
Introduction
Thyroid cancer is the most common endocrine-related cancer, highly heterogeneous in clinical and genetic aspects. Well-differentiated papillary thyroid carcinoma (PTC) constitutes about 85% of all thyroid malignancies [1]. Perturbation of Mitogen-activated Protein Kinase (MAPK) pathway has been frequently described in PTC patients [2]. Mutations in BRAF and RAS genes (HRAS, KRAS and NRAS) and genomic rearrangements involving RET gene (RET/PTC) account for about 70% of PTC cases [3]. In a small percentage of cases (~5%), TRK oncogenes rearrangements have been observed [4]. However, despite the presence of tumor-initiating genetic alterations, cancer results from the progressive accumulation of mutations in genes that confer growth advantage over surrounding cells [5]. Innovative sequencing technologies (Next-Generation Sequencing, NGS, 6) have revolutionized cancer research [7], improving our ability to investigate tumor mutations’ landscape. PTC genetic characterization will improve clinicians’ ability to establish diagnosis and to predict prognosis and individual response to treatments. Notably, during the writing of the manuscript, a large-scale study exploring single nucleotide variants (SNVs), gene expression and epigenetic features in PTC has been published [8].
Here we describe the identification of a fusion gene and new somatic mutations in PTC patients by RNA-Sequencing. We found, and validated by targeted sequencing, a new chimeric transcript generated by the fusion of WNK lysine deficient protein kinase 1 (WNK1) and beta-1,4-N-acetyl-galactosaminyl transferase 3 (B4GALNT3) genes. We also discovered new missense mutations in CBL, NOTCH1, PIK3R4 and SMARCA4 genes in PTC patients. Moreover, we report, for the first time in PTC, somatic mutations in cancer driver genes (DICER1, MET and VHL) described in other tumors. Finally, our analysis confirmed the frequencies of genetic/genomic alterations in BRAF, RET and RAS genes, revealing that these alterations are mutually exclusive and that give rise to distinct gene expression signatures.
Results
The experimental and computational workflow of our study is schematized in Supplementary Figure 1.
Detection of known driver PTC mutations
To verify if RNA-Seq can reliably identify mutations in driver genes we did not prescreen PTC samples for known mutations/gene fusions. We sequenced paired-end libraries from a discovery cohort of 4 healthy individuals and 18 patients randomly chosen from well-characterized cohort of 80 PTC patients (Supplementary Table 1). As expected by epidemiology and literature data, ~65-70% of tumors had at least one known driver mutation or gene rearrangement. Known mutations identified by RNA-Seq - and validated by targeted sequencing - are summarized in Figure 1A (upper panel). Most of them (~38%) were RET gene fusions. Six patients had CCDC6-RET (RET/PTC1, ~33%) and one NCOA4-RET (RET/PTC3, ~5%) gene fusions, with a PTC1/PTC3 ratio quite similar to that described in literature for patients not exposed to ionizing radiations. RNA-Seq data confirmed RET overexpression in these patients (FDR <0.01; Supplementary Figure 2). We also found PAX8-PPARG and ETVN6-NTRK3 gene fusions (~5% frequency each). RNA-Seq data confirmed the overexpression of PPARG and NTRK3 (FDR <0.01; Supplementary Figure 2). Notably, PAX8-PPARG chimeric gene, associated to follicular carcinomas, has been reported with low frequency in PTC [8]. RT-PCR and targeted sequencing on cDNAs validated all gene fusions detected by RNA-Sequencing.
Additionally, we found BRAFV600E and HRASQ61R (~16% frequency each; Figure 1A), whereas we did not detect NRAS/KRAS mutations nor TRK gene rearrangements. Notably, BRAF and HRAS mutations, as well as RET/PTC and other rearrangements, were mutually exclusive in PTC patients. The presence of mutations was confirmed on patients’ DNA by targeted sequencing. Such analysis was extended also to negative patients, confirming again the bona fide of the SNP calling procedure on RNA-Seq data.
Taking advantage of RNA-Seq data, we correlated global gene expression profiles to known mutations and rearrangements. We found that BRAF-mutated and RET/PTC samples have very similar gene expression patterns and that they differ from RAS-mutated patients (~1400 differentially expressed genes; FDR <0.05). Extending the analysis to PTC patients without any known mutation we found RAS- and BRAF-like gene signatures (Figure 1B). These findings are in agreement with the notion the BRAFV600E and RET over-expression activate MAPK pathway more than HRASQ61R and with the recent results of TCGA Consortium [8]. Indeed, we found a significant over-expression - in BRAF- vs RAS-like PTCs - of DUSP genes (DUSP2, DUSP5 and DUSP6) that are induced through the stimulation of ERK signaling via MAPK. Conversely, RAS-mutated patients over-expressed anti-apoptotic genes, including BCL2.
Finally, we identified known somatic mutations in E-cadherin (CDH1A592T), in thyroid stimulating hormone receptor (TSHRI568F), in isocitrate dehydrogenase 1 (IDH1V178I) and in fms-related tyrosine kinase 3 (FLT3D324N) genes (Figure 1A). CDH1 and IDH1 mutations co-occur with BRAF and HRAS mutations, respectively (Figure 1A). Mutation frequencies are in line with COSMIC database (~2-5%).
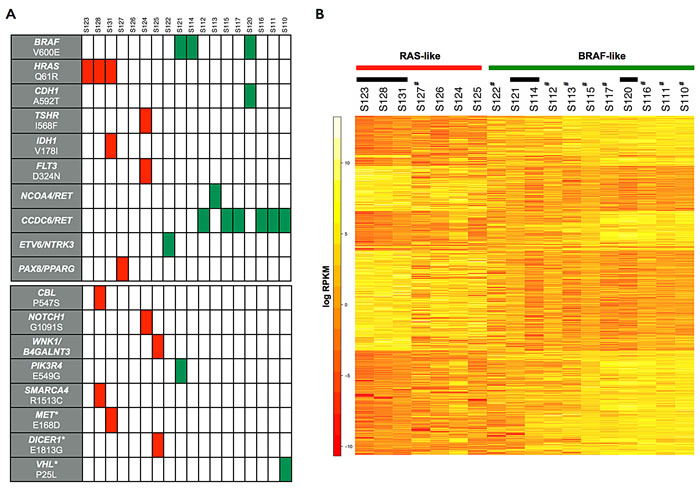
Figure 1: Point mutations, gene fusions and gene expression signatures in papillary thyroid carcinoma. A, Schematic representation of protein-altering mutations and gene fusions identified in PTC samples. Each vertical column represents a PTC patient. In the upper panel, known missense mutations and fusion transcripts associated with papillary thyroid carcinoma are shown. In the lower panel are depicted newly identified somatic mutations and other somatic alterations in cancer driver genes reported in other tumors but described for the first time in PTC (indicated by asterisks). Red boxes indicate HRAS-mutated patients or those with a RAS-like transcriptional profile. Green boxes indicate BRAF-mutated or RET/PTC patients with a BRAF-like transcriptional profile. B, Heatmap of the hierarchical clustering of differentially expressed genes between BRAF-like and RAS-like PTC samples. Black bars indicate samples with point mutations in HRAS and BRAF genes. # indicate samples with gene fusions.
CBL, NOTCH1, PIK3R4 and SMARCA4 genes are mutated in PTC
The reliable identification of known driver mutations using RNA-Seq encouraged us to search for new mutations in cancer-related genes. Combining RNA-Seq data with IntoGen [9] and COSMIC databases [10], we identified - and selected for validation - the following missense mutations (and amino acid changes): c.C1639T (P547S) in CBL (proto-oncogene E3 ubiquitin protein ligase), c.G3271A (G1091S) in NOTCH1, c.A1646G (E549G) in PIK3R4 (phosphoinositide-3-kinase regulatory subunit 4 gene) and c.C4537T (R1513C) in SMARCA4 (SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily a, member 4). Low-frequency mutations in CBL and SMARCA4 co-occur with HRASQ61R, PIK3R4 with BRAFV600E, whereas NOTCH1 with TSHRI568F and FLT3D324N in one patient (Figure 1A). Targeted sequencing on DNAs of positive and negative samples validated the RNA-Seq findings. Mutation frequencies are shown in Supplementary Table 2. Notably, new mutations in CBL, NOTCH1, PIK3R4 and SMARCA4 genes are not annotated as single nucleotide polymorphisms (SNPs) in dbSNP v138 (http://www.ncbi.nlm.nih.gov/SNP/), and in the 1000GenomeProject (http://www.1000genomes.org), nor in COSMIC database. To strengthen these findings, we screened by targeted sequencing 80 alleles from healthy donors and we did not detect any of these mutations.
Noteworthy, RNA-Seq data and targeted sequencing demonstrated that all new mutations are present both on DNA and mRNA, therefore they are not generated by RNA editing. In addition, mutated genes are expressed and it is likely they are translated into mutated proteins. Through in silico analyses we attempted to predict the role of these new mutations on protein functionality.
Possible functional/structural consequences of CBL, NOTCH1, PIK3R4 and SMARCA4 mutations
The proline-to-serine amino acid change in E3 ubiquitin protein ligase CBL occurs in a proline stretch (PPPPPPDR) of the Proline-rich domain (Figure 2A). Since the mutated residue is smaller and less hydrophobic than the wild-type it is predicted to affect local folding. Notably, proline residues in this domain are highly conserved and neither the mutant nor other residues with similar biochemical properties have been observed at this position in homologous sequences. Thus, conservation and structural scores indicate the mutation as damaging.
The glycine-to-serine mutation in Notch1 falls in a conserved glycine of a highly-conserved functional region, the EGF-like domain 28 (Figure 2B). Mutant and wild-type amino acids differ in size, charge and hydrophobic properties. Sift and Polyphen scores indicate this mutation as damaging.
The glutamic-acid-to-glycine mutation identified in the phosphoinositide-3-kinase regulatory subunit 4 (PI3KR4) falls in a highly conserved “Armadillo-like helical” domain (Figure 2C). This multi-helical fold, with extensive solvent-accessible surface, is suited to bind large substrates such as proteins and nucleic acids. The mutant and wild-type amino acids differ in electric charge and hydrophobic properties, and the presence of glycine - instead of glutamic acid - is predicted to significantly reduce chain rigidity. In silico data indicate that the mutation is potentially damaging to PIK3R4 activity.
The arginine-to-cystein mutation in SMARCA4 occurs in a critical functional region (Figure 2D), the bromodomain (BRD). Structural 3D analysis revealed that the wild-type residue (arginine) forms salt bridges with Asp1506, Glu1512 and Asp1528 and that the mutated residue loses these interactions (details in Figure 2D).
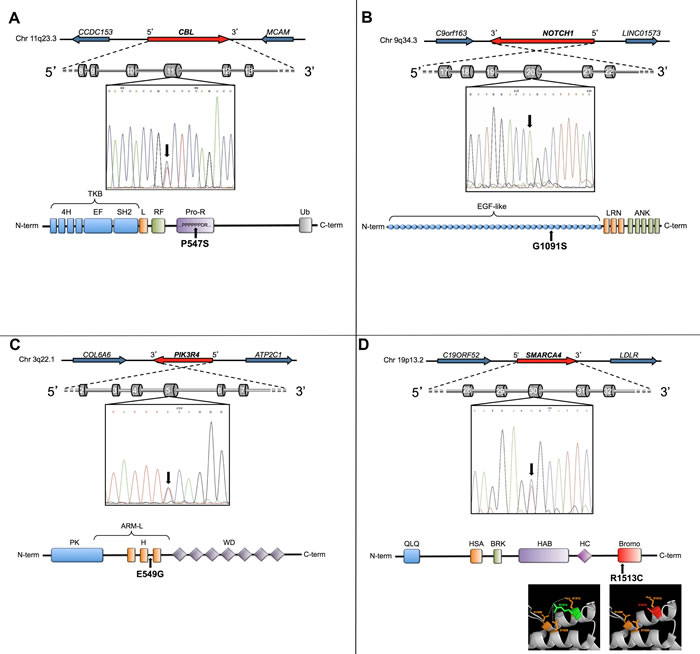
Figure 2: New mutations identified in papillary thyroid carcinoma. A-D, the genomic localization and the exon/intron structure of each mutated gene are schematized (A, CBL; B, NOTCH1; C, PIK3R4; D, SMARCA4). In each panel, the ectropherogram shows the nucleotide variation identified by RNA-Seq, and the protein graphic representation shows the functional domains affected. In panel D, a detail of the three-dimensional structure of SMARCA4 bromodomain highlights the salt interactions among wild-type residue (colored in green) and the surrounding amino acids (colored in orange). These interactions are lost in the mutated protein (the mutated residue colored in red).
WNK1-B4GALNT3: identification of a novel gene fusion
Computational analysis of chimeric transcripts revealed the presence of known gene fusions (CCDC6-RET, NCOA4-RET, PAX8-PPARG and ETVN6-NTRK3; Figure 1A). Moreover, we identified a new WNK1-B4GALNT3 chimeric transcript in one patient, negative for known PTC-causing genetic alterations. RNA-Seq data indicated that the new chimeric transcript originates by fusion of the exon 1 of WNK1 and the exon 2 of B4GALNT3 (Figure 3A). Fusion-specific RT-PCR and sequencing confirmed the fusion breakpoint in the transcript, revealing also that it undergoes alternative splicing (Figure 3B). Notably, sequence and ORF analysis revealed that the longest fusion transcript is out-of-frame whereas the alternative isoform keeps the ORF intact. We demonstrated that this patient does not carry reciprocal gene fusions. In addition, since RNA-Seq indicated that the canonical mRNAs of WNK1 and B4GALNT3 genes are transcribed in this patient, we confirmed it by RT-PCR (Figure 3C). Using the same approach, we validated the absence of gene fusions in negative patients (Figure 3D). In line with the over-expression of RET, PPARG and NTRK3 in patients with gene rearrangements, RNA-Seq data showed B4GALNT3 over-expression in this patient (p <0.05; Supplementary Figure 2). The expression of WNK1 was not affected. Fusion partners map on chromosome 12 (chr12p13.33), are transcribed from the same strand (5’-3’ orientation) and are separated by about 220 Kb (Figure 3A). It indicates that the fusion derives from an intrachromosomal paracentric rearrangement. We could not identify in this patient, or in other patients of the discovery cohort, additional fusions involving genes mapping in the same genomic region.
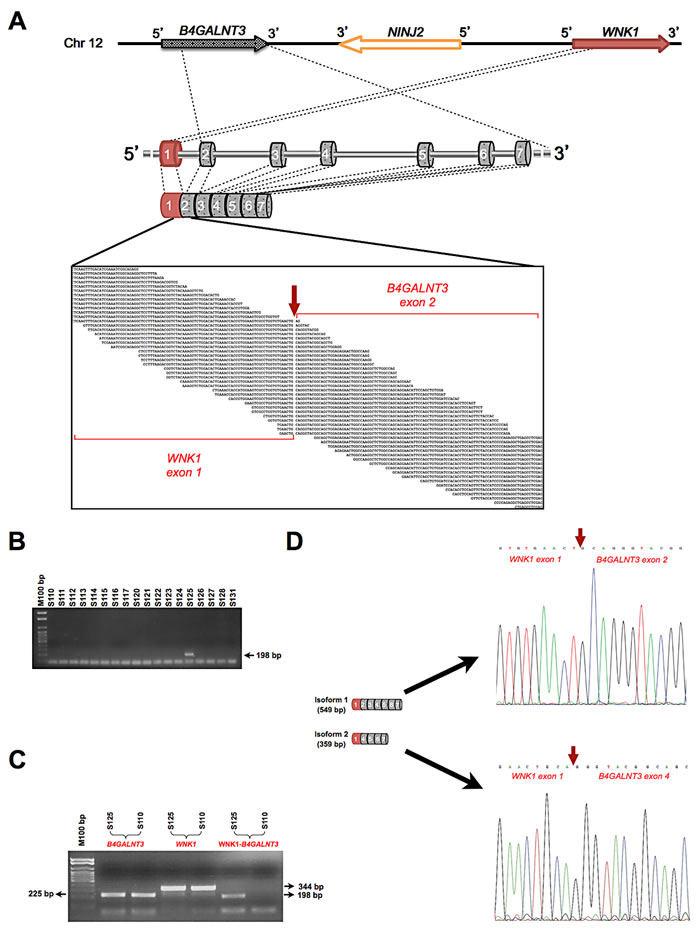
Figure 3: New WNK1-B4GALNT3 gene fusion in papillary thyroid carcinoma. A, Schematic representation of the localization of the fusion partners, WNK1 and B4GALNT3, on chromosome 12. The exons of WNK1 and B4GALNT3 genes that are involved in the fusion are indicated in red and grey, respectively. The RNA-Sequencing reads that map across the fusion breakpoint are shown in the black box. The red arrow indicates the exact fusion breakpoint. B, RT-PCR validation of the WNK1-B4GALNT3 fusion performed on the RNA of 18 PTC samples of the discovery cohort. C, Qualitative RT-PCR assay on the mRNAs of WNK1 and B4GALNT3 parent genes. Agarose gel picture shows the PTC sample with the fusion and a negative sample. D, Schematic mRNA structure of the two isoforms of WNK1-B4GALNT3 fusion gene. The electropherograms show the nucleotide sequences of the breakpoint (indicated by red arrows).
Mutations in known cancer driver genes
The filtering procedures during the SNP calling retrieved ~7430 missense, stop gain/loss point mutations and INDELs. We focused on 125 “Mut-driver” genes, defined by Vogelstein and colleagues [5] as those containing a sufficient number of driver mutations to unambiguously distinguish them from other genes. We found 44 variants in 32 genes (Supplementary File 1). Notably, most of the genes that we found mutated for the first time in PTC (i.e. CBL, NOTCH1, SMARCA4, MET and VHL) are “Mut-driver” genes. Figure 1 shows whether mutations in these genes co-occur with known oncogenic mutations, gene rearrangements and fusions.
Moreover, we investigated known cancer driver genes and their interacting partners in pathways commonly associated to tumorigenesis. We extensively searched damaging mutations affecting JAK-STAT signaling, MAPK, apoptosis, cell cycle, Hedgehog, oncosuppressors and oncogenes, DNA-repair and spliceosome pathways (Figure 4). We found 61 mutated genes in these pathways, some of them annotated as cancer driver in IntoGen [9]. We identified damaging mutations in ATR, BRCA1/2, MAP4K1, CUL3 and MAX, already reported in other cancer types but not described yet as mutated in PTC. Although most of them are not classified as cancer drivers, some mutations discovered here in PTC for the first time - MST1R703C and BOCQ915H - are somatic in COSMIC [10].

Figure 4: Genomic landscape of PTC mutations and related pathways. Co-occurrence of protein-altering nonsense and missense mutations identified in PTC patients (n=18) by RNA-Sequencing. Most relevant shared mutations in biological pathways associated to tumorigenesis are shown. Each vertical column represents a PTC patient. HRASQ61R, BRAFV600E and RET/PTC patients are indicated by †, * and #, respectively. The severity of the amino acid change is proportional to the intensity of red and green boxes (according to MA, “Mutation Assessor”, and Sift scores).
Discussion
Our study describes the identification in PTC of a novel gene fusion, new somatic mutations in established cancer driver genes and known mutations (reported in other cancer types) not described yet in PTC. Taking advantage of next-generation RNA-Seq, we found that BRAF- and RAS-mutated tumors have distinct gene expression profiles, and that RET/PTC samples resemble tumors with BRAFV600E mutation, in line with the very recent results of TCGA [8]. We found that gene expression is mostly correlated to specific genetic alterations rather than tumor stage, suggesting the importance to genetically characterize PTC patients.
We confirmed that driving somatic mutations and rearrangements are mutually exclusive in PTC. Accordingly, the new WNK1-B4GALNT3 fusion has been discovered in a patient negative for known PTC-inducing alterations. Noteworthy, a significant over-expression of B4GALNT3 gene was found in this patient, whereas the fusion partner was not affected (Supplementary Figure 2). B4GALNT3 can act both as tumor suppressor in neuroblastoma [11] and as oncogene in the colon, increasing the malignant phenotype of colon cancer cells through enhanced integrin and MAPK signaling [12]. Its expression positively correlates with metastasis and poor survival in patients with colorectal cancer [13]. Our data suggest a new role of B4GALNT3 as oncogene in PTC. Notably, WNK1-B4GALNT3 positive patient also carries a somatic mutation in DICER1, cancer driver gene. The mutation affects the metal binding site of RNase IIIb domain, recently found mutated in non-epithelial ovarian cancers [14]. To the best of our knowledge, DICER1E1813G mutation has not been previously described in PTC.
Furthermore, we discovered a new missense NOTCH1G1091S mutation in a patient negative for BRAF/RAS mutations and RTKs rearrangements. Many NOTCH1 driver mutations have been reported in hematopoietic tumors, head and neck squamous cell carcinoma and other malignancies [15]. The presence of inactivating mutations indicates this gene as tumor suppressor, rather than oncogene, in solid tumors [15], [16]. Interestingly, most of the mutations in solid tumors are clustered within EGF-like repeats. Accordingly, the NOTCH1G1091S mutation falls in the EGF-like domain 28 and affects a highly conserved residue. Although 3D model revealed this domain does not directly bind Notch ligands, Sharma and colleagues reported it to interact with EGF-like 11-15 domains, crucial for receptor activity [17]. Thus, this mutation may affect Notch1 protein functionality and Notch signaling that is directly linked to PTC cell proliferation [18]. These results suggest that this pathway should be taken into account as adjuvant therapy for treating PTC. The same mutation co-occurs with two low-frequency mutations (TSHRI568F and FLT3D324N) previously reported in PTC.
Additional missense mutations have been discovered in BRAF- and RAS-mutated patients. Among these, CBLP547S affects the proline-rich region, responsible of the binding with SH3 domain of Grb2 that indirectly recruits it to RTKs via GRB2 adaptor protein [19]. In lung cancer, mutations in CBL and in other driver genes usually co-occur [19]. Indeed, the new CBL mutation was found in a HRAS-mutated patient. Interestingly, the same patient carried a new missense mutation in the tumor suppressor gene SMARCA4, frequently mutated in lung cancer and small cell ovarian carcinoma [20], [21]. Such mutation disrupts salt interactions with charged residues in the BRD, a functional domain that allows the recognition of acetyl lysine marks on H3 and H4 tails [22]. SMARCA4 protein, associating with Rb proteins, induces cell cycle arrest through HDAC-dependent transcriptional repression. Mutations, rearrangements or over-expression of BRD-containing proteins have been reported in tumors, and BRD inhibitors have been developed to induce cycle arrest and apoptosis of carcinoma cells [22]. Therefore, a similar pharmacological approach could be adopted in the treatment of PTC cases with SMARCA4 mutations.
Finally, other mutations - confirmed somatic in other cancer types - were identified for the first time in PTC. METE168D in the SEMA domain, crucial for the interaction with plexins, has been previously described in small cell lung cancer. Interestingly, this mutation impairs the affinity for HGF and alters MET functionality [23]. We also found nucleotide variations in ATM gene. This finding is relevant since PTC is the most frequent radiation-sensitive tumor, and ATM is a fundamental kinase that triggers the DNA damage checkpoint, determining cell cycle arrest, DNA repair or apoptosis.
In this study we have confirmed that next generation RNA-Sequencing is a powerful approach to simultaneously identify gene expression profiles, gene fusions and mutations in cancer samples. One of the major limitations in this kind of approach is that only mutations in expressed genes can reliably be detected. However, although it may limit the SNP calling in RNA-Seq data, it is proven that most of mutations responsible of tumor phenotypes fall in the protein coding regions of actively transcribed genes and lead to mutated proteins. In conclusion, our data confirm the genetic heterogeneity of PTC and reveal the accumulation of mutations in new driver candidate genes. These findings pave the way to novel potential pharmacological therapies in PTC treatment, based on the presence of new affected pathways, such as Notch signaling and chromatin remodeling.
MATERIALS AND METHODS
Patients and RNA samples preparation
Thyroid biopsies of 18 PTC and 4 control thyroids constituted the discovery cohort for RNA-Sequencing. Tumor specimens were not prior genetically characterized. Biopsies from other 32 PTC patients and 26 healthy thyroids constituted the validation cohort. All biopsies were obtained from the Service d’Anatomo-Pathologie, Centre Hospitalier Lyon Sud, France. Informed written consent was obtained from patients of both cohorts. Approval of the ethic committee of University of Naples “Federico II” and Lyon Sud Hospital Center was obtained to use these samples for RNA-sequencing, Sanger sequencing and validation purposes. Total RNA was extracted from biopsies using Trizol standard procedure and RNA integrity was assessed using digital gel electrophoresis (Experion) and spectrophotometry as previously described [24].
Library preparation and RNA-Sequencing data analysis
Paired-end libraries were prepared using TruSeq RNA Sample Preparation Kit (Illumina) and sequenced on Illumina HiSeq2000 platform according to manufacturer’s instructions. Details of RNA-Seq data analysis are given in Supplementary Methods Section. Briefly, reads mapping was performed using TopHat v.2.0.7 [25], PCR duplicates were removed using Picard tools v1.117 (http://broadinstitute.github.io/picard/) and SNP calling was carried out using GATK v3.3 workflow optimized for RNA-Seq reads [26]. Nucleotide variants were annotated using ANNOVAR [27] and COSMIC database [10] was used to label variants as “somatic in cancer”. Common germ-line variants annotated in dbSNP v138, 1000 genomes as well as those identified in healthy thyroids were filtered out. SNVs in super-duplicated regions were also removed. For gene expression analysis, read counts per gene was obtained with HTSeq [28] and RPKM (Reads per kilobase transcript per million reads) normalization was used [32]. Fusion transcripts were identified using TopHat Fusion [30] and Chimerascan v0.4.5 [31].
Targeted DNA sequencing and mutation detection
Genomic DNA was isolated from 80 thyroid biopsies (50 PTC and 30 healthy thyroids) and 40 peripheral blood samples (healthy donors) using standard lysis/phenol extraction protocols as previously described [32]. Known and newly identified point mutations (Supplementary Table 2) were validated in tumor DNA using gene-specific PCR assays and Sanger sequencing. New mutations in CBL, NOTCH1, PIK3R4 and SMARCA4 genes were screened on genomic DNA of healthy individuals. Oligonucleotides sequences are provided in Supplementary Table 3.
Acknowledgements
This work was supported by grants from Ministero dell’Istruzione, dell’Università e della Ricerca MIUR (PRIN 2011), POR Campania FSE 2007-2013 (CREMe), PNR-CNR Aging Program 2012-2014, Associazione Italiana per la Ricerca sul Cancro (AIRC IG 11477), CNR Flagship Projects (Epigenomics-EPIGEN, Nanomax-DESIRED), PON01-02782 (Nuove strategie nanotecnologiche per la messa a punto di farmaci e presidi diagnostici diretti verso cellule cancerose circolanti) to Alfredo Fusco.
The work was also supported by grants from Associazione Italiana per la Ricerca sul Cancro (AIRC IG 2013-14689), the FLAGSHIP “InterOmics” Project (PB.P05), PON01_02460 (DIAGEN) to Alfredo Ciccodicola. The IGB-CNR laboratory is member of the COST-Action (BM1006) “Next Generation Sequencing Data Analysis Network”. We thank Marianna Aprile, Margherita Scarpato and Vincenzo Mercadante for RNA-Sequencing library preparation, and the NGS Facility of TIGEM for the Illumina platform.
Competing interests
The authors declare that they have no competing interests.
References
1. Kondo T, Ezzat S, Asa SL. Pathogenetic mechanisms in thyroid follicular-cell neoplasia. Nat Rev Cancer 2006;6:292-306.
2. Nikiforov Yuri E. Thyroid carcinoma: molecular pathways and therapeutic targets. Modern Pathology 2008;21:S37–S43.
3. Santoro M, Carlomagno F. Central role of RET in thyroid cancer. Cold Spring Harb Perspect Biol 2013;5:a009233.
4. Greco A, Miranda C, Pierotti MA. Rearrangements of NTRK1 gene in papillary thyroid carcinoma. Mol Cell Endocrinol 2010;321:44-49.
5. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science 2013;339:1546-1558.
6. Costa V, Angelini C, De Feis I, Ciccodicola A. Uncovering the complexity of transcriptomes with RNA-Seq. J Biomed Biotechnol 2010;2010:853916.
7. Costa V, Aprile M, Esposito R, Ciccodicola A. RNA-Seq and human complex diseases: recent accomplishments and future perspectives. Eur J Hum Genet 2013;21:134-142.
8. Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014; 159: 676-690.
9. Gonzalez-Perez A, Perez-Llamas C, Deu-Pons J, Tamborero D, Schroeder MP, Jene-Sanz A, Santos A, Lopez-Bigas N. IntOGen-mutations identifies cancer drivers across tumor types. Nat Methods 2013;10:1081-1082.
10. Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, Kok CY, Jia M, De T, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2014;pii:gku1075.
11. Hsu WM, Che MI, Liao YF, Chang HH, Chen CH, Huang YM, Jeng YM, Huang J, Quon MJ, Lee H, Huang HC, Huang MC. B4GALNT3 expression predicts a favorable prognosis and suppresses cell migration and invasion via β1 integrin signaling in neuroblastoma. Am J Pathol 2011;179:1394-1404.
12. Huang J, Liang JT, Huang HC, Shen TL, Chen HY, Lin NY, Che MI, Lin WC, Huang MC. Beta1,4-N-acetylgalactosaminyltransferase III enhances malignant phenotypes of colon cancer cells. Mol Cancer Res 2007;5:543-552.
13. Che MI, Huang J, Hung JS, Lin YC, Huang MJ, Lai HS, Hsu WM, Liang JT, Huang MC. β1, 4-N-acetylgalactosaminyltransferase III modulates cancer stemness through EGFR signaling pathway in colon cancer cells. Oncotarget 2014;5:3673-3684.
14. Heravi-Moussavi A, Anglesio MS, Cheng SW, Senz J, Yang W, Prentice L, Fejes AP, Chow C, Tone A, Kalloger SE, Hamel N, Roth A, Ha G, et al. Recurrent somatic DICER1 mutations in nonepithelial ovarian cancers. N Engl J Med 2012;366:234-242.
15. Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, Fakhry C, Xie TX, Zhang J, Wang J, Zhang N, El-Naggar AK, Jasser SA, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 2011;333:1154-1157.
16. Klinakis A, Lobry C, Abdel-Wahab O, Oh P, Haeno H, Buonamici S, van De Walle I, Cathelin S, Trimarchi T, Araldi E, Liu C, Ibrahim S, Beran M, et al. A novel tumour-suppressor function for the Notch pathway in myeloid leukaemia. Nature 2011;473:230-233.
17. Sharma A, Rangarajan A, Dighe RR. Antibodies against the extracellular domain of human Notch1 receptor reveal the critical role of epidermal-growth-factor-like repeats 25-26 in ligand binding and receptor activation. Biochem J 2013;449:519-530.
18. Yamashita AS, Geraldo MV, Fuziwara CS, Kulcsar MA, Friguglietti CU, da Costa RB, Baia GS, Kimura ET. Notch pathway is activated by MAPK signaling and influences papillary thyroid cancer proliferation. Transl Oncol 2013;6:197-205.
19. Tan YH, Krishnaswamy S, Nandi S, Kanteti R, Vora S, Onel K, Hasina R, Lo FY, El-Hashani E, Cervantes G, Robinson M, Hsu HS, Kales SC, et al. CBL is frequently altered in lung cancers: its relationship to mutations in MET and EGFR tyrosine kinases. PLoS One 2010;5:e8972.
20. Jelinic P, Mueller JJ, Olvera N, Dao F, Scott SN, Shah R, Gao J, Schultz N, Gonen M, Soslow RA, Berger MF, Levine DA. Recurrent SMARCA4 mutations in small cell carcinoma of the ovary. Nat Genet 2014;46:424-426.
21. Medina PP, Romero OA, Kohno T, Montuenga LM, Pio R, Yokota J, Sanchez-Cespedes M. Frequent BRG1/SMARCA4-inactivating mutations in human lung cancer cell lines. Hum Mutat 2008;29:617-622.
22. Muller S, Filippakopoulos P, Knapp S. Bromodomains as therapeutic targets. Expert Rev Mol Med 2011;13:e29.
23. Ma PC, Kijima T, Maulik G, Fox EA, Sattler M, Griffin JD, Johnson BE, Salgia R. c-MET mutational analysis in small cell lung cancer: novel juxtamembrane domain mutations regulating cytoskeletal functions. Cancer Res 2003;63:6272-6281.
24. Costa V, Angelini C, D’Apice L, Mutarelli M, Casamassimi A, Sommese L, Gallo MA, Aprile M, Esposito R, Leone L, Donizetti A, Crispi S, Rienzo M, et al. Massive-scale RNA-Seq analysis of non ribosomal transcriptome in human trisomy 21. PLoS One 2011;6:e18493.
25. Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 2013;14:R36.
26. Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, Banks E, Garimella KV, Altshuler D, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 2013;1110:11.10.1-11.10.33.
27. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010;38:e164.
28. Anders S, Pyl PT, Huber W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 2014;pii:btu638.
29. Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 2008;5:621-628.
30. Kim D, Salzberg SL. TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol 2011;12:R72.
31. Iyer MK, Chinnaiyan AM, Maher CA. ChimeraScan: a tool for identifying chimeric transcription in sequencing data. Bioinformatics 2011;27:2903-2904.
32. Costa V, Conte I, Ziviello C, Casamassimi A, Alfano G, Banfi S, Ciccodicola A. Identification and expression analysis of novel Jakmip1 transcripts. Gene 2007;402:1-8.