
INTRODUCTION
As the most-common noncutaneous cancer in men worldwide [1], mechanisms contributing to development of prostate cancer at all stages of disease remain of high interest for both diagnosis and treatment of clinically-relevant disease. Consistent with the multi-hit hypothesis, several genes that control critical growth, survival, and/or apoptotic pathways [2] must be altered to lead to fully penetrant prostate cancer [3–5]. A wide body of literature has identified many complex genetic alterations involved in neoplastic transformation, including TMPRSS2-ETS (ERG) chromosomal translocations and deletion of tumor suppressor genes (PTEN, TP53, RB1, NKX3–1, and CDKN1B) [3–10]. Of these, selection for the deletion of PTEN (phosphatase and tensin homolog) occurs in approximately 30% of prostate cancers, as lowered levels of PTEN increase the availability of phosphatidyl-inositol 3, 4, 5 triphosphate (PIP3) for driving PI 3-kinase (PI3K)-dependent signaling, such as cellular growth pathways downstream of AKT (protein kinase B) [7, 11]. In addition, we previously implicated ABI1 (SSH3BP1) as a putative tumor suppressor in prostate cancer [12] and have demonstrated that disruption of Abi1 in the mouse prostate leads to prostatic intraepithelial neoplasia (PIN), but not to invasive prostate cancer [13]. We have therefore sought to find additional genes that cooperate with ABI1 in prostate tumor progression.
In cells Abi1 is incorporated in the WAVE complex. In mammalian cells, several distinct WAVE complexes can form, depending on the isoform of proteins that is expressed [14]. Each WAVE complex assembles from ubiquitously expressed variants of 5 polypeptides: WAVE, Abelson interactor (Abi or SSH3BP1), Rac1-associated protein (Sra-1), Nck-associated protein (Nap), and Brk1 (HSPC300) [15–17]. Except for Brk1 [18], for which only one isoform exists, each of these proteins is a member of a protein family consisting of 2–3 genes in mammals including alternatively spliced isoforms of 10 genes [14, 19]: (i) WASF1, WAVE2 (WASF2), and WAVE3 (WASF3), (ii) ABI1, ABI2, and ABI3, (iii) Sra-1 and PIR121 (CYFIP1 and CYFIP2), and (iv) Nap1 (NCKAP1) and Hem1 (NCKAP1L1). Functionally, WAVE complexes are major actin cytoskeleton regulatory factors that promote actin polymerization through an Arp2/3 dependent mechanism [20]. Other functions of WAVE complex or its components involve binding to variety of membrane receptors [21], intracellular signaling [22, 23], and transcription [22, 24]. WAVE complexes are involved in cell motility and migration, cellular adhesion, cell-to-cell communication, cell division, and immunological responses [17, 25–31]. Thus, WAVE complexes are involved numerous cellular functions requiring actin cytoskeleton reorganization and dynamics, processes with roles in tumor progression and metastasis [32].
Consistent with this, dysregulation of WAVE complex or its components is associated with human cancer [33–35] including prostate cancer [12, 36–39], as the WAVE pathway cross talks with the PI3K pathway. It has been shown previously that PIP3 can bind directly to WAVE protein [40], and we and others have demonstrated that the p85 regulatory subunit of PI3K interacts with Abi1 [23, 41]. Importantly, we have also shown that genetic inactivation of Abi1 led to down-regulation of WASF1 (and WASF2 [42]) which in turn resulted in increased phosphorylation of Akt in murine prostate tissue [13]. To this effect, altered levels of WAVE complex genes would result in disruption of the WAVE complex’s ability to regulate p85, and thus would phenocopy deletion of PTEN in mediating cancer progression. Therefore we hypothesize that prostate cancers select for cells with lower levels of WAVE complex proteins.
In patients with metastatic PCa, standard treatments involve either surgical or medical castration, which effectively prevents testicular testosterone from driving androgen receptor (AR) activation in the tumor cell [43]. The fact that most patients invariably relapse has led to profound interest in identifying mechanisms contributing to the development of castration-resistant prostate cancer (CRPC) and commercial production of inhibitors of adrenal sources of androgens and AR pathway inhibitors such as galeterone, abiraterone and enzalutamide [44–46]. However, the fact that PTEN is deleted or down-regulated in these cancers has intensified investigation of how PI3K contributes to androgen independence via a PTEN-dependent mechanism. Not surprisingly, simultaneous inhibition of both AR and PI3K effectively blocked growth of human PCa xenografts [47–49]. These data further reinforce the need to identify causative mechanisms of CRPC development, especially in the context of PTEN deletion.
Therefore, to assess whether prostate cancer progression to castration resistance is mediated by changes to WAVE complex, we performed bioinformatic meta-analyses on several published datasets, including mutation, copy number, and gene expression data accumulated as part of The Cancer Genome Atlas (TCGA), as well as other publicly-available datasets deposited in GEO from experiments performed at Beth Israel Deaconess Medical Center (BIDMC), Memorial Sloan Kettering Medical Center (MSKCC) and the University of Michigan (UMICH) [7, 10, 50]. By comparing results across independent cohorts, we observed shifts in the frequency of WAVE complex gene alterations, which suggest that WAVE complex disruption may be a putative driver of prostate tumorigenesis. Importantly, we observed that deletion of WASF1, the gene that codes for WAVE1, occurs more frequently with PTEN deletion in metastatic lethal vs. primary disease, suggesting that WASF1 loss represents an aggressive of subtype of prostate cancer. It is thus possible that patients who harbor tumors with WASF1 deletion may benefit from either earlier or more aggressive intervention.
RESULTS AND DISCUSSION
We had previously assessed the status of ABI1 in a small cohort of primary prostate tissue [51], so we expanded our interrogation of WAVE complex alterations to The Cancer Genome Atlas (TCGA) and molecular analyses performed on these samples. We hypothesized that alterations to the WAVE complex may contribute to increased tumorigenicity and therefore poorer long-term outcome. When we interrogated the dataset for cases with mutations, copy number alterations, or abnormal expression, we found that alterations to WAVE complex genes were significantly associated with a 13.43% increase in the rate of biochemical recurrence (BCR) within 5 years (Figure 1a). Interrogating other datasets for which biochemical recurrence rates were available, we observed a similar trend in the MKSCC dataset, where alterations to WAVE complex genes were associated with a 11.14% increase in the five-year BCR rate (Supplementary Figure 1). Strikingly, the most common alterations were not mutations (Figure 1b) nor recurrent up- or down-regulation (Figure 1c), but rather copy number variation to WAVE complex genes, preeminently WAVE1 (WASF1) (Figure 1d), which was deleted hemizygously in 54 cases (16.9%) and homozygously in 32 cases (10%).
These observations are consistent with several recent reports that describe many recurrent translocations and deletions in prostate cancer and surprisingly few recurrent mutations. Indeed, mutations to SPOP, MED12 and FOXA1 comprise less than 10% (each) recurrence in primary PCa exomes sequenced as part of TCGA and previous studies [52], but large chromosomal events occur far more frequently, with translocations (or interstitial deletions) involving TMPRSS2 and ETS-family transcription factors (such as ERG) in approximately 50% of cases, or tumor suppressor deletions (PTEN, NKX3–1) in approximately 30–50% of cases [7]. Our observation of WASF1 deletion was not as frequent (approximately 25%). Although these deletions were identified, these same cases did not indicate lower levels of WASF1 mRNA. While we cannot rule out the possibility that cells can compensate by up-regulating the remaining copy, recent evidence suggests that the WAVE complex is regulated mostly at the protein level [42, 53].
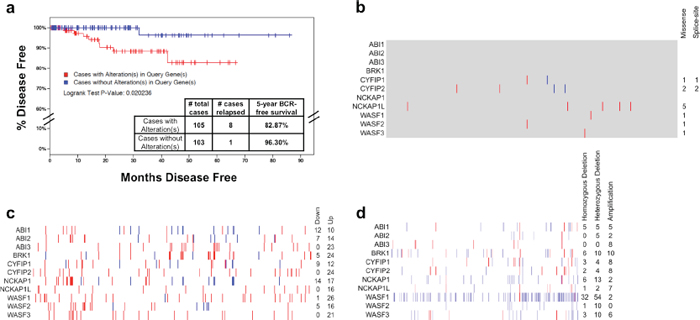
Figure 1: Spectrum of alterations of WAVE complex in primary prostate cancer. Data on prostate adenocarcinoma analyses performed as part of The Cancer Genome Atlas (TCGA) as of the time of writing was downloaded from cBioPortal [64], the NCBI TCGA data portal (https://tcga-data.nci.nih.gov) and CGHub [65]. a. Kaplan-Meier survival distribution showing the time until biochemical recurrence for patients with alterations in WAVE complex genes (ABI1, ABI2, ABI3, BRK1, CYFIP1, CYFIP2, NCKAP1, NCKAP1L, WASF1, WASF2, and WASF3). Log rank P value: 0.020 suggesting a significant difference between groups. b. Distribution of nonsynonymous somatic mutations to WAVE complex genes from 493 cases of PCa whole exome sequencing data generated by the TCGA. Blue: splice-site mutation; red: missense mutation; gray: unchanged. c. Gene expression changes in tumor samples relative to the mean of the sample set’s 75th percentile-normalized RSEM values (z-score/standard deviations, s.d.) from 493 cases of TCGA PCa RNA-seq data [66]. Normalized RSEM values are given in Supplementary Table 1, and z-score values are given in Supplementary Table 2. Blue: ≥ 2 s.d. down-regulated; Red: ≥ 2 s.d. up-regulated. Note that due to variability in the purity of the tumor sample (Supplementary Table 3) as determined by ESTIMATE [67], some up- or down-regulation of WAVE complex genes may be due to stromal contamination. d. Somatic copy number analysis of WAVE complex genes from 319 cases of TCGA PCa Affymetrix 6.0 SNP arrays, where 1 (white) = two copies. Red: > 1.2, inferring amplification; Blue: < 0.8, inferring deletion. Homozygous deletion: copy number less than 0.6. Hemizygous deletion: copy number 0.6–0.8. Cases with tumor cell purity less than 90% are excluded from totals, but values for all cases are given in Supplementary Table 4.
Nonetheless, the increased frequency of biochemical recurrence of tumors with WAVE complex gene alterations and our observation of recurrent deletion of WASF1 in primary prostate cancer therefore led us to ask whether WAVE complex was similarly disrupted in advanced PCa, in a castration-resistant (CRPC) setting. Examination of gene expression microarrays from 131 primary PCa (MSKCC dataset [7]) and 33 CRPC (BIDMC dataset [50]) revealed marked down-regulation of ABI1, ABI2, and NCKAP1 in the CRPC samples (Figure 2a). WASF1 expression displayed modest variation (within and between sets), which is consistent with the observation from TCGA (see Figure 1c) that WASF1 down-regulation by itself is not a mechanism of progression in PCa, although down-regulation of ABI1, ABI2, or NCKAP1 may lead to destabilization of WAVE complex and thus lower levels of WAVE1 protein.
Importantly, the WAVE complex acts as a mediator PI-3 kinase-directed cell motility, and thus may also contribute to overall tumorigenenicity when dysregulated [54, 55]. Given its function downstream of PI3K, we therefore sought to explore WASF1 copy number status in the context of PTEN deletion, a frequent genomic lesion found in primary and advanced PCa [56–58]. To make comparisons, we selected two published datasets (from MSKCC [7] and UMICH [10]) in which copy number analysis was performed simultaneously in both primary PCa and CRPC. As anticipated, PTEN and WASF1 were frequently deleted (hemizygously and homozygously) in primary PCa from both datasets (Figures 2b and 2c). To our surprise however, PTEN and WASF1 deletion were mutually exclusive in primary PCa, but hemizygous WASF1 deletion co-occurred with both hemizygous and homozygous PTEN deletion in CRPC (P < 0.0001 for the MSKCC set, P = 0.0006, for the UMICH set). As PTEN and WASF1 are on different chromosomes (10q23 and 6q21) these events are not physically linked, and because in CPRC nearly all WASF1 deletions were in cases that also harbored at least one copy of PTEN (but not vice versa), these data suggest that WASF1 deletion is an earlier event preceding tumor cells’ acquisition of PTEN deletions that likely occur later as tumors progress.
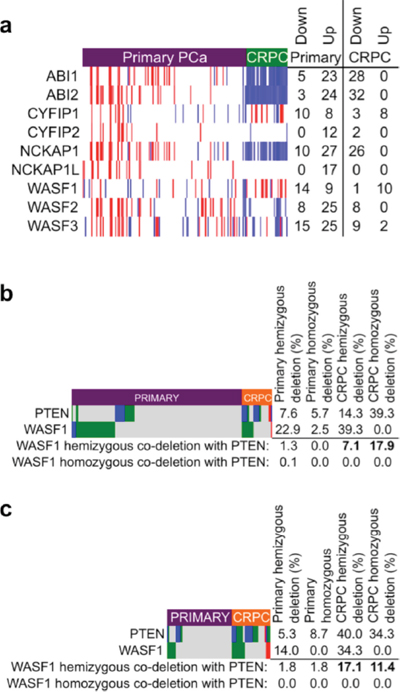
Figure 2: WAVE complex disruption in castration-resistant prostate cancer. a. Heatmap of gene expression changes in WAVE complex genes relative to the mean of the sample. Microarray data for primary PCa (Affymetrix Human Exon 1.0 array, accession ID GSE21034) and CRPC (Affymetrix U133A array, accession ID GSE32269) were downloaded from the Gene Expression Omnibus (GEO) and normalized to the same scale using SCAN for Bioconductor [68]. Gene expression values in each row are displayed according to their z-score (number of standard deviations (s.d.) greater or lower than the mean for the sample set). Note that probes to BRK1 and ABI3 were not present on the Affymetrix U133A microarray and thus are excluded from this analysis. Red: high expression (greater than 1 s.d. up-regulated); blue: low expression (greater than 1 s.d. downregulated). Normalized intensity values are given in Supplementary Table 5 and z-scores are values are given in Supplementary Table 6. b. Somatic copy number depiction of PTEN and WASF1 of 157 primary PCa and 28 CRPC from the MKSCC dataset [7] (Agilent 244A aCGH array, accession ID GSE21032). P < 0.0001 by Fisher’s exact test for the frequency of co-occurrence of hemizygous WASF1 deletion with combined frequencies of hemizygous and homozygous deletion of PTEN in CRPC vs. primary PCa. Numeric copy number calls are given in Supplementary Table 7. c. Somatic copy number depiction of PTEN and WASF1 of 59 primary PCa and 35 lethal CRPC from the UMICH dataset [10] (Agilent 44K aCGH array, accession ID GSE35988). P = 0.0006 by Fisher’s exact test for the frequency of co-occurrence of hemizygous WASF1 deletion with combined frequencies of hemizygous and homozygous deletion of PTEN in CRPC vs. primary PCa. Numeric copy number calls are given in Supplementary Table 8. For (b) and (c) data were downloaded from GEO and loci copy number were assessed with Nexus Copy Number software (Biodiscovery, Hawthorne, CA); dark blue: homozygous deletion; green: hemizygous deletion; red: amplification; gray: unchanged.
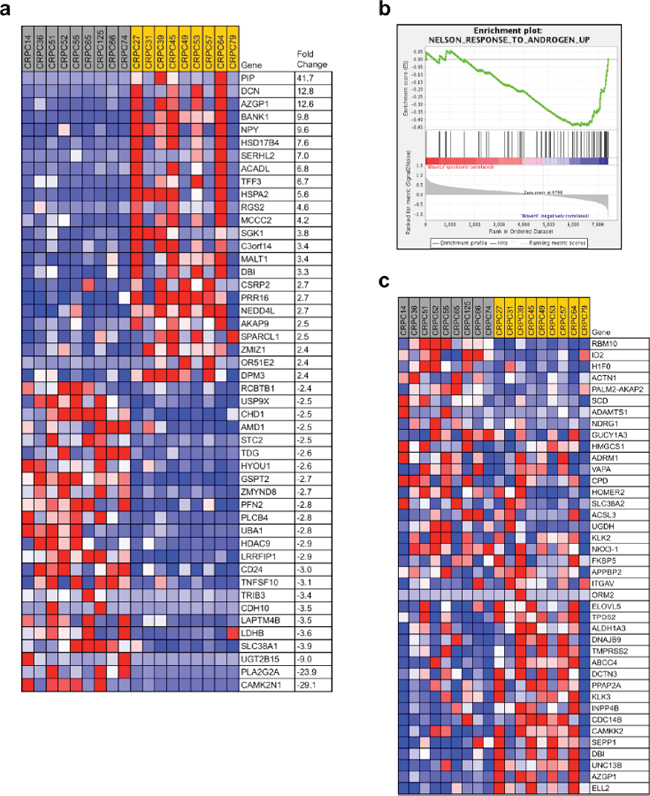
Figure 3: WASF1 deletion may identify an aggressive subtype of prostate cancer. a. Of the 36 CRPC samples in the BIDMC dataset (Affymetrix U133A array, accession ID GSE32269), the 9 samples with the highest quartile of WASF1 expression (gray, mean = 2.67) were compared against the 9 samples with the lowest quartile of WASF1 expression (yellow, mean = 1.41), and the genes contributing most to the distinguishing of these groups were identified with the ComparativeMarkerSelection [69] module in GenePattern [70] (fold-change ≥ ± 2.4 ×, P < 0.05 by Student’s t-test). Red: high expression; blue: low expression. Expression intensity values are given in Supplementary Table 9. b. Enrichment plot of WASF1-high vs. WASF1-low gene expression and correlation with Androgen Receptor (AR) activity. Normalized enrichment score: –1.50; P = 0.038. c. Heatmap of gene expression associated with AR activity positively correlated with the WASF1-low group. Red: high expression; blue: low expression. Expression intensity values are given in Supplementary Table 10.
Importantly, as PTEN and WASF1 were rarely deleted simultaneously in the same case in primary PCa (see Figures 2b and 2c), selection for dual loss may be associated with processes that mediate disease progression. Specifically, the frequency of WASF1 deletion in lethal prostate cancer and its co-deletion with PTEN raised the possibility that advanced PCa cells select for PTEN deletion in cases with WASF1 already deleted in order to drive PI 3-kinase signaling in castration-resistant cases with stronger AR reactivation. Furthermore, because we did not observe frequent homozygous deletion of WASF1, tumors with lower WAVE1 levels (rather than total loss of WAVE1) may similarly confer a selective advantage at later stages of disease. Indeed, WASF1 expression within the BIDMC dataset of CRPC was varied to permit stratification of samples by the top quartile of WASF1 expression (WASF1-high) vs. the lower quartile (WASF1-low) to predict the potential effects of WASF1 genomic deletion (Figure 3a). Consistent with our hypothesis that low WASF1 expression represents an aggressive phenotype, we observed significant down-regulation of the putative tumor suppressors CHD1 and CDH10 (cadherin-10) [59, 60]. Moreover, amongst the genes most up-regulated in the WASF1-low group are HSD17B4, which codes for an androgen inactivating enzyme [61], and PIP (prolactin-induced protein), whose expression was recently reported as a readout of AR (Androgen Receptor) activity [62]. Therefore, to test whether AR activity is indeed increased in the WASF1-low cases, we performed Geneset Enrichment Analysis (high vs. low) using the expression values for all genes. In the WASF1-low phenotype, we observed negative enrichment for up-regulated genes derived from a dataset in which LNCaP cells were treated with methyltrienolone/R1881 (Figure 3b) [63]. Indeed, these genes include the AR targets KLK3 (PSA), TMPRSS2, and FKBP5, which are up-regulated in the WASF1-low group (Figure 3c), suggesting that CRPC with lower levels of WASF1 have increased tumorigenicity.
While metastatic CRPC circumvents androgen deprivation by intratumoral testosterone synthesis [50], increased expression of AR target genes suggests higher stability of ligand-bound AR and thus higher levels of androgen synthesis. Because the reactivation of AR activity and down-regulation of additional tumor suppressors in these cases (see Figure 3a) represents an aggressive prostate cancer phenotype, deletion of WASF1 and lower levels of ABI1 and ABI2 likely cooperate with other perturbations in these CRPC for overcoming androgen deprivation, providing a selective advantage for those tumors harboring these genetic changes. Thus, the normal function of the WAVE complex may be to serve as a tumor suppressor. Therefore, WASF1 deletion co-occurring with PTEN in advanced prostate cancers may result in even stronger PI3K signaling, having removed inhibitory forces from p85 (WASF1 deletion) and increasing levels of PIP3 (PTEN deletion). Further investigation is needed to determine the biological consequences of WASF1 deletion, the interactions between WAVE pathway and the AR signaling axis, and the role of WAVE pathway in establishing tumors with long-term aggressive potential.
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (DF/HCC-Prostate Cancer SPORE P50 CA090381 to AGS, R01 CA161018 to LK) and the Department of Defense Prostate Cancer Research Program (Postdoctoral Training Award W81XWH-13–1-0267 to AGS). Portions of this research were conducted on the Orchestra High Performance Compute Cluster at Harvard Medical School. The results published here are in whole or part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/.
FUNDING SOURCES
National Cancer Institute, Department of Defense.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to declare.
REFERENCES
1. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics. CA Cancer J Clin. 2014; 64:9–29.
2. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–674.
3. Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, Cordon-Cardo C, Pandolfi PP. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005; 436:725–730.
4. Kim MJ, Bhatia-Gaur R, Banach-Petrosky WA, Desai N, Wang Y, Hayward SW, Cunha GR, Cardiff RD, Shen MM, Abate-Shen C. Nkx3.1 mutant mice recapitulate early stages of prostate carcinogenesis. Cancer research. 2002; 62:2999–3004.
5. Di Cristofano A, De Acetis M, Koff A, Cordon-Cardo C, Pandolfi PP. Pten and pKIP1 cooperate in prostate cancer tumor suppression in the mouse. Nat Genet. 2001; 27:222–224.
6. Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Kuefer R, Lee C, Montie JE, Shah RB, Pienta KJ, Rubin MA, Chinnaiyan AM. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. New York, NY. 2005; 310:644–648.
7. Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, Antipin Y, Mitsiades N, Landers T, Dolgalev I, Major JE, Wilson M, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010; 18:11–22.
8. Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet. 1998; 19:348–355.
9. Hill R, Song Y, Cardiff RD, Van Dyke T. Heterogeneous tumor evolution initiated by loss of pRb function in a preclinical prostate cancer model. Cancer research. 2005; 65:10243–10254.
10. Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, Asangani IA, Ateeq B, Chun SY, Siddiqui J, Sam L, Anstett M, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012; 487:239–243.
11. Hemmings BA, Restuccia DF. PI3K-PKB/Akt pathway. Cold Spring Harb Perspect Biol. 2012; 4:a011189.
12. Macoska JA, Xu J, Ziemnicka D, Schwab TS, Rubin MA, Kotula L. Loss of expression of human spectrin src homology domain binding protein 1 is associated with 10p loss in human prostatic adenocarcinoma. Neoplasia. New York, NY. 2001; 3:99–104.
13. Xiong X, Chorzalska A, Dubielecka PM, White JR, Vedvyas Y, Hedvat CV, Haimovitz-Friedman A, Koutcher JA, Reimand J, Bader GD, Sawicki JA, Kotula L. Disruption of Abi/Hssh3bp expression induces prostatic intraepithelial neoplasia in the conditional Abi/Hssh3bp KO mice. Oncogenesis. 2012; 1:e26.
14. Stovold CF, Millard TH, Machesky LM. Inclusion of Scar/WAVE3 in a similar complex to Scar/WAVE1 and 2. BMC cell biology. 2005; 6:11.
15. Eden S, Rohatgi R, Podtelejnikov AV, Mann M, Kirschner MW. Mechanism of regulation of WAVE1-induced actin nucleation by Rac1 and Nck. Nature. 2002; 418:790–793.
16. Stradal TE, Rottner K, Disanza A, Confalonieri S, Innocenti M, Scita G. Regulation of actin dynamics by WASP and WAVE family proteins. Trends in cell biology. 2004; 14:303–311.
17. Takenawa T, Suetsugu S. The WASP-WAVE protein network: connecting the membrane to the cytoskeleton. Nature reviews. 2007; 8:37–48.
18. Derivery E, Fink J, Martin D, Houdusse A, Piel M, Stradal TE, Louvard D, Gautreau A. Free Brick1 is a trimeric precursor in the assembly of a functional wave complex. PloS one. 2008; 3:e2462.
19. Dubielecka PM, Cui P, Xiong X, Hossain S, Heck S, Angelov L, Kotula L. Differential regulation of macropinocytosis by Abi1/Hssh3bp1 isoforms. PloS one. 2010; 5:e10430.
20. Derivery E, Gautreau A. Generation of branched actin networks: assembly and regulation of the N-WASP and WAVE molecular machines. Bioessays. 2010; 32:119–131.
21. Chen B, Brinkmann K, Chen Z, Pak CW, Liao Y, Shi S, Henry L, Grishin NV, Bogdan S, Rosen MK. The WAVE regulatory complex links diverse receptors to the actin cytoskeleton. Cell. 2014; 156:195–207.
22. Scita G, Nordstrom J, Carbone R, Tenca P, Giardina G, Gutkind S, Bjarnegard M, Betsholtz C, Di Fiore PP. EPS8 and E3B1 transduce signals from Ras to Rac. Nature. 1999; 401:290–293.
23. Dubielecka PM, Machida K, Xiong X, Hossain S, Ogiue-Ikeda M, Carrera AC, Mayer BJ, Kotula L. Abi1/Hssh3bp1 pY213 links Abl kinase signaling to p85 regulatory subunit of PI-3 kinase in regulation of macropinocytosis in LNCaP cells. FEBS letters. 2010; 584:3279–3286.
24. Miyamoto K, Teperek M, Yusa K, Allen GE, Bradshaw CR, Gurdon JB. Nuclear Wave1 is required for reprogramming transcription in oocytes and for normal development. Science. New York, NY. 2013; 341:1002–1005.
25. Stradal TE, Scita G. Protein complexes regulating Arp2/3-mediated actin assembly. Current opinion in cell biology. 2006; 18:4–10.
26. Ladwein M, Rottner K. On the Rho’d: the regulation of membrane protrusions by Rho-GTPases. FEBS letters. 2008; 582:2066–2074.
27. Zhuang C, Tang H, Dissanaike S, Cobos E, Tao Y, Dai Z. CDK1-mediated phosphorylation of Abi1 attenuates Bcr-Abl-induced F-actin assembly and tyrosine phosphorylation of WAVE complex during mitosis. The Journal of biological chemistry. 2011; 286:38614–38626.
28. King JS, Veltman DM, Georgiou M, Baum B, Insall RH. SCAR/WAVE is activated at mitosis and drives myosin-independent cytokinesis. Journal of cell science. 2010; 123:2246–2255.
29. Suetsugu S, Yamazaki D, Kurisu S, Takenawa T. Differential roles of WAVE1 and WAVE2 in dorsal and peripheral ruffle formation for fibroblast cell migration. Developmental cell. 2003; 5:595–609.
30. Legg JA, Bompard G, Dawson J, Morris HL, Andrew N, Cooper L, Johnston SA, Tramountanis G, Machesky LM. N-WASP involvement in dorsal ruffle formation in mouse embryonic fibroblasts. Molecular biology of the cell. 2007; 18:678–687.
31. Zipfel PA, Bunnell SC, Witherow DS, Gu JJ, Chislock EM, Ring C, Pendergast AM. Role for the Abi/wave protein complex in T cell receptor-mediated proliferation and cytoskeletal remodeling. Curr Biol. 2006; 16:35–46.
32. Campellone KG, Welch MD. A nucleator arms race: cellular control of actin assembly. Nature reviews. 2010; 11:237–251.
33. Taki T, Shibuya N, Taniwaki M, Hanada R, Morishita K, Bessho F, Yanagisawa M, Hayashi Y. ABI-1, a human homolog to mouse Abl-interactor 1, fuses the MLL gene in acute myeloid leukemia with t(10, 11)(p11.2;q23). Blood. 1998; 92:1125–1130.
34. Wang C, Tran-Thanh D, Moreno JC, Cawthorn TR, Jacks LM, Wang DY, McCready DR, Done SJ. Expression of Abl interactor 1 and its prognostic significance in breast cancer: a tissue-array-based investigation. Breast Cancer Res Treat. 2011; 129:373–386.
35. Chen H, Wu X, Pan ZK, Huang S. Integrity of SOS1/EPS8/ABI1 tri-complex determines ovarian cancer metastasis. Cancer research. 2010; 70:9979–9990.
36. Silva JM, Ezhkova E, Silva J, Heart S, Castillo M, Campos Y, Castro V, Bonilla F, Cordon-Cardo C, Muthuswamy SK, Powers S, Fuchs E, Hannon GJ. Cyfip1 is a putative invasion suppressor in epithelial cancers. Cell. 2009; 137:1047–1061.
37. Fernando HS, Sanders AJ, Kynaston HG, Jiang WG. WAVE1 is associated with invasiveness and growth of prostate cancer cells. J Urol. 2008; 180:1515–1521.
38. Fernando HS, Sanders AJ, Kynaston HG, Jiang WG. WAVE3 is associated with invasiveness in prostate cancer cells. Urol Oncol. 2010; 28:320–327.
39. Hossain S, Dubielecka PM, Sikorski AF, Birge RB, Kotula L. Crk and ABI1: binary molecular switches that regulate abl tyrosine kinase and signaling to the cytoskeleton. Genes & cancer. 2012; 3:402–413.
40. Oikawa T, Yamaguchi H, Itoh T, Kato M, Ijuin T, Yamazaki D, Suetsugu S, Takenawa T. PtdIns(3, 5)P3 binding is necessary for WAVE2-induced formation of lamellipodia. Nature cell biology. 2004; 6:420–426.
41. Innocenti M, Frittoli E, Ponzanelli I, Falck JR, Brachmann SM, Di Fiore PP, Scita G. Phosphoinositide 3-kinase activates Rac by entering in a complex with Eps8, Abi1, and Sos-1. The Journal of cell biology. 2003; 160:17–23.
42. Dubielecka PM, Ladwein KI, Xiong X, Migeotte I, Chorzalska A, Anderson KV, Sawicki JA, Rottner K, Stradal TE, Kotula L. Essential role for Abi1 in embryonic survival and WAVE2 complex integrity. Proceedings of the National Academy of Sciences of the United States of America. 2011; 108:7022–7027.
43. Yuan X, Cai C, Chen S, Yu Z, Balk SP. Androgen receptor functions in castration-resistant prostate cancer and mechanisms of resistance to new agents targeting the androgen axis. Oncogene. 2014; 33:2815–2825.
44. Chen E, Sowalsky AG, Gao S, Cai C, Voznesensky O, Schaefer R, Loda M, True LD, Ye H, Troncoso P, Lis RT, Kantoff P, Montgomery B, Nelson PS, Bubley GJ, Balk SP, et al. Abiraterone Treatment in Castration-Resistant Prostate Cancer Selects for Progesterone Responsive Mutant Androgen Receptors. Clin Cancer Res. 2014.
45. Yu Z, Cai C, Gao S, Simon NI, Shen HC, Balk SP. Galeterone prevents androgen receptor binding to chromatin and enhances degradation of mutant androgen receptor. Clin Cancer Res. 2014; 20:4075–4085.
46. Beer TM, Tombal B. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med. 2014; 371:1755–1756.
47. Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, Arora VK, Le C, Koutcher J, Scher H, Scardino PT, Rosen N, Sawyers CL. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011; 19:575–586.
48. Lin HK, Hu YC, Lee DK, Chang C. Regulation of androgen receptor signaling by PTEN (phosphatase and tensin homolog deleted on chromosome 10) tumor suppressor through distinct mechanisms in prostate cancer cells. Mol Endocrinol. 2004; 18:2409–2423.
49. Nan B, Snabboon T, Unni E, Yuan X-Y, Whang YE, Marcelli M. The PTEN tumor suppressor is a negative modulator of androgen receptor transcriptional activity. J Mol Endocrinol. 2003; 31:169–183.
50. Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin MA, Penning TM, Febbo PG, Balk SP. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006; 66:2815–2825.
51. Xiong X, Chorzalska A, Dubielecka PM, White JR, Vedvyas Y, Hedvat CV, Haimovitz-Friedman A, Koutcher JA, Reimand J, Bader GD, Sawicki JA, Kotula L. Disruption of Abi1/Hssh3bp1 expression induces prostatic intraepithelial neoplasia in the conditional Abi1/Hssh3bp1 KO mice. Oncogenesis. 2012; 1:e26.
52. Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, White TA, Stojanov P, Van Allen E, Stransky N, Nickerson E, Chae SS, Boysen G, Auclair D, Onofrio RC, Park K, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012; 44:685–689.
53. Kunda P, Craig G, Dominguez V, Baum B. Abi, Sra1, and Kette control the stability and localization of SCAR/WAVE to regulate the formation of actin-based protrusions. Curr Biol. 2003; 13:1867–1875.
54. Kölsch V, Charest PG, Firtel RA. The regulation of cell motility and chemotaxis by phospholipid signaling. J Cell Sci. 2008; 121:551–559.
55. Kotula L. Abi1, a critical molecule coordinating actin cytoskeleton reorganization with PI-3 kinase and growth signaling. FEBS Lett. 2012; 586:2790–2794.
56. Schmitz M, Grignard G, Margue C, Dippel W, Capesius C, Mossong J, Nathan M, Giacchi S, Scheiden R, Kieffer N. Complete loss of PTEN expression as a possible early prognostic marker for prostate cancer metastasis. Int J Cancer. 2007; 120:1284–1292.
57. Feilotter HE, Nagai MA, Boag AH, Eng C, Mulligan LM. Analysis of PTEN and the 10q23 region in primary prostate carcinomas. Oncogene. 1998; 16:1743–1748.
58. Bismar TA, Yoshimoto M, Vollmer RT, Duan Q, Firszt M, Corcos J, Squire JA. PTEN genomic deletion is an early event associated with ERG gene rearrangements in prostate cancer. BJU Int. 2011; 107:477–485.
59. Walker MM, Ellis SM, Auza MJ, Patel A, Clark P. The intercellular adhesion molecule, cadherin-10, is a marker for human prostate luminal epithelial cells that is not expressed in prostate cancer. Mod Pathol. 2008; 21:85–95.
60. Huang S, Gulzar ZG, Salari K, Lapointe J, Brooks JD, Pollack JR. Recurrent deletion of CHD1 in prostate cancer with relevance to cell invasiveness. Oncogene. 2012; 31:4164–4170.
61. Rasiah KK, Gardiner-Garden M, Padilla EJ, Möller G, Kench JG, Alles MC, Eggleton SA, Stricker PD, Adamski J, Sutherland RL, Henshall SM, Hayes VM. HSD17B4 overexpression, an independent biomarker of poor patient outcome in prostate cancer. Mol Cell Endocrinol. 2009; 301:89–96.
62. Baniwal SK, Little GH, Chimge NO, Frenkel B. Runx2 controls a feed-forward loop between androgen and prolactin-induced protein (PIP) in stimulating T47D cell proliferation. J Cell Physiol. 2012; 227:2276–2282.
63. Nelson PS, Clegg N, Arnold H, Ferguson C, Bonham M, White J, Hood L, Lin B. The program of androgen-responsive genes in neoplastic prostate epithelium. Proc Natl Acad Sci U S A. 2002; 99:11890–11895.
64. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012; 2:401–404.
65. Wilks C, Cline MS, Weiler E, Diehkans M, Craft B, Martin C, Murphy D, Pierce H, Black J, Nelson D, Litzinger B, Hatton T, Maltbie L, Ainsworth M, Allen P, Rosewood L, et al. The Cancer Genomics Hub (CGHub): overcoming cancer through the power of torrential data. Database (Oxford). 2014; 2014.
66. Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011; 12:323.
67. Yoshihara K, Shahmoradgoli M, Martinez E, Vegesna R, Kim H, Torres-Garcia W, Trevino V, Shen H, Laird PW, Levine DA, Carter SL, Getz G, Stemke-Hale K, Mills GB, Verhaak RG. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013; 4:2612.
68. Piccolo SR, Withers MR, Francis OE, Bild AH, Johnson WE. Multiplatform single-sample estimates of transcriptional activation. Proc Natl Acad Sci U S A. 2013; 110:17778–17783.
69. Gould J, Getz G, Monti S, Reich M, Mesirov JP. Comparative gene marker selection suite. Bioinformatics. 2006; 22:1924–1925.
70. Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP. GenePattern 2.0. Nat Genet. 2006; 38:500–501.