injected with shDIAPH3 DU145 cells in comparison to mice injected with control cells (Figure 6A). Importantly, the tumor tissue of the lung metastatic foci of the same animals expressed high levels of CK18, and exhibited LO-like features, strongly supporting a tumor origin for the large EVs detected in the plasma (Figure 6B).
Finally, as a first test for the potential use of CK18 as circulating marker in humans, total EVs were isolated from human plasma from prostate cancer patients using a commercial kit (ExoQuickTM) that does not distinguish between LO and exosomes [14, 16]. The rationale for using this kit is that it has the potential to be employed clinically to isolate EVs and other circulating tumor products. Levels of CK18 in ExoQuickTM preparations from prostate cancer patients (n=6) were significantly higher than in healthy control subjects (n=5) (Figure 6C), suggesting that CK18 in EVs could potentially be used as a circulating cancer biomarker.
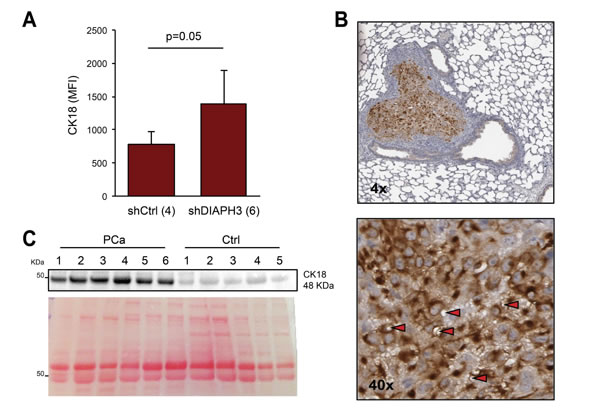
Figure 6: CK18 is a marker of large oncosomes in vivo. (A) FACS analysis of CK18 positive EVs from the plasma of mice with lung metastasis injected with DIAPH3-silenced (n=6) or control (n=4) DU145 cells. The plot shows the mean fluorescent intensity (MFI) relative to CK18 positive EVs >1 µm. (B) Representative lung tissue section immunostained with CK18 (4X). The tumor is strongly positive for CK18, and LO features can be identified at higher magnification (40X). (C) CK18 western blotting of EVs isolated by ExoQuickTM from the plasma of 6 patients with prostate cancer (PCa), and 5 healthy male subjects (Ctrl). Ponceau staining is displayed as a loading control.
Discussion
This study reports the first comparative, large scale, quantitative analysis of the protein cargo of large and nano-sized EVs derived from a metastatic model of prostate cancer. We identified unique and differentially expressed proteins in the two EV populations. Glutamine and glucose metabolism emerged as biological pathways enriched in large versus nano-sized EVs. Large EVs with the appearance of particles described previously as LO could be purified using density gradient centrifugation [14, 15]. Furthermore, treatment with LO but not with exosomes altered aspartate transaminase activity in recipient cancer cells. Among the proteins enriched in LO, CK18 emerged as a marker for tumor-derived LO in tissues and in plasma.
Cancer cell-derived EVs can be heterogeneous in content, size, and density, and have been functionally implicated in the regulation of several processes during tumor growth and metastasis [5, 6, 36-39]. Despite the expectation that EVs derived from plasma membrane budding might carry different proteins than EVs derived from MVB, recent proteomic studies show a large overlap in proteins identified in different types of EVs from the same system [17, 40]. Therefore, the substantial overlap (69.9%) in proteins identified in this study in large and nano-sized EVs is corroborated by previous findings. However, the mass spectrometry findings reported here were obtained using a quantitative approach, and they indicate that levels of proteins identified in both EV types are quantitatively distinct. Importantly, although some of the proteins we identified in large EVs have been described as mitochondrial, the lack of cytochrome P450 argues against significant subcellular organelle contamination in our preparations [7]. Furthermore, membrane proteins constituted 30% of the proteins identified in both EV types, consistent with previous reports on EVs [24, 32]. Our findings that tetraspanins, growth factors (i.e. TGFβ1) and proteins related to cell adhesion are enriched in nano-sized EVs are consistent with previous reports on exosomes [41-43]. Similarly, cathepsin proteases, previously reported as EV cargo and involved in ECM degradation and remodeling, tumor progression, invasion and resistance to chemotherapeutic drugs, was identified in nano-sized EVs [29]. Additional studies are necessary to determine whether these cathepsin-containing EVs are tumor type-specific, and if they could be used as markers to purify distinct subtypes of EVs.
Our study newly demonstrates that we can purify large EV populations by floating medium-speed pellets on discontinuous density gradients. These particles localize to two discrete fractions, floating at 1.10-1.15 g/ml density, and shows morphology similar to LO described previously [8, 10, 18]. Notably, our density gradient of high-speed pellets allowed purification of pure populations of exosomes, floating at the original proposed density for this well-characterized EV population [14, 17, 44]. Further investigation and more granular gradient resolution will clarify whether LO have distinct physical properties in comparison with exosomes. On a functional point of view, while the relevance of exosomes to tumor progression is well established, data on LO are still limited. Our findings of a stronger association of LO cargo with aggressive cancer using GO, KEGG and iHOP literature mining softwares, along with the result that metabolism emerges as a LO-enriched biological process, suggest a distinct role for LO in tumor progression and indicate that these EV populations might play a selective metabolic function over other EV subclasses.
Altered glutamine metabolism in tumor cells, including a phenotype known as glutamine addiction, has been described in aggressive tumors such as glioma, melanoma and pancreas carcinoma [34, 45, 46]. We newly identified GOT1 as an enriched enzyme in LO and that LO can promote glutamine metabolism in recipient cancer cells by transferring the protein. However, this may not be the only mechanism. In fact, large EVs are also enriched in other proteins that affect glutamine metabolism, such as HSPA5 (GRP78), which was recently demonstrated to promote c-Myc-mediated glutamine flux and proliferation of cancer cells [31]. Additionally, HSPA5 expression has been correlated with tumor grade and metastatic events in triple negative breast carcinoma (TNBC) and is functionally relevant to clinical progression [47, 48]. Furthermore, GLS, a cytoplasmic enzyme that converts glutamine to glutamate is identified in large EVs as a unique protein. Alterations in glutamine and glucose metabolism are key aspects of the metabolic derangement in tumor cells and tumor-associated stroma [49], which can promote cancer cell proliferation. Additional implications of our findings are that LO may be involved in metabolic reprogramming of cells within the tumor microenvironment. This hypothesis will be the subject of future studies by our laboratory.
Our identification of CK18 as a protein significantly enriched in LO is in line with our previous identification of LO-like structures using CK18 IHC in human prostate cancer tissues. CK18 was recently detected in the circulation of patients with gastric and colorectal cancer [50, 51], and linked to a particular subtype of prostate cancer that aberrantly expresses p63, lacks the androgen receptor (AR) and harbors rearrangements of the ERG gene [52]. Expression of CK18 was also reported to be maintained in cancer cells after castration, in association with loss of AR and appearance of neuroendocrine markers, suggesting clinical utility for this luminal type cytokeratin [53]. However, this is the first report that identifies CK18 in plasma of patients with prostate cancer. This result is promising and suggests the possibility that this marker might be adapted for clinical assessment of disease status by employing quantitative, high-sensitivity detection methods (e.g., ELISA). Taken together, the results of this study support further investigation into the heterogeneous nature of tumor-derived EVs. They also suggest that the characterization of LO, as a distinct population from nano-sized exosomes, could result in the development of circulating diagnostic or prognostic signatures. Ongoing experiments in our laboratory are testing the feasibility of employing multiplex labeling of LO directly in body fluids.
Materials and Methods
Cell culture
DU145 cells, expressing DIAPH3 hRNA or control vector, were cultured as previously described [10]. For SILAC labeling, DIAPH3-silenced DU145 cells were grown for at least six doublings in arginine- and lysine-depleted DMEM medium supplemented with 10% (v/v) dialyzed fetal bovine serum (Invitrogen, Grand Island, NY) and L-arginine (Arg0) and L-lysine (Lys0) or 13C615N4-L-arginine (Arg10) and 13C615N2-L-lysine (Lys8) (Cambridge Isotope Laboratories, Andover, MA), before undergoing isolation of large EVs and nano-sized EVs, respectively.
Protein separation and digestion
Proteins were separated and digested as described [54]. SILAC-labeled proteins were mixed at a 1:1 (w/w) ratio, separated by 10% SDS-PAGE, and stained with Coomassie Brilliant Blue solution (Bio-Rad). Each lane was cut into ten gel slices of similar size and further cut into about 1 mm3 particles. Proteins were reduced by 10 mM dithiothreitol (DTT), alkylated by 55 mM iodoacetamide, and digested with mass spectrometry-grade trypsin (Promega) at 37°C for 16 h. Tryptic peptides were successively extracted with 100 μL of 5% acetic acid, 100 μL of 2.5% acetic acid and 50% acetonitrile, and 100 μL of 100% acetonitrile. Samples were dried in a SpeedVac concentrator (Thermo Scientific) and stored at -80°C until MS analysis.
Liquid chromatography-tandem mass spectrometry
Tryptic peptides were redissolved with 20 μL 1.5% acetic acid and 5% acetonitrile. Samples (10 μL each) were analyzed by online C18 nanoflow reversed-phase HPLC (Eksigent nanoLC∙2D™) connected to an LTQ Orbitrap XL mass spectrometer (Thermo Scientific) as described previously [55, 56]. Briefly, samples were loaded onto a 15 cm nanospray column (75 μm inner diameter, Magic C18 AQ, 3 μm particle size, 200Å pore size, Precision Capillary Columns) and separated at 300 nL/min with a 60-min gradient from 5 to 35% acetonitrile in 0.1% formic acid. MS data were acquired in a data-dependent manner selecting the fragmentation events based on the precursor abundance in the survey scan (350-1600 Th). The resolution of the survey scan was set at 30,000 at m/z 400 Th. Dynamic exclusion was 90 s. After each survey scan, up to ten collision-induced dissociation (CID) MS/MS spectra were acquired in the linear ion trap. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository [57] with the dataset identifier PXD000776.
Protein identification and quantitation
The MS data were analyzed using MaxQuant (v1.3.0.5) [58]. Proteins were identified by searching MS/MS spectra against the UniProt database for Human (released on 09/11/2012, 84,680 entries) combined with 262 common contaminants by the Andromeda search engine [59], with second peptide identification enabled. Carbamidomethylation of cysteines was set as a fixed modification, oxidation of methionines and acetylation of the protein N-terminus as variable modifications. Trypsin allowing for cleavage N-terminal to proline was chosen as the enzyme specificity. A maximum of two missed cleavages were allowed. The minimum peptide length was specified to be seven amino acids. The maximal mass tolerance in MS mode was set to 20 ppm for first search and 6 ppm for main search, and in MS/MS mode 0.7 Da. The maximum false discovery rates (FDR) for peptide and protein identifications were specified as 0.01.
Identification of differentially expressed proteins (DEPs) and functional enrichment analysis
Lists of DEPs (FDR < 0.05 and fold change ≥ 2) were identified by integrative hypothesis testing method as previously described [21, 60] using quantile normalized SILAC intensities [19]. Briefly, for each protein, we performed 1) two-tailed t-test and median ratio test; 2) computation of false discovery rates (FDRs) from the two tests using an empirical distribution of the null hypothesis (that the means of the genes are not different), which was obtained from random permutations of the samples; 3) combination of the two FDRs for the individual peptides using Stouffer’s method [20]. In addition, to identify proteins uniquely detected in large or nano-sized EVs, we employed the following criteria: 1) being identified in at least one replicate with more than two sibling peptides and 2) being quantified in one single condition, not quantified in the other condition. The proteome of large and nano-sized EVs was analyzed by using functional enrichment analysis software FunRich (http://www.funrich.org).
Isolation of large oncosomes and nano-sized EVs
Large EVs were purified by differential centrifugation (Beckman SW28 rotor) from conditioned medium or 300 µl of mouse platelet-poor plasma as previously described [10, 18]. Briefly, cells and debris were eliminated by centrifugation at 2,800 g for 10 min. The supernatant was then centrifuged at 10,000 g for 30 min to precipitate large EVs. For isolation of nano-sized EVs, the supernatant remaining after the 10,000 g spin was subjected to additional centrifugation at 100,000 g for 60 min. For discontinuous centrifugation gradient we used a modified version of a previously applied protocol [61]. Briefly 60%, 50%, 40%, 30%, 25%, 15%, 10% and 5% solutions were made by diluting a stock solution of OptiPrep™ (60% aqueous iodixanol from Sigma) in 0.25 M Sucrose/0.9 M NaCl/ 120 mM HEPES, pH 7.4. The 10,000 x g and 100,000 x g pellets were mixed in the bottom layer and the following solutions carefully layered. Centrifugation was performed at 100,000 x g for 3h and 50 min at 4 °C with a SW28 Beckman rotor. Eight individual fractions were collected, washed with PBS, and after centrifugation at 100,000 x g for 1 h at 4 °C, the pellet from each fraction suspended in either PBS or lysis buffer.
FACS analysis of large EVs
Purified large EVs from conditioned media or mouse plasma samples (6 shDIAPH3 and 4 control) were washed in PBS, fixed and permeabilized with 0.5% Tween 20 and then stained with rabbit monoclonal CK18 (Abcam, 1:50). Samples were processed on a LSRII Flow Cytometer (BD) using 1, 2 and 10 µm bead standards [10, 18]. A minimum of 3000 events per experiment was recorded and the data analysis was performed with the FlowJo software (Treestar). The plot shows the mean fluorescent intensity (MFI). Statistical significance was calculated using a 2-tailed unpaired Student’s t test.
Isolation of EVs from human plasma
EVs were isolated from human plasma (in accordance with Ethics Board Approval Cert. H09-01010 obtained from the University of British Columbia, Canada) using precipitation solution ExoQuick™ (System biosciences) according to the manufacturer’s manual with a few modifications. Briefly, 500 µl of plasma samples from prostate cancer patients and disease-free normal controls were diluted with 1X PBS (1:1). ExoQuick™ solution was added to the plasma, gently mixed and stored at 4º C overnight. The plasma samples were centrifuged at 1,500 x g for 30 min and the resulting exosome pellet was suspended in PBS. EVs pellet was stored at -80ºC until further analysis.
Fluorescence microscopy
Cells were stained with FITC-conjugated cholera toxin B (CTxB) subunit (Sigma) and imaged using an Axioplan 2 microscope (Zeiss), as previously described [8, 10, 18]. Alternatively, control or DIAPH3-silenced cells were imaged by a 20x objective on an Ultravox Spinning Disc Confocal microscope at the Boston Children’s Hospital Intellectual and Developmental Disabilities Research Imaging Core facility (IDDRC, NIH-P30-HD-18655). The 25% iodixanol fraction, corresponding to the 1.15 g/ml fraction were stained with the lipophilic tracer DiO (Invitrogen) and imaged using a Leica TCS SP spectral confocal microscope.
Transmission electron microscopy
EVs from the 15% iodixanol fraction 100,000 x g, corresponding to the 1.10 g/ml fraction were washed in HEPES, fixed with 1% glutaraldehyde and layered onto a mesh copper grids. Grids were then stained with the Negative Staining (NS) technique. Imaging was performed at an acceleration voltage by a transmission
electron microscope JEM1200EX (JEOL, Japan) provided with a BioScan 600W digital camera (Gatan, USA).
Immunoblot analysis
Cells, purified large EVs and nano-sized EVs were lysed and analyzed by western blotting using the following antibodies: rabbit monoclonal GOT1 (Sigma, 1:1000 dilution), rabbit polyclonal CD63, clone H-193 (Santa Cruz, 1:1000 dilution), rabbit polyclonal CK18 (Abcam, 1:1000 dilution); rabbit polyclonal CD81, clone M38 (Abcam, 1:1000 dilution), mouse monoclonal TSG101, clone C-2 (Santa Cruz, 1:500 dilution), rabbit monoclonal HSPA5 (Cell Signaling, 1:1000), and GAPDH (Cell Signaling 1:2000).
Immunoblot and qRT-PCR analysis for GOT1 in cells exposed to EVs
DU145 cells, exposed to vehicle or large EVs for the indicated times, were blotted with GOT1 and GAPDH antibodies. mRNA from DU145 cells, exposed to vehicle or large EVs for the indicated times, was analyzed by qRT-PCR with commercially available specific primers for GOT1 and GAPDH (Hs.PT.58.45409452, and Hs.PT.39a22214836, respectively) (Integrated DNA Technologies).
Aspartate Aminotransferase Activity Assay
Parental DU145 cells were treated with either 1.15 g/mL (large EVs) or 1.10 g/mL (exosomes) density fractions (20µg/ml of protein lysate) for 24 h in presence of 1% or 5% glutamine and then analyzed by using an Aspartate Aminotransferase Activity Assay kit following the manufacturer’s instructions (Sigma). Statistical significance was calculated using a 2-tailed unpaired Student’s t test among the relative biological replicates.
Cell cycle analysis of DU145 cells exposed to large oncosomes
1x105 DU145 cells were plated in 6 well plates overnight and then treated with large EVs or vehicle for 24 h, in presence of 1% or 5% glutamine. Cells were fixed, permeabilized, treated with RNAses (50µg/ml), and labeled with Propidium Iodide (5µg/ml). At least 10,000 events per experiment were recorded using a LSRII cytometer (BD Biosciences) and a BD FACSDiva software. Data analysis was performed with the FlowJo software (Treestar).
Lung mouse tissue staining
Lung mouse tissues from animals injected with shDIAPH3 DU145 cells into the tail vein were immunostained with CK18 [9] and imaged by light microscopy.
Acknowledgments
The authors thank Drs. Stefano Pluchino, Clotilde Thery, Richard Simpson, Leonora Balaj, Xandra Breakefield, Julie Yang, Mirja Rotinen, Kenneth Steadman, and Jayoung Kim for helpful discussions and comments. This study was supported by grants from the National Institutes of Health (NCI NIH R00 CA131472 (to D.D.V.); NIH UCLA SPORE in Prostate Cancer award P50 CA092131 (to D.D.V.); Avon Foundation Fund 02-2013-043 (to D.D.V.); R01 CA143777, CA112303, R37 DK47556 (to M.R.F.); the Steven Spielberg Discovery Fund in Prostate Cancer Research (to M.R.F. and D.D.V.); the Martz Translational Breast Cancer Research Fund (to M.R.F. and D.D.V.); and by a Ladies Auxiliary VFW Cancer Research Postdoctoral Fellowship (to S.M.).
CONFLICTS OF INTEREST
D.D.V.: Inventor of two patents on EVs as circulating biomarkers of cancer (pending). M.R.F., V.R.M., M.M.: co-inventors of two patents on EVs as circulating biomarkers of cancer (pending).
References
1. Redzic JS, Balaj L, van der Vos KE and Breakefield XO. Extracellular RNA mediates and marks cancer progression. Seminars in cancer biology. 2014; 28:14-23.
2. Raposo G, Nijman HW, Stoorvogel W, Liejendekker R, Harding CV, Melief CJ and Geuze HJ. B lymphocytes secrete antigen-presenting vesicles. J Exp Med. 1996; 183(3):1161-1172.
3. Muralidharan-Chari V, Clancy J, Plou C, Romao M, Chavrier P, Raposo G and D’Souza-Schorey C. ARF6-regulated shedding of tumor cell-derived plasma membrane microvesicles. Curr Biol. 2009; 19(22):1875-1885.
4. D’Souza-Schorey C and Di Vizio D. Biology and proteomics of extracellular vesicles: harnessing their clinical potential. Expert Rev Proteomics. 2014; 11(3):251-253.
5. Antonyak MA, Li B, Boroughs LK, Johnson JL, Druso JE, Bryant KL, Holowka DA and Cerione RA. Cancer cell-derived microvesicles induce transformation by transferring tissue transglutaminase and fibronectin to recipient cells. Proc Natl Acad Sci U S A. 2011; 108(12):4852-4857.
6. Cocucci E, Racchetti G and Meldolesi J. Shedding microvesicles: artefacts no more. Trends Cell Biol. 2009; 19(2):43-51.
7. Tauro BJ, Greening DW, Mathias RA, Mathivanan S, Ji H and Simpson RJ. Two distinct populations of exosomes are released from LIM1863 colon carcinoma cell-derived organoids. Mol Cell Proteomics. 2013; 12(3):587-598.
8. Di Vizio D, Kim J, Hager MH, Morello M, Yang W, Lafargue CJ, True LD, Rubin MA, Adam RM, Beroukhim R, Demichelis F and Freeman MR. Oncosome formation in prostate cancer: association with a region of frequent chromosomal deletion in metastatic disease. Cancer Res. 2009; 69(13):5601-5609.
9. Di Vizio D, Morello M, Dudley AC, Schow PW, Adam RM, Morley S, Mulholland D, Rotinen M, Hager MH, Insabato L, Moses MA, Demichelis F, Lisanti MP, Wu H, Klagsbrun M, Bhowmick NA, et al. Large oncosomes in human prostate cancer tissues and in the circulation of mice with metastatic disease. The American journal of pathology. 2012; 181(5):1573-1584.
10. Hager MH, Morley S, Bielenberg DR, Gao S, Morello M, Holcomb IN, Liu W, Mouneimne G, Demichelis F, Kim J, Solomon KR, Adam RM, Isaacs WB, Higgs HN, Vessella RL, Di Vizio D, et al. DIAPH3 governs the cellular transition to the amoeboid tumour phenotype. EMBO Mol Med. 2012; 4(8):743-760.
11. Kim J, Morley S, Le M, Bedoret D, Umetsu DT, Di Vizio D and Freeman MR. Enhanced shedding of extracellular vesicles from amoeboid prostate cancer cells: Potential effects on the tumor microenvironment. Cancer Biol Ther. 2014; 15(4).
12. Minciacchi VR, Freeman MR, Di Vizio D. Extracellular vesicles in cancer: Exosomes, microvesicles and the emerging role of large oncosomes. Semin Cell Dev Biol. 2015; http://dx.doi:10.1016/j.semcdb.2015.02.010.
13. Tahir SA, Ren C, Timme TL, Gdor Y, Hoogeveen R, Morrisett JD, Frolov A, Ayala G, Wheeler TM and Thompson TC. Development of an immunoassay for serum caveolin-1: a novel biomarker for prostate cancer. Clin Cancer Res. 2003; 9(10 Pt 1):3653-3659.
14. Bobrie A, Colombo M, Krumeich S, Raposo G and Thery C. Diverse subpopulations of vesicles secreted by different intracellular mechanisms are present in exosome preparations obtained by differential ultracentrifugation. Journal of extracellular vesicles. 2012; 1:18397
15. Tauro BJ, Greening DW, Mathias RA, Ji H, Mathivanan S, Scott AM and Simpson RJ. Comparison of ultracentrifugation, density gradient separation, and immunoaffinity capture methods for isolating human colon cancer cell line LIM1863-derived exosomes. Methods. 2012; 56(2):293-304.
16. Lötvall J, Hill AF, Hochberg F, Buzás EI, Di Vizio D, Gardiner C, Gho YS, Kurochkin IV, Mathivanan S, Quesenberry P, Sahoo S, Tahara H, Wauben MH, Witwer KW and Théry C. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. Journal of Extracellular Vesicles. 2014; 3:26913
17. Colombo M, Raposo G and Thery C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annual review of cell and developmental biology. 2014; 30:255-289.
18. Morello M, Minciacchi VR, de Candia P, Yang J, Posadas E, Kim H, Griffiths D, Bhowmick N, Chung LW, Gandellini P, Freeman MR, Demichelis F and Di Vizio D. Large oncosomes mediate intercellular transfer of functional microRNA. Cell Cycle. 2013; 12(22):3526-3536.
19. Bolstad BM, Irizarry RA, Astrand M and Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003; 19(2):185-193.
20. Hwang D, Rust AG, Ramsey S, Smith JJ, Leslie DM, Weston AD, de Atauri P, Aitchison JD, Hood L, Siegel AF and Bolouri H. A data integration methodology for systems biology. Proc Natl Acad Sci U S A. 2005; 102(48):17296-17301.
21. Lee HJ, Suk JE, Patrick C, Bae EJ, Cho JH, Rho S, Hwang D, Masliah E and Lee SJ. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J Biol Chem. 2010; 285(12):9262-9272.
22. Mathivanan S, Ji H and Simpson RJ. Exosomes: extracellular organelles important in intercellular communication. Journal of proteomics. 2010; 73(10):1907-1920.
23. Mathivanan S, Lim JW, Tauro BJ, Ji H, Moritz RL and Simpson RJ. Proteomics analysis of A33 immunoaffinity-purified exosomes released from the human colon tumor cell line LIM1215 reveals a tissue-specific protein signature. Mol Cell Proteomics. 2010; 9(2):197-208.
24. Simpson RJ, Kalra H and Mathivanan S. ExoCarta as a resource for exosomal research. Journal of extracellular vesicles. 2012; 1; 1:18374
25. Franco OE, Jiang M, Strand DW, Peacock J, Fernandez S, Jackson RS, 2nd, Revelo MP, Bhowmick NA and Hayward SW. Altered TGF-beta signaling in a subpopulation of human stromal cells promotes prostatic carcinogenesis. Cancer Res. 2011; 71(4):1272-1281.
26. Tuxhorn JA, Ayala GE, Smith MJ, Smith VC, Dang TD and Rowley DR. Reactive stroma in human prostate cancer: induction of myofibroblast phenotype and extracellular matrix remodeling. Clin Cancer Res. 2002; 8(9):2912-2923.
27. Webber JP, Spary LK, Sanders AJ, Chowdhury R, Jiang WG, Steadman R, Wymant J, Jones AT, Kynaston H, Mason MD, Tabi Z and Clayton A. Differentiation of tumour-promoting stromal myofibroblasts by cancer exosomes. Oncogene. 2015; 34(3):290-302.
28. Clayton A, Mitchell JP, Court J, Mason MD and Tabi Z. Human tumor-derived exosomes selectively impair lymphocyte responses to interleukin-2. Cancer Res. 2007; 67(15):7458-7466.
29. Premzl A, Zavasnik-Bergant V, Turk V and Kos J. Intracellular and extracellular cathepsin B facilitate invasion of MCF-10A neoT cells through reconstituted extracellular matrix in vitro. Experimental cell research. 2003; 283(2):206-214.
30. Coller HA. Is cancer a metabolic disease? The American journal of pathology. 2014; 184(1):4-17.
31. Li Z, Wang Y, Wu H, Zhang L, Yang P and Li Z. GRP78 enhances the glutamine metabolism to support cell survival from glucose deficiency by modulating the beta-catenin signaling. Oncotarget. 2014; 5(14):5369-5380.
32. Kim DK, Kang B, Kim OY, Choi DS, Lee J, Kim SR, Go G, Yoon YJ, Kim JH, Jang SC, Park KS, Choi EJ, Kim KP, Desiderio DM, Kim YK, Lotvall J, et al. EVpedia: an integrated database of high-throughput data for systemic analyses of extracellular vesicles. Journal of extracellular vesicles. 2013; 2:20384.
33. Hanahan D and Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144(5):646-674.
34. Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, Kang Y, Fleming JB, Bardeesy N, Asara JM, Haigis MC, DePinho RA, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013; 496(7443):101-105.
35. Cossetti C, Iraci N, Mercer TR, Leonardi T, Alpi E, Drago D, Alfaro-Cervello C, Saini HK, Davis MP, Schaeffer J, Vega B, Stefanini M, Zhao C, Muller W, Garcia-Verdugo JM, Mathivanan S, et al. Extracellular Vesicles from Neural Stem Cells Transfer IFN-gamma via Ifngr1 to Activate Stat1 Signaling in Target Cells. Molecular cell. 2014; 56(2):193-204.
36. Al-Nedawi K, Meehan B, Micallef J, Lhotak V, May L, Guha A and Rak J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell Biol. 2008; 10(5):619-624.
37. D’Asti E, Garnier D, Lee TH, Montermini L, Meehan B and Rak J. Oncogenic extracellular vesicles in brain tumor progression. Front Physiol. 2012; 3:294.
38. Peinado H, Aleckovic M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, Hergueta-Redondo M, Williams C, Garcia-Santos G, Ghajar C, Nitadori-Hoshino A, Hoffman C, Badal K, Garcia BA, Callahan MK, Yuan J, et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med. 2012; 18(6):883-891.
39. Rak J and Guha A. Extracellular vesicles--vehicles that spread cancer genes. Bioessays. 2012; 34(6):489-497.
40. Turiak L, Misjak P, Szabo TG, Aradi B, Paloczi K, Ozohanics O, Drahos L, Kittel A, Falus A, Buzas EI and Vekey K. Proteomic characterization of thymocyte-derived microvesicles and apoptotic bodies in BALB/c mice. Journal of proteomics. 2011; 74(10):2025-2033.
41. Marthick JR and Dickinson JL. Emerging putative biomarkers: the role of alpha 2 and 6 integrins in susceptibility, treatment, and prognosis. Prostate Cancer. 2012; 2012:298732.
42. Ricci E, Mattei E, Dumontet C, Eaton CL, Hamdy F, van der Pluije G, Cecchini M, Thalmann G, Clezardin P and Colombel M. Increased expression of putative cancer stem cell markers in the bone marrow of prostate cancer patients is associated with bone metastasis progression. Prostate. 2013; 73(16):1738-1746.
43. Hosseini-Beheshti E, Pham S, Adomat H, Li N and Tomlinson Guns ES. Exosomes as biomarker enriched microvesicles: characterization of exosomal proteins derived from a panel of prostate cell lines with distinct AR phenotypes. Mol Cell Proteomics. 2012; 11(10):863-885.
44. Ji H, Greening DW, Barnes TW, Lim JW, Tauro BJ, Rai A, Xu R, Adda C, Mathivanan S, Zhao W, Xue Y, Xu T, Zhu HJ and Simpson RJ. Proteome profiling of exosomes derived from human primary and metastatic colorectal cancer cells reveal differential expression of key metastatic factors and signal transduction components. Proteomics. 2013; 13(10-11):1672-1686.
45. Long Y, Tsai WB, Wangpaichitr M, Tsukamoto T, Savaraj N, Feun LG and Kuo MT. Arginine deiminase resistance in melanoma cells is associated with metabolic reprogramming, glucose dependence, and glutamine addiction. Molecular cancer therapeutics. 2013; 12(11):2581-2590.
46. Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB and Thompson CB. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A. 2008; 105(48):18782-18787.
47. Chen HA, Chang YW, Tseng CF, Chiu CF, Hong CC, Wang W, Wang MY, Hsiao M, Ma JT, Chen CH, Jiang SS, Wu CH, Hung MC, Huang MT and Su JL. E1A-Mediated Inhibition of HSPA5 Suppresses Cell Migration and Invasion in Triple-Negative Breast Cancer. Annals of surgical oncology. 2015; 22(3):889-898.
48. Huang M, Li Z, Li D, Walker S, Greenan C and Kennedy R. Structure-based design of HSPA5 inhibitors: from peptide to small molecule inhibitors. Bioorganic & medicinal chemistry letters. 2013; 23(10):3044-3050.
49. Freeman MR, Kim J, Lisanti MP and Di Vizio D. A metabolic perturbation by U0126 identifies a role for glutamine in resveratrol-induced cell death. Cancer Biol Ther. 2011; 12(11):966-977.
50. Oyama K, Fushida S, Kinoshita J, Okamoto K, Makino I, Nakamura K, Hayashi H, Inokuchi M, Nakagawara H, Tajima H, Fujita H, Takamura H, Ninomiya I, Kitagawa H, Fujimura T and Ohta T. Serum cytokeratin 18 as a biomarker for gastric cancer. Clin Exp Med. 2013; 13(4):289-295.
51. Koelink PJ, Lamers CB, Hommes DW and Verspaget HW. Circulating cell death products predict clinical outcome of colorectal cancer patients. BMC Cancer. 2009; 9:88.
52. Tan HL, Haffner MC, Esopi DM, Vaghasia AM, Giannico GA, Ross HM, Ghosh S, Hicks JL, Zheng Q, Sangoi AR, Yegnasubramanian S, Osunkoya AO, De Marzo AM, Epstein JI and Lotan TL. Prostate adenocarcinomas aberrantly expressing p63 are molecularly distinct from usual-type prostatic adenocarcinomas. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2015; 28(3):446-456.
53. Germann M, Wetterwald A, Guzman-Ramirez N, van der Pluijm G, Culig Z, Cecchini MG, Williams ED and Thalmann GN. Stem-like cells with luminal progenitor phenotype survive castration in human prostate cancer. Stem cells. 2012; 30(6):1076-1086.
54. Yang W, Chung YG, Kim Y, Kim TK, Keay SK, Zhang CO, Ji M, Hwang D, Kim KP, Steen H, Freeman MR and Kim J. Quantitative proteomics identifies a beta-catenin network as an element of the signaling response to Frizzled-8 protein-related antiproliferative factor. Mol Cell Proteomics. 2011; 10(6):M110 007492.
55. Yang W, Di Vizio D, Kirchner M, Steen H and Freeman MR. Proteome scale characterization of human S-acylated proteins in lipid raft-enriched and non-raft membranes. Mol Cell Proteomics. 2010; 9(1):54-70.
56. Dowal L, Yang W, Freeman MR, Steen H and Flaumenhaft R. Proteomic analysis of palmitoylated platelet proteins. Blood. 2011; 118(13):e62-73.
57. Vizcaino JA, Cote RG, Csordas A, Dianes JA, Fabregat A, Foster JM, Griss J, Alpi E, Birim M, Contell J, O’Kelly G, Schoenegger A, Ovelleiro D, Perez-Riverol Y, Reisinger F, Rios D, et al. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic acids research. 2013; 41(Database issue):D1063-1069.
58. Cox J and Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008; 26(12):1367-1372.
59. Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV and Mann M. Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res. 2011; 10(4):1794-1805.
60. Moon PG, Lee JE, You S, Kim TK, Cho JH, Kim IS, Kwon TH, Kim CD, Park SH, Hwang D, Kim YL and Baek MC. Proteomic analysis of urinary exosomes from patients of early IgA nephropathy and thin basement membrane nephropathy. Proteomics. 2011; 11(12):2459-2475.
61. Choi DS, Choi DY, Hong BS, Jang SC, Kim DK, Lee J, Kim YK, Kim KP and Gho YS. Quantitative proteomics of extracellular vesicles derived from human primary and metastatic colorectal cancer cells. Journal of extracellular vesicles. 2012; 1:18704.