
Introduction
Monoclonal antibodies which inhibit downstream pathway signaling by targeting the extracellular ligand binding domain have become one of the mainstays of EGFR inhibition. For the treatment of metastatic colorectal cancer (mCRC) the chimeric EGFR antibody cetuximab and the fully human antibody panitumumab were approved as single agents or in combination with chemotherapy [1-11]. Both antibodies were also used in patients with gastric or pancreatic cancer, cholangiocellular carcinoma (CCC) or other gastrointestinal cancers in clinical trials [12-17]. Resistance to these antibodies is mediated by mutations in downstream signaling molecules [18-21], with mutated RAS, which is currently the only validated and widely accepted molecular marker that predicts lack of response to EGFR antibodies and, therefore, guides treatment decisions in mCRC [20, 22-25]. Therefore, patients are routinely screened for KRAS exon 2/3/4 and NRAS exon2/3/4 mutations before the initiation of EGFR targeted therapy [26, 27]. However, even patients without RAS mutations who primarily respond well to EGFR antibodies will eventually develop secondary resistance limiting the clinical benefit of these drugs.
Some recent studies have addressed the molecular mechanisms underlying acquired resistance. Accumulating evidence suggests that RAS wt tested tumors may harbor small RAS mutated subclones at diagnosis that emerge and thus mediate secondary resistance under the selective pressure of treatment with EGFR antibodies [28-30]. Moreover, very recently a mutation in the ectodomain of EGFR leading to the substitution of serine by arginine in position 492 has been described. This mutation can be acquired during therapy with cetuximab and mediates resistance to this antibody (but not to panitumumab) by abrogating its binding to the EGFR [31, 32]. Differential resistance in this mutant is not surprising as we could recently show that the large conformational EGFR domain III epitopes of both antibodies only partially overlap and position S492 belongs exclusively to the cetuximab binding site [33].
Here, we investigated EGFR ectodomain and RAS mutations in patients with gastrointestinal cancer treated with EGFR-targeting antibodies and describe for the first time a panitumumab-induced EGFR mutation that mediates cross-resistance to both panitumumab and cetuximab by critically changing an amino acid position localized within the overlap of both antibody epitopes. Perspectively, screening of ctDNA for EGFR ectodomain mutations may be helpful in monitoring patients for resistance-mediating tumor subclones.
Results
Clinical characteristics of the “tumor tissue” patient cohort
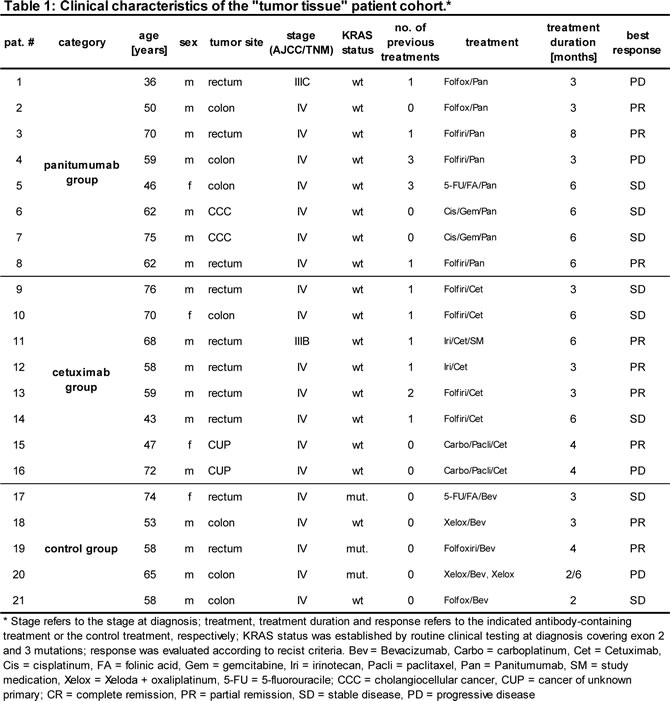
16 EGFR antibody-naïve patients of the “tumor tissue” patient cohort were treated with cetuximab or panitumumab in combination with chemotherapy as shown in Table 1. EGFR antibodies were applied after an average of one prior therapy and the majority of patients showed at least stable disease. The mean duration of EGFR antibody treatment prior to secondary surgery and thus post-treatment sample acquisition was 4.8 months. Five patients treated with the VEGF antibody bevacizumab in combination with chemotherapy were used as control group.
Targeted NGS of EGFR and RAS in samples from the “tumor tissue” cohort
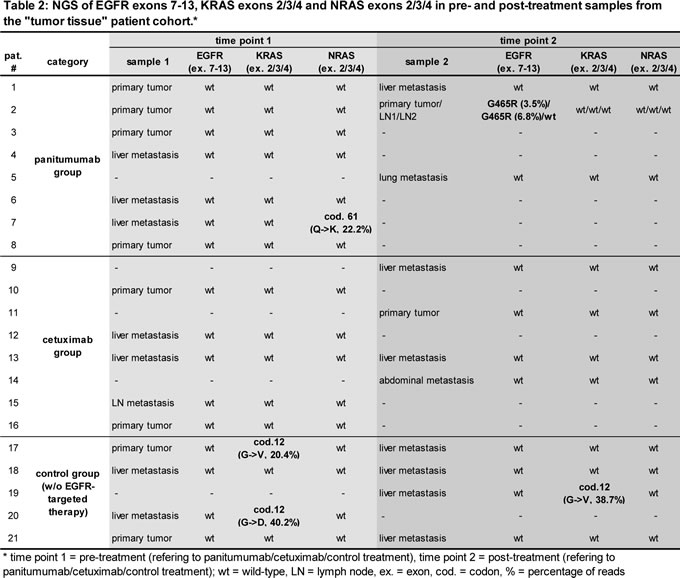
KRAS 2/3 status of baseline samples (determined by routine clinical testing) was confirmed by targeted NGS of these exons. In addition, the mutational status of KRAS exon 4 and NRAS exon 2/3/4 was determined by NGS at baseline (Table 2). Interestingly, tissue samples from patients tested as RAS wt at baseline showed no evidence for RAS mutated minimal subclones after treatment.
In addition, we performed NGS to identify mutations in the EGFR ectodomain potentially interfering with antibody binding. None of the pre-treatment or control samples showed EGFR ectodomain mutations in exons 7-13. In 1 out of 3 patients treated with panitumumab we found an acquired EGFR G465R ectodomain mutation after treatment with panitumumab and FOLFOX in post-treatment tumor material (patient 2, Table 2). This novel exon 12 mutation was localized in proximity to the previously described cetuximab-induced S492R mutation (Figure 1A) [31]. It constituted 3.5% of all exon 12 reads and could also be detected in a tumor-infiltrated lymph node resected together with the local tumor after treatment with panitumumab and FOLFOX at a frequency of 6.8%. Deep sequencing of pre-treatment primary tumor (Table 2) as well as peripheral blood leukocyte DNA from this patient resulted in 100% germline sequence for exon 12 (data not shown) confirming the acquired nature of the mutation.
Binding profile of EGFR mutation G465R
Based on structure analysis position, G465 is located right in the center of the overlap region of the large conformational cetuximab and panitumumab epitopes previously characterized by our group (Figure 1A). We therefore hypothesized that the introduction of a positive charge at this position due to the glycine to arginine exchange in the malignant cells may abrogate not only panitumumab, but also cetuximab binding. To address this question, we generated a recombinant EGFR variant containing the same amino acid substitution (glycine to arginine: G465R). The mutation was introduced by site-directed mutagenesis in a vector suitable for protein expression of the EGFR ectodomain as Fc-fusion protein (EGFR-Fc) and a vector suitable for membrane expression of the whole receptor. Furthermore, the previously published EGFR S492R mutant was included in the experiment. Correct expression and immobilization of EGFR-Fc wt and mutant proteins was assessed by ELISA (Supplementary Figure 2). Whilst panitumumab did not bind to immobilized EGFR-Fc G465R protein, its binding to wt EGFR-Fc and EGFR-Fc S492R was preserved (Figure 1B). Cetuximab only bound to wt EGFR-Fc, but to neither of the mutant receptors (Figure 1B). Moreover, EGFR wt and mutant constructs were transfected into EGFR-negative CHO cells and binding of panitumumab and cetuximab to membrane-expressed wt and mutant receptors was analyzed by flow cytometry (Figure 1C and D). A polyclonal EGFR antibody served as a control for receptor expression. As previously shown, cetuximab binding was significantly inhibited in the EGFR S492R mutant, while panitumumab binding was preserved. In the EGFR G465R mutant, binding of both antibodies was almost completely abrogated (binding reduction of 85-90%), in concordance with the central localization of the mutation within the epitopes of both antibodies.
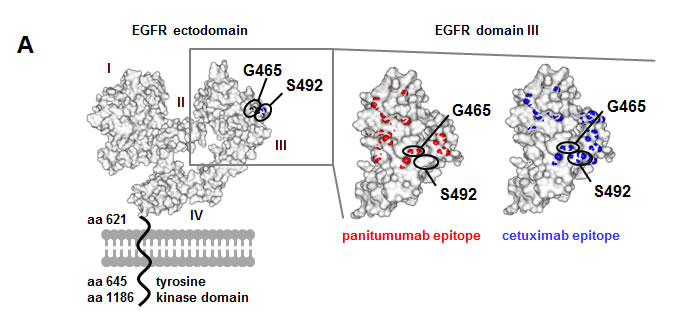
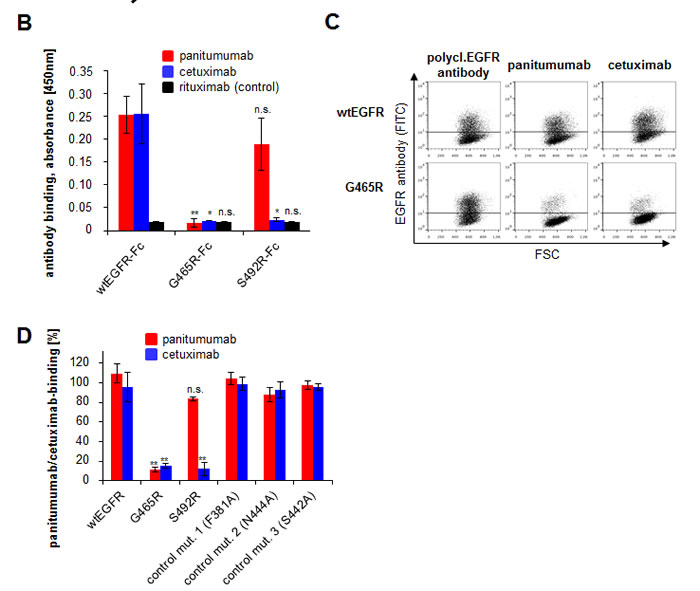
Figure 1: EGFR G465R mutant reveals almost complete abrogation of panitumumab and cetuximab binding. A: Localization of EGFR mutations G465R and S492R on the ectodomain of the EGFR. The 3-dimensional EGFR model was created from pdb file 1NQL from RCSB Protein Data Bank. The S492 position is shown in blue, the G465 position in black. The panel on the right shows the EGFR domain III alone with panitumumab epitope in red and cetuximab epitope in blue. Overlaps of mutated positions G465 and S492 with antibody epitopes are shown. B: EGFR mutation G465R abrogates binding of panitumumab and cetuximab at the protein level. Wile-type and mutant EGFR-Fc proteins were expressed and the binding of therapeutic antibodies to immobilized proteins was assessed by ELISA. Data are means from 3 experiments +/- SEM. * p<0.05, ** p<0.01, n.s. = not significant (student’s T-test comparing binding of respective antibody to mutant versus wt EGFR-Fc) C and D: Panitumumab and cetuximab binding is abrogated in CHO cells transfected with EGFR G465R. EGFR negative CHO cells were transfected with wild type EGFR or mutants thereof. Binding of panitumumab, cetuximab or a control polyclonal EGFR antibody was assessed by FACS analysis 48h after transfection. FSC = forward scatter. Panel C shows exemplary FACS plots, panel D shows mean data from 5 experiments with binding of the polyclonal EGFR antibody set to 100% +/- SEM. * p<0.05, ** p<0.01, n.s. = not significant (student’s T-test comparing binding of respective antibody to mutant versus wt EGFR).
Functional validation of EGFR mutation G465R in Ba/F3 cells
Next, we asked i) if receptor function was still preserved in the EGFR G465R mutant and ii) if the significant inhibition of antibody binding to EGFR G465R translated into resistance to panitumumab and cetuximab in a cellular model. To address this experimentally, we stably transfected murine EGFR-negative, IL-3-dependent Ba/F3 pro-B cells with the EGFR wt or mutant G465R and S492R constructs. After selection with G418, ectopic expression of wt and mutant EGFRin these cells conferred IL-3 independence in the presence of EGF, but not if erlotinib was added (Supplementary Figure 3). This indicated that EGF binding to the EGFR mutants was still preserved and receptor function intact. Stable Ba/F3 cell lines expressing wt or mutant EGFR were then treated with panitumumab, cetuximab, control antibody rituximab or erlotinib for 2 hours (Figure 2A). Whilst in EGFR wt cells cetuximab, panitumumab and erlotinib completely blocked EGFR phosphorylation, only panitumumab showed this effect in the S492R mutant. In the G465R mutant, none of the antibodies blocked EGFR phosphorylation. EGFR wt and G465R mutant Ba/F3 cells were then cultured for 108 hours in the presence of panitumumab, cetuximab or control antibody rituximab. Whilst EGFRwt transfected cells were sensitive to treatment with panitumumab and cetuximab, proliferation of EGFR G465R transfected cells was unaffected by treatment with either of these antibodies (Figure 2B). This data suggested that the G465R mutation, acquired under treatment with panitumumab, mediates cross-resistance to panitumumab and cetuximab by disrupting the antibody-EGFR interaction.
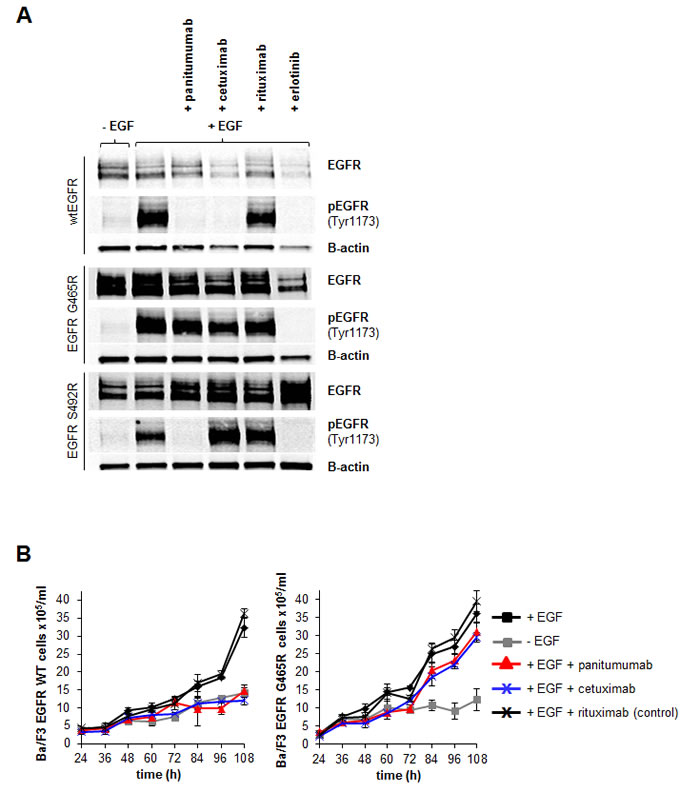
Figure 2: EGFR G465R mutation induces cross-resistance to panitumumab and cetuximab in an EGF-dependent Ba/F3 cellular model. A: EGFR signaling in EGF-dependent Ba/F3 model. Wt and S492R or G465R mutant EGFR-expressing Ba/F3 cells were cultured in the presence or absence of EGF and with addition of cetuximab, panitumumab, rituximab or erlotinib. After 2 hours, cells were harvested and EGFR/pEGFR expression analyzed by western blot analysis. B: Sensitivity of EGFR wt or EGFR G465R mutant-transfected Ba/F3 cells to treatment with EGFR-targeted antibodies. Ba/F3 cells were transformed to IL-3 independence with EGFR wt or mutant constructs and subsequently cultured in the presence or absence of EGF or with EGF in combination with panitumumab, cetuximab or control antibody rituximab. The number of viable cells was determined by trypan blue exclusion every 12 hours beginning 24 hours after seeding and plotted. Data are means from triplicate experiments +/-SEM.
Targeted NGS of EGFR and RAS in ctDNA of independent “liquid biopsy” patient cohort
To validate our findings, ctDNA from an independent “liquid biopsy” cohort of 22 patients treated with EGFR antibodies and 6 control cases was subjected to NGS to detect acquired EGFR exon 12 and RAS mutations (Table 3). In contrast to the “tumor tissue” cohort, these patients had clinically more advanced disease with on average 2.4 prior therapies.
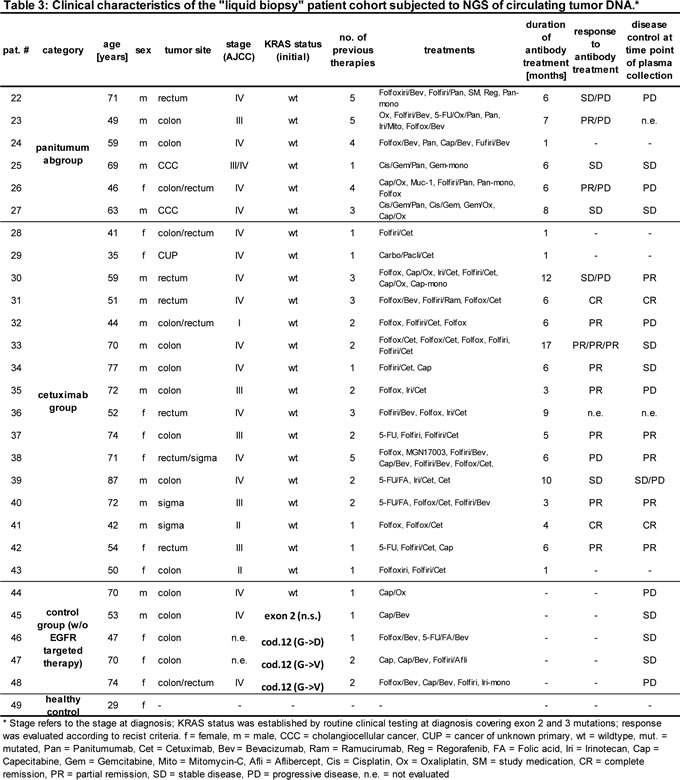
As expected, the ctDNA analysis confirmed the RAS status of the control group without EGFR antibody treatment. Well in line with previous studies, we found additional acquired RAS mutations in about one third of patients (Table 3) [28, 29]. RAS mutations were exclusively found in the cetuximab patient group. However, this apparent overrepresentation did not result statistically significant by χ2 testing, corresponding to previously published work on acquired RAS mutations after both cetuximab and panitumumab treatment [28, 29].
Interestingly, 1 out of 6 panitumumab-treated patients showed evidence of the EGFR G465R mutation with a frequency of 7.7% (patient 25, Table 4). This patient with cholangiocellular carcinoma was unmutated for EGFR exon 12 at baseline as evidenced by NGS of his primary tumor tissue (data not shown) and had stable disease after 6 months of chemotherapy in combination with panitumumab. The blood sample was drawn 3 months after cessation of panitumumab. Of note, the previously described S492R mutation was not detected in our patient cohort.

Discussion
Treatment and survival of patients with mCRC has improved over the past decade, largely due to the advent of new drugs, in particular targeted therapies. Around 60% of patients with RAS wt tumors respond to first-line chemotherapy plus EGFR-directed antibodies [2]. However, secondary resistance emerges in most patients at a median of 9 to 12 months [34]. Whilst drug resistance to EGFR-directed small molecule tyrosine kinase inhibitors has been well characterized in oncology and mutational analyses trigger therapeutic decisions in specific disease settings, resistance to monoclonal antibodies is less well understood. In the context of EGFR targeting, recent publications shed light on some of the molecular mechanisms underlying clinical resistance to cetuximab and panitumumab. These incriminated the selection of subclones with activating RAS mutations [28, 29] as well as an epitope-changing point mutation [31] in the EGFR ectodomain acquired during cetuximab treatment. The latter mechanism is of particular interest since it represents the first mutation described to confer resistance by destroying a therapeutic antibody’s epitope.
These data raised several questions: How frequently do EGFR ectodomain mutations contribute to clinical resistance to EGFR antibody treatment in relation to acquired RAS mutations? How early do they occur in the course of therapy? Are other EGFR ectodomain mutations than the previously described S492R mutation acquired during EGFR inhibition (particularly during treatment with panitumumab)?
Here, we investigated EGFR domain III and activating RAS mutations in a “tumor tissue” cohort of 21 patients with gastrointestinal cancers, mainly mCRC, including 16 patients with EGFR-targeted therapy. These patients were – in their majority – clinically responsive to the antibody-containing treatment. In 1 out of 3 patients with available post-treatment samples after panitumumab-containing treatment we found a novel acquired mutation in exon 12 of the EGFR ectodomain. Since this mCRC patient had undergone surgery at partial remission after 3 months of FOLFOX in combination with panitumumab, we had post-treatment tumor material as well as tumor-infiltrated lymph nodes available for mutational analysis by NGS (unfortunately, no follow-up clinical data was available as this patient died due to post-surgery complications). Interestingly, the mutation was not only present in the post-treatment tumor, but also in one of the resected tumor-infiltrated lymph nodes in 3.5 and 6.8% of reads (excluding a potential sequencing artifact). The actual percentage of cells carrying the mutated receptor can, however, not directly be inferred from the percentage of reads showing the point mutation since signals of non-neoplastic cells are admixed in both materials and the sequencing of genomic DNA always yields sequences from the unmutated allele as well. Since the G465R mutation centrally introduces a positive charge in both epitopes, it was not surprising to find abrogation of panitumumab and cetuximab binding leading to cross-resistance to both antibodies in a cellular model. NGS of ctDNA in an independent “liquid biopsy” patient cohort showed that this mutation was present in one additional patient with cholangiocellular carcinoma. Altogether, this sums up to 2/9 patients with this resistance-mediating mutation after treatment with panitumumab. Correlating clinical outcomes of patients with the EGFR G465 mutational status is, however, limited by the overall patient number (n=2 patients harboring the G465R mutation) in this study as well as the fact that these patients received antibody-chemotherapy combinations which makes it difficult to estimate the net clinical effect of the antibody. Since the mutation was found in patients who had stable disease or a partial response to chemotherapy in combination with panitumumab, we conclude that our technique may allow us to detect mutations before overt clinical resistance occurs [30, 35-38].
It remains an open question, however, if the addition of an EGFR targeting antibody to chemotherapy is entirely useless in a patient harboring a subclonal mutation or if patients may derive some benefit since the unmutated tumor cells may still be targeted. This issue needs to be prospectively addressed by future studies.
In addition to the newly discovered EGFR exon 12 mutation, we detected acquired RAS mutations in about one third of patients treated with EGFR antibodies in the “liquid biopsy” cohort, in line with previously published work [28, 29]. In contrast, sequencing performed on post-treatment tumors of the “tumor tissue” cohort did not show any acquired RAS mutations. This difference may be in part due to the fact that the “tumor tissue” cohort included less advanced disease stages than the “liquid biopsy” cohort. More importantly our data suggests that ctDNA sequencing may be more suitable for the identification of small resistant subclones since ctDNA reflects to a greater extent the genetic heterogeneity of the tumor.
Putting this investigation in relation to what other studies have shown, we most be aware that different patient cohorts were investigated. While this study was based on an unselected patient cohort with the majority of patient not considered clinically refractory to EGFR inhibition, other studies on acquired RAS and EGFR S492R mutations have been conducted in patient cohorts considered resistant to the antibody-containing regimen [28, 31]. This represents the most obvious difference between the studies and may help to explain disparities between mutational frequencies regarding the EGFR S492R mutation as well as acquired RAS mutations.
Taken together, our data shapes our understanding of epitope-changing EGFR mutations in gastrointestinal tumors showing that two different mutations can arise early on during EGFR-targeted treatment. These mutations are less frequently acquired as compared to RAS mutations and they can induce resistance or even cross-resistance depending on their localization within the panitumumab/cetuximab epitopes. Liquid biopsy strategies allow therapy monitoring and may perspectively help to guide treatment decisions in patients during EGFR targeted therapy. Validation in samples from large clinical studies of defined patient cohorts receiving chemotherapy in combination with EGFR-inhibiting antibodies are clearly warranted. We expect that these studies will help to define cut-offs for clinically significant mutational loads.
Materials and Methods
Study design, patient cohorts and ethics statement
Data, tumor and blood samples of 48 patients with gastrointestinal cancer (41 CRC, 7 non-CRC) treated at our institution between February 2012 and August 2014 as well as one healthy donor were included in this prospective, longitudinal study after patients’ written informed consent and approval by the ethics commission. In the first training cohort, tissue samples were analyzed pre- and post-treatment (if available) for EGFR and RAS genes as described below ("tumor tissue“ patient cohort). In the second cohort, ctDNA post-treatment was analyzed for EGFR and RAS genes in an independent set of patients ("liquid biopsy“ patient cohort).
Targeted next-generation sequencing (NGS)
EGFR exons 7-13, KRAS exons 2/3/4 and NRAS exons 2/3/4 were amplified from genomic DNA isolated from paraffin embedded tumor tissue or leukocyte DNA as described in the Supplementary Detailed NGS Methods Section using the primers shown in Supplementary Table 1. NGS was performed with a median number of 27694 reads per exon per patient to detect even small tumor subclones. Sequences were aligned with the reference sequences shown in Supplementary Table 2. A schematic overview of the amplification strategies is shown in Supplementary Figure 1A.
Generation of human EGFR wt and mutant constructs
cDNAs coding for the human wt EGFR ectodomain (aa1 to 645) and the human IgG1 Fc-fragment (for EGFR-Fc fusion protein expression) or the complete human wt EGFR (for membrane expression of the receptor in eukaryotic cells) were inserted into the vector pcDNA3.1(+) (Life Technologies, Carlsbad, USA). EGFR mutants were generated with the QuikChange XL Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, USA) as described [39] using individually designed oligonucleotides (Supplementary Table 3).
Recombinant expression and purification of EGFR-Fc proteins
HEK293 cells (CRL-1573, ATCC, Manassas, USA) were transfected with EGFR -Fc wt and mutant constructs using Lipofectamin 2000 (Life Technologies, Carlsbad, USA). Conditioned serum-free medium was collected and EGFR-Fc proteins were purified via Protein A-Sepharose (Pierce, Appleton, USA).
ELISA with EGFR-Fc proteins
96-well ELISA plates were coated with recombinant wt or mutant EGFR-Fc proteins. Correct expression and immobilization of EGFR-Fc proteins was assessed by ELISA using a biotinylated anti-human EGFR antibody (R&D Systems, Minneapolis, USA) and streptavidin-peroxidase conjugate (Roche, Basel, Switzerland).
To study binding of therapeutic antibodies to the fusion proteins, immobilized EGFR-Fc proteins were incubated with cetuximab (Merck, Darmstadt, Germany), panitumumab (Amgen, Thousand Oaks, USA) or rituximab (Roche, Basel, Switzerland) at 100ng/ml and detected with a biotinylated goat anti-human kappa-specific antibody (Southern Biotech, Birmingham, USA) followed by secondary detection as above.
Transfection with EGFR wt and mutant constructs
EGFR -negative CHO cells (CCL-61, ATCC, Wesel, Germany) were chemically transfected with wt, G465R or S492R EGFR encoding vector. Ba/F3 cells (CSC-C2045, Creative Bioarray, New York, USA) kindly provided by Stefan Horn (Research Department Cell and Gene Therapy, Department of Stem Cell Transplantation, University Medical Center Hamburg-Eppendorf, Hamburg, Germany) were maintained in medium containing 10ng/ml murine IL-3 (Peprotech, Rocky Hill, USA, [40]). Electroporation-transfected cells expressing wt or mutant EGFR were G418-selected (1mg/ml) and subsequently cultured in the absence of IL-3 and in the presence of 10ng/ml EGF. Stable cells transformed to IL-3 independence were screened for EGFR functionality by treatment with EGF or EGF + erlotinib at 5µM (Roche, Basel, Switzerland) and subsequently used for drug-sensitivity experiments.
Flow cytometry of transfected cells
Transfected CHO or Ba/F3 cells were stained with a polyclonal goat anti-human EGFR antibody (R&D Systems, Minneapolis, USA), panitumumab or cetuximab followed by secondary detection with FITC-labeled rabbit anti-human (Sigma-Aldrich, St.Gallen, Switzerland), or rabbit anti-goat antibodies (Dako Cytomation, Copenhagen, Denmark). Cells were analyzed on a FACS Calibur (BD Biosciences, Franklin Lakes, USA).
EGFR signaling and drug-sensitivity assays
EGFR wt or mutant transfected Ba/F3 cells were cultured for 2 hours with or without EGF (10ng/ml), in combination with panitumumab, cetuximab, rituximab (10µg/ml), or erlotinib. Lysates were subjected to western blot analysis using the following antibodies: EGFR and phospho-EGFR (Tyr1173) (53A5) (Cell Signaling, Danvers, USA), mouse anti-β-actin (Sigma-Aldrich, St. Louis, USA), IRDye 800CW goat anti-rabbit IgG, IRDye 800CW goat anti-mouse IgG and IRDye 680RD goat anti-rabbit IgG (LI-COR Biosciences, Lincoln, USA). For drug-sensitivity assays, EGFR wt and G465R transfected Ba/F3 cells were cultured +/-EGF or with EGF/panitumumab, EGF/cetuximab or EGF/rituximab, as above. Viable cells were quantified by counting trypan blue excluding cells for 108 hours every 12 hours.
NGS of ctDNA
ctDNA was extracted from plasma using the QIAamp Circulating Nucleic Acid Kit (Qiagen, Hilden, Germany). EGFR exon 12 regions surrounding positions coding for S492 and G465, KRAS exon 2/3/4 and NRAS exon 2/3/4 were amplified using the primer pairs shown in Supplementary Table 1 (ctDNA). Illumina-adapter sequences / sample-specific barcodes were added as schematically shown in Supplementary Figure 1B. Sequencing (with a median number of 27 694 reads per exon per patient) as well as data analysis was performed as mentioned in the in the Supplementary Detailed NGS Methods Section.
Statistics
Overrepresentation of RAS mutations in one of the two EGFR antibody-treated groups (panitumumab versus cetuximab) of both patient cohorts (“tumor tissue” and “liquid biopsy” cohort) was evaluated by chi-square-test Χ2 testing.
Acknowledgements
The authors thank Anita Schulenkorf and Konstantin Hoffer for expert technical assistance.
References
1. Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Verslype C, Chau I, Van Cutsem E. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. The New England journal of medicine. 2004; 351: 337-345.
2. Bokemeyer C, Bondarenko I, Makhson A, Hartmann JT, Aparicio J, de Braud F, Donea S, Ludwig H, Schuch G, Stroh C, Loos AH, Zubel A, Koralewski P. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. Journal of clinical oncology. 2009; 27: 663-671.
3. Jonker DJ, O’Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, Au HJ, Berry SR, Krahn M, Price T, Simes RJ, Tebbutt NC, van Hazel G, Wierzbicki R, et al. Cetuximab for the treatment of colorectal cancer. The New England journal of medicine. 2007; 357: 2040-2048.
4. Van Cutsem E, Peeters M, Siena S, Humblet Y, Hendlisz A, Neyns B, Canon JL, Van Laethem JL, Maurel J, Richardson G, Wolf M, Amado RG. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. Journal of clinical oncology. 2007; 25: 1658-1664.
5. Peeters M, Price TJ, Cervantes A, Sobrero AF, Ducreux M, Hotko Y, Andre T, Chan E, Lordick F, Punt CJ, Strickland AH, Wilson G, Ciuleanu TE, et al. Randomized phase III study of panitumumab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as second-line treatment in patients with metastatic colorectal cancer. Journal of clinical oncology. 2010; 28: 4706-4713.
6. Douillard JY, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, Rivera F, Kocakova I, Ruff P, et al. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. Journal of clinical oncology. 2010; 28: 4697-4705.
7. Van Cutsem E, Kohne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, D’Haens G, Pinter T, Lim R, Bodoky G, Roh JK, Folprecht G, Ruff P, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. The New England journal of medicine. 2009; 360: 1408-1417.
8. Sobrero AF, Maurel J, Fehrenbacher L, Scheithauer W, Abubakr YA, Lutz MP, Vega-Villegas ME, Eng C, Steinhauer EU, Prausova J, Lenz HJ, Borg C, Middleton G, et al. EPIC: phase III trial of cetuximab plus irinotecan after fluoropyrimidine and oxaliplatin failure in patients with metastatic colorectal cancer. Journal of clinical oncology. 2008; 26: 2311-2319.
9. Lang I, Kohne CH, Folprecht G, Rougier P, Curran D, Hitre E, Sartorius U, Griebsch I, Van Cutsem E. Quality of life analysis in patients with KRAS wild-type metastatic colorectal cancer treated first-line with cetuximab plus irinotecan, fluorouracil and leucovorin. European journal of cancer. 2013; 49: 439-448.
10. Tabernero J, Van Cutsem E, Diaz-Rubio E, Cervantes A, Humblet Y, Andre T, Van Laethem JL, Soulie P, Casado E, Verslype C, Valera JS, Tortora G, Ciardiello F, et al. Phase II trial of cetuximab in combination with fluorouracil, leucovorin, and oxaliplatin in the first-line treatment of metastatic colorectal cancer. Journal of clinical oncology. 2007; 25: 5225-5232.
11. Heinemann V, von Weikersthal LF, Decker T, Kiani A, Vehling-Kaiser U, Al-Batran SE, Heintges T, Lerchenmuller C, Kahl C, Seipelt G, Kullmann F, Stauch M, Scheithauer W, et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): a randomised, open-label, phase 3 trial. The lancet oncology. 2014; 15: 1065-1075.
12. Jensen LH, Lindebjerg J, Ploen J, Hansen TF, Jakobsen A. Phase II marker-driven trial of panitumumab and chemotherapy in KRAS wild-type biliary tract cancer. Annals of oncology. 2012; 23: 2341-2346.
13. Crosby T, Hurt CN, Falk S, Gollins S, Mukherjee S, Staffurth J, Ray R, Bashir N, Bridgewater JA, Geh JI, Cunningham D, Blazeby J, Roy R, et al. Chemoradiotherapy with or without cetuximab in patients with oesophageal cancer (SCOPE1): a multicentre, phase 2/3 randomised trial. The lancet oncology. 2013; 14: 627-637.
14. Philip PA, Benedetti J, Corless CL, Wong R, O’Reilly EM, Flynn PJ, Rowland KM, Atkins JN, Mirtsching BC, Rivkin SE, Khorana AA, Goldman B, Fenoglio-Preiser CM, et al. Phase III study comparing gemcitabine plus cetuximab versus gemcitabine in patients with advanced pancreatic adenocarcinoma: Southwest Oncology Group-directed intergroup trial S0205. Journal of clinical oncology. 2010; 28: 3605-3610.
15. Waddell T, Chau I, Cunningham D, Gonzalez D, Okines AF, Okines C, Wotherspoon A, Saffery C, Middleton G, Wadsley J, Ferry D, Mansoor W, Crosby T, et al. Epirubicin, oxaliplatin, and capecitabine with or without panitumumab for patients with previously untreated advanced oesophagogastric cancer (REAL3): a randomised, open-label phase 3 trial. The lancet oncology. 2013; 14: 481-489.
16. Lordick F, Kang YK, Chung HC, Salman P, Oh SC, Bodoky G, Kurteva G, Volovat C, Moiseyenko VM, Gorbunova V, Park JO, Sawaki A, Celik I, et al. Capecitabine and cisplatin with or without cetuximab for patients with previously untreated advanced gastric cancer (EXPAND): a randomised, open-label phase 3 trial. The lancet oncology. 2013; 14: 490-499.
17. Malka D, Cervera P, Foulon S, Trarbach T, de la Fouchardiere C, Boucher E, Fartoux L, Faivre S, Blanc JF, Viret F, Assenat E, Seufferlein T, Herrmann T, et al. Gemcitabine and oxaliplatin with or without cetuximab in advanced biliary-tract cancer (BINGO): a randomised, open-label, non-comparative phase 2 trial. The lancet oncology. 2014; 15: 819-828.
18. Bardelli A, Siena S. Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. Journal of clinical oncology. 2010; 28: 1254-1261.
19. Wheeler DL, Dunn EF, Harari PM. Understanding resistance to EGFR inhibitors-impact on future treatment strategies. Nature reviews clinical oncology. 2010; 7: 493-507.
20. Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD, Robitaille S, Price TJ, Shepherd L, Au HJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. The New England journal of medicine. 2008; 359: 1757-1765.
21. Giampieri R, Scartozzi M, Del Prete M, Maccaroni E, Bittoni A, Faloppi L, Bianconi M, Cecchini L, Cascinu S. Molecular biomarkers of resistance to anti-EGFR treatment in metastatic colorectal cancer, from classical to innovation. Critical reviews in oncology/hematology. 2013; 88: 272-283.
22. Lievre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, Cote JF, Tomasic G, Penna C, Ducreux M, Rougier P, Penault-Llorca F, Laurent-Puig P. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer research. 2006; 66: 3992-3995.
23. Douillard JY, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, Rivera F, Kocakova I, Ruff P, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. The New England journal of medicine. 2013; 369: 1023-1034.
24. Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, Patterson SD, Chang DD. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. Journal of clinical oncology. 2008; 26: 1626-1634.
25. Kumar SS, Price TJ, Mohyieldin O, Borg M, Townsend A, Hardingham JE. KRAS G13D Mutation and Sensitivity to Cetuximab or Panitumumab in a Colorectal Cancer Cell Line Model. Gastrointestinal cancer research. 2014; 7: 23-26.
26. Van Cutsem E, Cervantes A, Nordlinger B, Arnold D. Metastatic colorectal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-updagger. Annals of oncology. 2014; 25: iii1-iii9.
27. Schirripa M, Cremolini C, Loupakis F, Morvillo M, Bergamo F, Zoratto F, Salvatore L, Antoniotti C, Marmorino F, Sensi E, Lupi C, Fontanini G, Gregorio VD, et al. Role of NRAS mutations as prognostic and predictive markers in metastatic colorectal cancer. International journal of cancer. 2015; 136: 83-90.
28. Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M, Siravegna G, Bencardino K, Cercek A, Chen CT, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012; 486: 532-536.
29. Diaz LA Jr, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, Kinzler KW, Oliner KS, Vogelstein B. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012; 486: 537-540.
30. Misale S, Arena S, Lamba S, Siravegna G, Lallo A, Hobor S, Russo M, Buscarino M, Lazzari L, Sartore-Bianchi A, Bencardino K, Amatu A, Lauricella C, et al. Blockade of EGFR and MEK intercepts heterogeneous mechanisms of acquired resistance to anti-EGFR therapies in colorectal cancer. Science translational medicine. 2014; 6: 224ra226.
31. Montagut C, Dalmases A, Bellosillo B, Crespo M, Pairet S, Iglesias M, Salido M, Gallen M, Marsters S, Tsai SP, Minoche A, Seshagiri S, Serrano S, et al. Identification of a mutation in the extracellular domain of the Epidermal Growth Factor Receptor conferring cetuximab resistance in colorectal cancer. Nature medicine. 2012; 18: 221-223.
32. Esposito C, Rachiglio AM, La Porta ML, Sacco A, Roma C, Iannaccone A, Tatangelo F, Forgione L, Pasquale R, Barbaro A, Botti G, Ciardiello F, Normanno N. The S492R EGFR ectodomain mutation is never detected in KRAS wild type colorectal carcinoma before exposure to EGFR monoclonal antibodies. Cancer biology & therapy. 2013; 14: 1143-1146.
33. Voigt M, Braig F, Gothel M, Schulte A, Lamszus K, Bokemeyer C, Binder M. Functional dissection of the epidermal growth factor receptor epitopes targeted by panitumumab and cetuximab. Neoplasia. 2012; 14: 1023-1031.
34. Bokemeyer C, Van Cutsem E, Rougier P, Ciardiello F, Heeger S, Schlichting M, Celik I, Kohne CH. Addition of cetuximab to chemotherapy as first-line treatment for KRAS wild-type metastatic colorectal cancer: pooled analysis of the CRYSTAL and OPUS randomised clinical trials. European journal of cancer. 2012; 48: 1466-1475.
35. Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM, Antonarakis ES, Azad NS, Bardelli A, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Science translational medicine. 2014; 6: 224ra224.
36. Murtaza M, Dawson SJ, Tsui DW, Gale D, Forshew T, Piskorz AM, Parkinson C, Chin SF, Kingsbury Z, Wong AS, Marass F, Humphray S, Hadfield J, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2013; 497: 108-112.
37. Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, Thornton K, Agrawal N, Sokoll L, Szabo SA, Kinzler KW, Vogelstein B, Diaz LA. Circulating mutant DNA to assess tumor dynamics. Nature medicine. 2008; 14: 985-990.
38. Taniguchi K, Uchida J, Nishino K, Kumagai T, Okuyama T, Okami J, Higashiyama M, Kodama K, Imamura F, Kato K. Quantitative detection of EGFR mutations in circulating tumor DNA derived from lung adenocarcinomas. Clinical cancer research. 2011; 17: 7808-7815.
39. Binder M, Vogtle FN, Michelfelder S, Muller F, Illerhaus G, Sundararajan S, Mertelsmann R, Trepel M. Identification of their epitope reveals the structural basis for the mechanism of action of the immunosuppressive antibodies basiliximab and daclizumab. Cancer research. 2007; 67: 3518-3523.
40. Karasuyama H, Melchers F. Establishment of mouse cell lines which constitutively secrete large quantities of interleukin 2, 3, 4 or 5, using modified cDNA expression vectors. European journal of immunology. 1988; 18: 97-104.