
Introduction
Epidemiological studies indicate that the population of people aged more than 65 years is constantly growing [1]. Therefore, the study of age-related diseases such as neurodegenerative or cardiovascular disorders is becoming more important. The number of patients suffering from Alzheimer´s disease (AD) is expected to triple by the year 2050 [2]. AD is a progressive disorder characterized by a loss of memory and cognitive functions with behavioural alterations. The key neuropathological hallmarks of AD are extracellular amyloid plaques in the brain and intracellular neurofibrillary tangles, accompanied by the loss of neurons, white matter and synapses [3]. The plaques are often surrounded by activated microglia and inflammation may also be important in the pathogenesis of AD since increased concentrations of several inflammatory mediators, including tumor necrosis factor-alpha (TNF-α), interleukin (IL)-6, and IL-1ß have also been detected in blood [4].
In this context, the growth factor VGF may provide a link between central nervous system pathology and systemic immune and energy metabolism changes in AD. VGF is synthesized by neurons in the brain where it promotes growth and survival of neurons [5], and is involved in neurogenesis, synaptogenesis and energy homeostasis [6]. VGF is synthesized initially as a larger precursor, which undergoes proteolytic processing to produce smaller bioactive peptides that are secreted into the cerebrospinal fluid (CSF) and blood. Expression of VGF is induced by other growth factors, such as nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin 3, fibroblast growth factors and insulin. Since previous studies have shown that altered secretion of these factors occurs in neurological and psychiatric disorders, VGF peptides may also be biomarkers for AD, frontotemporal dementia or schizophrenia [7].
Similar to other brain-associated factors, VGF has also been detected in peripheral human leukocytes [8]. Recently, we investigated VGF expression in T cells of mentally healthy persons aged between 22 and 88 years and detected an age-dependent increase in the number of VGF+CD3+ T cells that correlated with glycated hemoglobin (HbA1c) and body mass index (BMI) [9]. T cells are known to be involved in healthy brain functions such as spatial learning, memory [10] and adult neurogenesis [11]. In addition, they have been implicated in neurodegenerative disorders like AD [12, 13] by regulating and maintaining inflammatory responses in the brain and periphery. Since currently used medications can only decelerate neurodegeneration for a certain time, new therapeutic treatment options for these disorders are needed. For example, studies have shown that the mTOR inhibitor rapamycin reduces amyloid-beta levels, abolishes cognitive deficits in mouse models of Alzheimer´s disease [14] and suppresses brain aging in rats [15].
T cells are known to produce acetylcholine (ACh) [16] and may therefore be affected by ACh inhibitors that are commonly used to treat AD patients. Therefore, we analyzed T cell VGF expression in AD patients and matched healthy controls and tested for effects of treatment with the Ach inhibitor rivastigmine on the number of VGF+CD3+ T cells.
Results
Expression of VGF and Hb1Ac levels are enhanced in AD patients
We determined the number of VGF+CD3+ T cells in parallel to HbA1c in AD patients, given the previous association found for these two factors in normal aging [9]. On average 8.45% of CD3+ T cells expressed VGF in healthy controls. However, the frequency of VGF+CD3+ T cells was significantly higher at 15.05% (p=0.032) in AD patients at the time of diagnosis (Fig. 1). The HbA1c level was determined during routine blood analysis as an 8–12 week integrated average blood glucose measurement. At the time of diagnosis, AD patients showed a trend towards higher HbA1c (6.7%) compared to controls (5.7%; p=0.060; Fig. 1).
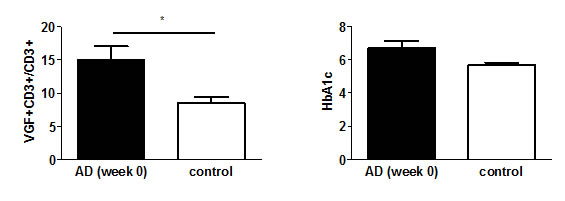
Figure 1: VGF+CD3+ T cells and HbA1C level are increased in AD patients. The number of VGF-expressing T cells (left) and levels of HbA1c (right) were determined in the peripheral blood of 24 AD patients (week 0) and 14 neuropsychiatric healthy age-matched controls.
Expression of VGF decreases during treatment of AD patients with rivastigmine
The number of VGF+CD3+ T cells was also determined after treatment of AD patients with rivastigmine patches for 12 and 30 weeks. This showed that the VGF+CD3+ T cell percentage decreased from 15.05% at week 0 (before treatment initiation) to 14.31% at week 12 and to 6.0% at week 30 (p=0.001; Fig. 2).
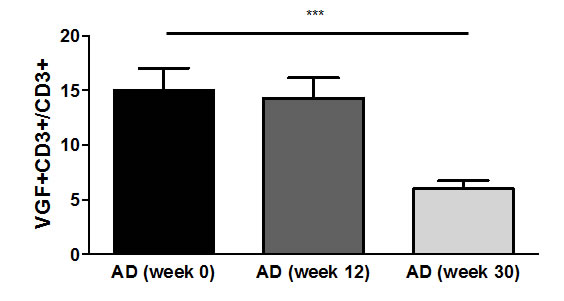
Figure 2: Frequency of VGF+CD3+ T cells decreases upon rivastigmine treatment. Upon diagnosis of AD (week 0), treatment with rivastigmine patches was initiated. VGF expression was determined in peripheral T cells after 12 weeks and 30 weeks of constant medication.
Proliferation of T cells is influenced by rivastigmine treatment
To measure the influence of rivastigmine treatment on T cell proliferation, cells were stimulated with either anti-CD3 or a combination of phorbol 12-myristate 13-acetate (PMA) and ionomycin. The proliferation index (PI) was calculated by dividing the value of ³H-thymidine incorporation in stimulated cell cultures by that in untreated cells (medium control). The PI of anti-CD3-stimulated T cells at the time of diagnosis was 361.2 and this was reduced after the 12 week rivastigmine treatment to 48.0 (p=0.024). Stimulation with PMA/ionomycin resulted in a PI of 294.8 which was reduced to 33.4 after the treatment period (p= 0.032; Fig. 3).
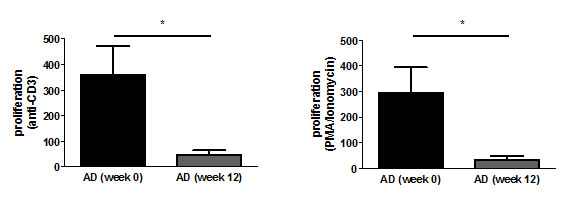
Figure 3: T cell proliferation decreases upon rivastigmine treatment. The proliferation index was calculated by dividing the mean of anti-CD3-stimulated cultures by that of medium controls (left) and by dividing the mean of PMA/Ionomycin-stimulated cultures by that of medium controls (right).
Rapamycin decreases the frequency of VGF+CD3+ cells in AD patients
Since Rapamycin is a suggested novel treatment for AD, we cultured PBMCs from AD patients for 24h in the presence or absence of rapamycin and determined the frequency of VGF+CD3+ T cells by flow cytometry. In cells treated without rapamycin, the percentage of VGF+CD3+ T cells was 9.56% in controls and13.65% in AD patients (p=0.0284). Treatment with rapamycin decreased the number of VGF+CD3+ T cells in AD patients to 9.1% (p=0.0197) (Fig. 4).
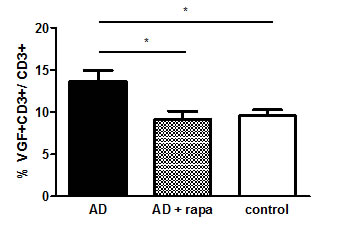
Figure 4: The frequency of VGF+ T cells decreases upon rapamycin treatment in vitro.
Discussion
In this study, we showed that VGF is expressed in a higher percentage of peripheral CD3+ T cells from AD patients compared to age-matched neuropsychiatric healthy volunteers. Upon treatment with rivastigmine, the percentage of VGF+CD3+ T cells decreased to the level found in control persons.
T lymphocytes are key components of the immune system which also exert pro-cognitive functions [17]. Brain-specific T cells could have beneficial roles in the protection against neurodegeneration by several mechanisms, such as enhancing uptake of cell debris by microglia [18, 19], release of anti-inflammatory cytokines [20, 21], increasing the capacity for buffering glutamate toxicity [22, 23], increased expression of neurotrophic factors [19, 24, 25] and enhanced neurogenesis [11, 26-28]. However, effector and regulatory functions of lymphocytes are compromised during aging [29, 30], a process called immunosenescence. Further immune manifestations accompany the progression of AD [4, 31], with negative effects on brain function and neuronal repair processes in general. This includes the deposition of extracellular amyloid plaques and intracellular neurofibrillary tangles and the associated induction of inflammatory reactions in the brain [32].
Previous studies have shown that Aβ-specific T cells are not suitable as biomarkers for AD, as the frequency of Aβ-specific T cells is significantly increased both in elderly healthy individuals and patients with AD [33]. Moreover, we have shown that an immune response against brain-specific proteins is found in patients with dementia as well as in aged people without neuropsychiatric disorders [9, 34, 35]. However, other studies have found differences in brain-specific miRNAs in blood from AD patients and healthy controls [36, 37]. Here, we found that VGF is expressed at a higher frequency in T cells from AD patients compared with those from aged-matched healthy controls. This is consistent with previous studies which found changes in the VGF level in brain or CSF from schizophrenia [38], depressive disorder [39], Parkinson’s and Alzheimer’s disease patients [40, 41], as compared to the respective healthy control populations. We also found previously that blocking VGF reduced the cytokine secretion and proliferation of T cells, suggesting that the enhanced frequency of VGF-expressing T cells from AD patients reflects a higher inflammatory state.
Neurotransmitters and neuropeptides can modulate the functions of immune cells such as T cells, myeloid cells or dendritic cells when released into the blood. Moreover, T cells themselves produce growth factors like NGF [42, 43], BDNF [44, 45], and insulin/ insulin-like growth factors (IGF-I and IGF-II) [46]Production of these factors is increased in activated T cells, as found in inflammation [43], and may induce VGF via an autocrine mechanism. Here, we showed that treatment with the acetlycholinesterase inhibitor rivastigmine led to decreased VGF expression and T cell proliferation to the levels detected in control T cells. Treatment of cognitive impairment in AD patients [47] with acetylcholinesterase inhibitors have also been found to decrease the production of pro-inflammatory cytokines and induced the secretion of the anti-inflammatory cytokine IL-4 [48]. T cells express both muscarinic and nicotinic acetylcholine receptors and choline acetytransferase [49]. The latter synthesizes ACh which is released from T cells and acts as an immunomodulator [50]. Rivastigmine treatment stimulates ACh release which subsequently induces cholinergic receptor activation. The drug suppresses α7 nAChR-dependent the T-cell proliferation [51], as we have shown here for anti-CD3- and PMA/Ionomycin-induced T cell proliferation. However, while T cell proliferation was reduced 12 weeks after beginning of rivastigmine treatment, VGF expression was diminished only after 30 weeks. This suggests that VGF is mainly expressed by activated T cells.
Previous studies have shown that VGF peptides are involved in regulation of several functions, including energy balance homeostasis [52]. We have already described a positive correlation between HbA1C and the frequency of VGF-positive T cells [9]. Here we show that AD patients have an increased HbA1C level compared to aged-matched controls. Increased Previous studies have shown that HbA1c levels are correlated with plasma Aβ1-42 concentrations [53] and mild cognitive impairment or dementia [54]. Moreover, elevated HbA1c levels are associated with insulin resistance [56], cardiovascular diseases [54], which could also be linked to the development of neurodegenerative disorders [55]. Our data identify VGF as a biomarker for AD that involves peripheral inflammation as well as long-term glucose levels.
As with insulin and insulin-like growth factor 1 (IGF-1) signalling, the mTOR (the molecular target of rapamycin) pathway, has been implicated in aging according to the hyperfunction theory [57, 58]. Specifically, mTOR regulates cellular biomass by promoting protein translation and inhibiting autophagy. Consequently, blockade of mTOR signalling via rapamycin treatment has anti-aging and neuroprotective effects, and has been proposed as a potential therapeutic pharmacological compound to restrict neurodegenerative disorders [59] such as AD [60-62].
It is generally accepted that aging is the greatest risk factor for AD. We have already described an age-associated increase in VGF-expressing T cells in healthy volunteers [8] and here we have shown that treatment of PBMCs with both rivastigmine and rapamycin in vitro reduces the number of these cells in AD patients. Halloran et al. have shown that chronic inhibition of mTOR by rapamycin treatment in mice enhances learning and memory and modulates their behavior throughout their lifespan [66], an effect mediated by IL-1β and NMDA signalling [67]. Therefore, rapamycin could be an effective cognition-improving drug in AD. This may also be achieved via antioxidant and anti-inflammatory effects of rapamycin [77-80], leading to a reduction of Aβ plaques and neurofibrillary tangles, which normally contribute to the progressive cognitive deficits of AD [70-72]. The present findings suggest that peripheral VGF levels could be used as a biomarker for detection of AD and for monitoring treatment response with rivastigmine and rapamycin.
MATERIALS AND METHODS
Study population
The study was performed in accordance with German laws, the Declaration of Helsinki and guidelines of the local institutional review board. Written consent was obtained from all patients and healthy controls. We collected 18 ml blood from 24 AD patients and 14 matched controls without neuropsychiatric disorders (suppl. table 1). Routine blood analysis were performed, including differential blood cell count, levels of C-reactive protein, glucose, lipids, liver enzymes and thyroid hormones. None of the subjects were excluded due to changes in these routine blood parameters. Also, no person had a history of autoimmune disorders, immunomodulating treatment, cancer, chronic terminal disease, severe cardiovascular disorder, substance abuse or severe trauma.
Treatment of AD patients
Within the first six weeks after diagnosis, AD patients received patches with 4.6 mg rivastigmine, followed by patches with 9.5 mg rivastigmineuntil the end of the study. Patches were changed each day.
Preparation of peripheral blood monocyte cells (PBMC)
Blood was collected in BD Vacutainer lithium heparin-treated tubes (BD Biosciences; San Jose, CA, USA), and diluted 1:1 with phosphate-buffered saline (PBS). Peripheral blood mononuclear cells (PBMCs) were prepared by density gradient centrifugation on Ficoll Paque (Biochrom AG, Berlin) at 375g for 20 min at room temperature. The cells were harvested and washed twice in PBS. Cells were then suspended in Fluorescence Assisted Cell Sorting (FACS) staining buffer (PBS w/ 0.5% BSA) and cell numbers determined.
Flow cytometry
Isolated PBMCs were washed in PBS and incubated with fluorescently-labeled antibodies for 20 min at 4°C in FACS buffer. Antibodies (Abs) used in this study were anti-CD3 (UCHT1) from BD Pharmingen (San Diego, CA, USA), and anti-VGF (D-20) and donkey anti-goat IgG FITC from Santa Cruz Biotechnology (Dallas, TX, USA). Data were collected on a FACS LSR-Fortessa (BD Biosciences, Mountain View, CA, USA) and analyzed using FlowJo software (Treestar Inc., Ashland, OR, USA). Data were analyzed using biexponential transformation for complete data visualization.
Measurement of cell proliferation
CD3+ T cells were isolated using the Pan T cell kit (Miltenyi Biotech, Bergisch Gladbach, Germany) and AutoMacs separation. CD3+ T cells were resuspended in RPMI 1640 medium supplemented with 10% FCS and 1% antibiotics and plated in triplicate at a density of 1x106 cells/ml in 96-well plates. T cells were stimulated with PMA/ Ionomycin (1mg/ml) and anti-CD3 (0.5µl/100µl; eBioscience, San Diego, CA, USA) for 3 days. ³H-Thymidine (0.2µCi/well) was added to the cultures for the last 17 hours Then cells were harvested and ³H-Thymidine incorporation was measured as an index of lymphocyte proliferation in a betaplate liqid scintillation counter (Wallac). Proliferation of T cells was calculated by division of the mean value of each triplicate of anti-CD3- or PMA/ Ionomycin-stimulated cell cultures by that of control medium cultures.
Treatment of PBMC with rapamycin in vitro
Isolated PBMCs from AD patients and controls were cultured for 24h in complete RPMI medium with or without addition of rapamycin (200 ng/ml). The cells were harvested and expression of VGF on CD3+ T cells was measured using flow cytometry.
Statistical analysis
Diagnostic group differences were calculated using Student’s t test or ANOVA. Significance was defined as p< 0.05, while a probability level of 0.05≤ p< 0.10 was considered as a statistical trend.
Acknowledgments
We are grateful to Bianca Jerzykiewicz for her skilful medical technical assistance.
Conflicts of Interest
None.
References
1. Spyridopoulos I, Hoffmann J, Aicher A, Brummendorf TH, Doerr HW, Zeiher AM and Dimmeler S. Accelerated telomere shortening in leukocyte subpopulations of patients with coronary heart disease: role of cytomegalovirus seropositivity. Circulation. 2009; 120(14):1364-1372.
2. Oeppen J and Vaupel JW. Demography. Broken limits to life expectancy. Science (New York, NY. 2002; 296(5570):1029-1031.
3. Reitz C and Mayeux R. Alzheimer disease: epidemiology, diagnostic criteria, risk factors and biomarkers. Biochemical pharmacology. 2014; 88(4):640-651.
4. Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, et al. Inflammation and Alzheimer’s disease. Neurobiology of aging. 2000; 21(3):383-421.
5. Sato H, Fukutani Y, Yamamoto Y, Tatara E, Takemoto M, Shimamura K and Yamamoto N. Thalamus-derived molecules promote survival and dendritic growth of developing cortical neurons. J Neurosci. 32(44):15388-15402.
6. Ferri GL, Noli B, Brancia C, D’Amato F and Cocco C. VGF: an inducible gene product, precursor of a diverse array of neuro-endocrine peptides and tissue-specific disease biomarkers. Journal of chemical neuroanatomy. 42(4):249-261.
7. Bartolomucci A, Pasinetti GM and Salton SR. Granins as disease-biomarkers: translational potential for psychiatric and neurological disorders. Neuroscience. 170(1):289-297.
8. Cattaneo A, Sesta A, Calabrese F, Nielsen G, Riva MA and Gennarelli M. The expression of VGF is reduced in leukocytes of depressed patients and it is restored by effective antidepressant treatment. Neuropsychopharmacology. 35(7):1423-1428.
9. Busse SS, J; Micheel, J; Dobrowolny, H; Mawrin, C; Krause, TJ; Adamaszek, M; Bogerts, B; Bommhardt, U; Hartig, R; Busse, M. Age-related increase of VGF-expression in T lymphocytes. Aging. 2014; 6(6):440-453.
10. Kipnis J, Cohen H, Cardon M, Ziv Y and Schwartz M. T cell deficiency leads to cognitive dysfunction: implications for therapeutic vaccination for schizophrenia and other psychiatric conditions. Proceedings of the National Academy of Sciences of the United States of America. 2004; 101(21):8180-8185.
11. Ziv Y, Ron N, Butovsky O, Landa G, Sudai E, Greenberg N, Cohen H, Kipnis J and Schwartz M. Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nature neuroscience. 2006; 9(2):268-275.
12. Panossian LA, Porter VR, Valenzuela HF, Zhu X, Reback E, Masterman D, Cummings JL and Effros RB. Telomere shortening in T cells correlates with Alzheimer’s disease status. Neurobiology of aging. 2003; 24(1):77-84.
13. Franco S, Blasco MA, Siedlak SL, Harris PL, Moreira PI, Perry G and Smith MA. Telomeres and telomerase in Alzheimer’s disease: epiphenomena or a new focus for therapeutic strategy? Alzheimers Dement. 2006; 2(3):164-168.
14. Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, Richardson A, Strong R and Galvan V. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PloS one. 5(4):e9979.
15. Kolosova NG, Vitovtov AO, Muraleva NA, Akulov AE, Stefanova NA and Blagosklonny MV. Rapamycin suppresses brain aging in senescence-accelerated OXYS rats. Aging. 5(6):474-484.
16. Rosas-Ballina M, Olofsson PS, Ochani M, Valdes-Ferrer SI, Levine YA, Reardon C, Tusche MW, Pavlov VA, Andersson U, Chavan S, Mak TW and Tracey KJ. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science (New York, NY. 334(6052):98-101.
17. Kipnis J, Gadani S and Derecki NC. Pro-cognitive properties of T cells. Nature reviews. 12(9):663-669.
18. Frenkel D, Maron R, Burt DS and Weiner HL. Nasal vaccination with a proteosome-based adjuvant and glatiramer acetate clears beta-amyloid in a mouse model of Alzheimer disease. The Journal of clinical investigation. 2005; 115(9):2423-2433.
19. Butovsky O, Koronyo-Hamaoui M, Kunis G, Ophir E, Landa G, Cohen H and Schwartz M. Glatiramer acetate fights against Alzheimer’s disease by inducing dendritic-like microglia expressing insulin-like growth factor 1. Proceedings of the National Academy of Sciences of the United States of America. 2006; 103(31):11784-11789.
20. Monsonego A, Imitola J, Petrovic S, Zota V, Nemirovsky A, Baron R, Fisher Y, Owens T and Weiner HL. Abeta-induced meningoencephalitis is IFN-gamma-dependent and is associated with T cell-dependent clearance of Abeta in a mouse model of Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2006; 103(13):5048-5053.
21. Frenkel D, Huang Z, Maron R, Koldzic DN, Moskowitz MA and Weiner HL. Neuroprotection by IL-10-producing MOG CD4+ T cells following ischemic stroke. Journal of the neurological sciences. 2005; 233(1-2):125-132.
22. Shaked I, Tchoresh D, Gersner R, Meiri G, Mordechai S, Xiao X, Hart RP and Schwartz M. Protective autoimmunity: interferon-gamma enables microglia to remove glutamate without evoking inflammatory mediators. Journal of neurochemistry. 2005; 92(5):997-1009.
23. Garg SK, Banerjee R and Kipnis J. Neuroprotective immunity: T cell-derived glutamate endows astrocytes with a neuroprotective phenotype. J Immunol. 2008; 180(6):3866-3873.
24. Aharoni R, Arnon R and Eilam R. Neurogenesis and neuroprotection induced by peripheral immunomodulatory treatment of experimental autoimmune encephalomyelitis. J Neurosci. 2005; 25(36):8217-8228.
25. Hohlfeld R, Kerschensteiner M, Stadelmann C, Lassmann H and Wekerle H. The neuroprotective effect of inflammation: implications for the therapy of multiple sclerosis. Neurol Sci. 2006; 27 Suppl 1:S1-7.
26. Baron R, Nemirovsky A, Harpaz I, Cohen H, Owens T and Monsonego A. IFN-gamma enhances neurogenesis in wild-type mice and in a mouse model of Alzheimer’s disease. Faseb J. 2008; 22(8):2843-2852.
27. Wolf SA, Steiner B, Akpinarli A, Kammertoens T, Nassenstein C, Braun A, Blankenstein T and Kempermann G. CD4-positive T lymphocytes provide a neuroimmunological link in the control of adult hippocampal neurogenesis. J Immunol. 2009; 182(7):3979-3984.
28. Mastrangelo MA, Sudol KL, Narrow WC and Bowers WJ. Interferon-{gamma} differentially affects Alzheimer’s disease pathologies and induces neurogenesis in triple transgenic-AD mice. The American journal of pathology. 2009; 175(5):2076-2088.
29. Maue AC, Yager EJ, Swain SL, Woodland DL, Blackman MA and Haynes L. T-cell immunosenescence: lessons learned from mouse models of aging. Trends in immunology. 2009; 30(7):301-305.
30. Panda A, Arjona A, Sapey E, Bai F, Fikrig E, Montgomery RR, Lord JM and Shaw AC. Human innate immunosenescence: causes and consequences for immunity in old age. Trends in immunology. 2009; 30(7):325-333.
31. Rocha NP, Teixeira AL, Coelho FM, Caramelli P, Guimaraes HC, Barbosa IG, da Silva TA, Mukhamedyarov MA, Zefirov AL, Rizvanov AA, Kiyasov AP, Vieira LB, Janka Z, Palotas A and Reis HJ. Peripheral blood mono-nuclear cells derived from Alzheimer’s disease patients show elevated baseline levels of secreted cytokines but resist stimulation with beta-amyloid peptide. Molecular and cellular neurosciences. 2012; 49(1):77-84.
32. Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL and Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nature medicine. 2008; 14(8):837-842.
33. Monsonego A, Zota V, Karni A, Krieger JI, Bar-Or A, Bitan G, Budson AE, Sperling R, Selkoe DJ and Weiner HL. Increased T cell reactivity to amyloid beta protein in older humans and patients with Alzheimer disease. The Journal of clinical investigation. 2003; 112(3):415-422.
34. Busse S, Busse M, Brix B, Probst C, Genz A, Bogerts B, Stoecker W and Steiner J. Seroprevalence of N-methyl-D-aspartate glutamate receptor (NMDA-R) autoantibodies in aging subjects without neuropsychiatric disorders and in dementia patients. European archives of psychiatry and clinical neuroscience. 2014.
35. Busse S, Brix B, Kunschmann R, Bogerts B, Stoecker W and Busse M. N-methyl-d-aspartate glutamate receptor (NMDA-R) antibodies in mild cognitive impairment and dementias. Neuroscience research. 2014.
36. Sheinerman KS, Tsivinsky VG, Crawford F, Mullan MJ, Abdullah L and Umansky SR. Plasma microRNA biomarkers for detection of mild cognitive impairment. Aging. 2012; 4(9):590-605.
37. Sheinerman KS, Tsivinsky VG, Abdullah L, Crawford F and Umansky SR. Plasma microRNA biomarkers for detection of mild cognitive impairment: biomarker validation study. Aging. 2013; 5(12):925-938.
38. Busse S, Bernstein HG, Busse M, Bielau H, Brisch R, Mawrin C, Muller S, Sarnyai Z, Gos T, Bogerts B and Steiner J. Reduced density of hypothalamic VGF-immunoreactive neurons in schizophrenia: a potential link to impaired growth factor signaling and energy homeostasis. European archives of psychiatry and clinical neuroscience. 2012; 262(5):365-374.
39. Thakker-Varia S, Jean YY, Parikh P, Sizer CF, Jernstedt Ayer J, Parikh A, Hyde TM, Buyske S and Alder J. The neuropeptide VGF is reduced in human bipolar postmortem brain and contributes to some of the behavioral and molecular effects of lithium. J Neurosci. 30(28):9368-9380.
40. Cocco C, D’Amato F, Noli B, Ledda A, Brancia C, Bongioanni P and Ferri GL. Distribution of VGF peptides in the human cortex and their selective changes in Parkinson’s and Alzheimer’s diseases. Journal of anatomy. 217(6):683-693.
41. Wijte D, McDonnell LA, Balog CI, Bossers K, Deelder AM, Swaab DF, Verhaagen J and Mayboroda OA. A novel peptidomics approach to detect markers of Alzheimer’s disease in cerebrospinal fluid. Methods (San Diego, Calif. 56(4):500-507.
42. Lambiase A, Bracci-Laudiero L, Bonini S, Bonini S, Starace G, D’Elios MM, De Carli M and Aloe L. Human CD4+ T cell clones produce and release nerve growth factor and express high-affinity nerve growth factor receptors. The Journal of allergy and clinical immunology. 1997; 100(3):408-414.
43. Aloe L, Bracci-Laudiero L, Bonini S and Manni L. The expanding role of nerve growth factor: from neurotrophic activity to immunologic diseases. Allergy. 1997; 52(9):883-894.
44. Hohlfeld R. Neurotrophic cross-talk between the nervous and immune systems: relevance for repair strategies in multiple sclerosis? Journal of the neurological sciences. 2008; 265(1-2):93-96.
45. Ruggieri M, Avolio C, Livrea P and Trojano M. Glatiramer acetate in multiple sclerosis: a review. CNS drug reviews. 2007; 13(2):178-191.
46. Geenen V. The thymic insulin-like growth factor axis: involvement in physiology and disease. Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolisme. 2003; 35(11-12):656-663.
47. Kaduszkiewicz H, Zimmermann T, Beck-Bornholdt HP and van den Bussche H. Cholinesterase inhibitors for patients with Alzheimer’s disease: systematic review of randomised clinical trials. BMJ (Clinical research ed. 2005; 331(7512):321-327.
48. Reale M, Iarlori C, Gambi F, Feliciani C, Salone A, Toma L, DeLuca G, Salvatore M, Conti P and Gambi D. Treatment with an acetylcholinesterase inhibitor in Alzheimer patients modulates the expression and production of the pro-inflammatory and anti-inflammatory cytokines. Journal of neuroimmunology. 2004; 148(1-2):162-171.
49. Kawashima K, Fujii T, Moriwaki Y and Misawa H. Critical roles of acetylcholine and the muscarinic and nicotinic acetylcholine receptors in the regulation of immune function. Life sciences. 2012; 91(21-22):1027-1032.
50. Kawashima K and Fujii T. The lymphocytic cholinergic system and its contribution to the regulation of immune activity. Life sciences. 2003; 74(6):675-696.
51. Nizri E, Irony-Tur-Sinai M, Faranesh N, Lavon I, Lavi E, Weinstock M and Brenner T. Suppression of neuroinflammation and immunomodulation by the acetylcholinesterase inhibitor rivastigmine. Journal of neuroimmunology. 2008; 203(1):12-22.
52. Salton SR, Ferri GL, Hahm S, Snyder SE, Wilson AJ, Possenti R and Levi A. VGF: a novel role for this neuronal and neuroendocrine polypeptide in the regulation of energy balance. Frontiers in neuroendocrinology. 2000; 21(3):199-219.
53. Fujiwara Y, Takahashi M, Tanaka M, Hoshi T, Someya T and Shinkai S. Relationships between plasma beta-amyloid peptide 1-42 and atherosclerotic risk factors in community-based older populations. Gerontology. 2003; 49(6):374-379.
54. Yaffe K, Blackwell T, Whitmer RA, Krueger K and Barrett Connor E. Glycosylated hemoglobin level and development of mild cognitive impairment or dementia in older women. The journal of nutrition, health & aging. 2006; 10(4):293-295.
55. Schneider JA and Bennett DA. Where vascular meets neurodegenerative disease. Stroke; a journal of cerebral circulation. 2010; 41(10 Suppl):S144-146.
56. Olefsky JM and Glass CK. Macrophages, inflammation, and insulin resistance. Annual review of physiology. 72:219-246.
57. Blagosklonny MV. Answering the ultimate question “what is the proximal cause of aging?” Aging. 4(12):861-877.
58. Gems D and de la Guardia Y. Alternative Perspectives on Aging in Caenorhabditis elegans: Reactive Oxygen Species or Hyperfunction? Antioxidants & redox signaling. 19(3):321-329.
59. Li S. The possible cellular mechanism for extending lifespan of mice with rapamycin. Biological procedures online. 2009; 11:1-2.
60. Cai Z, Zhao B, Li K, Zhang L, Li C, Quazi SH and Tan Y. Mammalian target of rapamycin: a valid therapeutic target through the autophagy pathway for Alzheimer’s disease? Journal of neuroscience research. 2012; 90(6):1105-1118.
61. Santos RX, Correia SC, Cardoso S, Carvalho C, Santos MS and Moreira PI. Effects of rapamycin and TOR on aging and memory: implications for Alzheimer’s disease. Journal of neurochemistry. 2011; 117(6):927-936.
62. Pierce A, Podlutskaya N, Halloran JJ, Hussong SA, Lin PY, Burbank R, Hart MJ and Galvan V. Over-expression of heat shock factor 1 phenocopies the effect of chronic inhibition of TOR by rapamycin and is sufficient to ameliorate Alzheimer’s-like deficits in mice modeling the disease. Journal of neurochemistry. 2013; 124(6):880-893.
63. Chong ZZ, Shang YC, Wang S and Maiese K. Shedding new light on neurodegenerative diseases through the mammalian target of rapamycin. Progress in neurobiology. 2012; 99(2):128-148.
64. Graziotto JJ, Cao K, Collins FS and Krainc D. Rapamycin activates autophagy in Hutchinson-Gilford progeria syndrome: implications for normal aging and age-dependent neurodegenerative disorders. Autophagy. 2012; 8(1):147-151.
65. Mendelsohn AR and Larrick JW. Rapamycin as an antiaging therapeutic?: targeting mammalian target of rapamycin to treat Hutchinson-Gilford progeria and neurodegenerative diseases. Rejuvenation research. 2011; 14(4):437-441.
66. Halloran J, Hussong SA, Burbank R, Podlutskaya N, Fischer KE, Sloane LB, Austad SN, Strong R, Richardson A, Hart MJ and Galvan V. Chronic inhibition of mammalian target of rapamycin by rapamycin modulates cognitive and non-cognitive components of behavior throughout lifespan in mice. Neuroscience. 2012; 223:102-113.
67. Majumder S, Caccamo A, Medina DX, Benavides AD, Javors MA, Kraig E, Strong R, Richardson A and Oddo S. Lifelong rapamycin administration ameliorates age-dependent cognitive deficits by reducing IL-1beta and enhancing NMDA signaling. Aging cell. 2012; 11(2):326-335.
68. Agostinho P, Cunha RA and Oliveira C. Neuroinflammation, oxidative stress and the pathogenesis of Alzheimer’s disease. Current pharmaceutical design. 2010; 16(25):2766-2778.
69. Cai Z, Yan LJ and Ratka A. Telomere shortening and Alzheimer’s disease. Neuromolecular medicine. 2013; 15(1):25-48.
70. Majumder S, Richardson A, Strong R and Oddo S. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PloS one. 2011; 6(9):e25416.
71. Rasool S, Albay R, 3rd, Martinez-Coria H, Breydo L, Wu J, Milton S, Misra S, Tran A, Pensalfini A, Laferla F, Kayed R and Glabe CG. Vaccination with a non-human random sequence amyloid oligomer mimic results in improved cognitive function and reduced plaque deposition and micro hemorrhage in Tg2576 mice. Molecular neurodegeneration. 2012; 7:37.
72. Lee KW, Lee SH, Kim H, Song JS, Yang SD, Paik SG and Han PL. Progressive cognitive impairment and anxiety induction in the absence of plaque deposition in C57BL/6 inbred mice expressing transgenic amyloid precursor protein. Journal of neuroscience research. 2004; 76(4):572-580.
73. Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, Richardson A, Strong R and Galvan V. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PloS one. 2010; 5(4):e9979.
74. Liu Y, Su Y, Wang J, Sun S, Wang T, Qiao X, Run X, Li H and Liang Z. Rapamycin decreases tau phosphorylation at Ser214 through regulation of cAMP-dependent kinase. Neurochemistry international. 2013; 62(4):458-467.
75. Maiese K, Chong ZZ, Wang S and Shang YC. Oxidant stress and signal transduction in the nervous system with the PI 3-K, Akt, and mTOR cascade. International journal of molecular sciences. 2012; 13(11):13830-13866.
76. Galimberti D and Scarpini E. Inflammation and oxidative damage in Alzheimer’s disease: friend or foe? Frontiers in bioscience (Scholar edition). 2011; 3:252-266.
77. Jiang J, Jiang J, Zuo Y and Gu Z. Rapamycin protects the mitochondria against oxidative stress and apoptosis in a rat model of Parkinson’s disease. International journal of molecular medicine. 2013; 31(4):825-832.
78. Marobbio CM, Pisano I, Porcelli V, Lasorsa FM and Palmieri L. Rapamycin reduces oxidative stress in frataxin-deficient yeast cells. Mitochondrion. 2012; 12(1):156-161.
79. Chen HC, Fong TH, Hsu PW and Chiu WT. Multifaceted effects of rapamycin on functional recovery after spinal cord injury in rats through autophagy promotion, anti-inflammation, and neuroprotection. The Journal of surgical research. 2013; 179(1):e203-210.
80. Chen WQ, Zhong L, Zhang L, Ji XP, Zhang M, Zhao YX, Zhang C and Zhang Y. Oral rapamycin attenuates inflammation and enhances stability of atherosclerotic plaques in rabbits independent of serum lipid levels. British journal of pharmacology. 2009; 156(6):941-951.