
INTRODUCTION
Acetylation of lysine residues is a widespread protein post-translational modification (PTM), and extensively relevant to modulation of cellular processes, including protein conformation and interaction [1]. Histone lysine acetylation was historically proposed to be a hallmark of transcriptionally active genes [2], and hitherto, deregulation of histone acetylation patterns often drives the aberrant expression of oncogenes resulting in proliferation and tumorigenesis [3]. Three types of proteins have been identified to regulate lysine acetylation: bromodomain (BRD) proteins [4, 5], histone acetyltransferases (HATs), histone deacetylases (HDACs) and sirtuins (SIRTs) [6-9]. BRD proteins bind to acetylated lysine (Kac) and thus acting as readers of lysine acetylation state; HATs effect lysine acetylation acting as writers; HDACs and SIRTs remove acetyl groups as erasers [9]. Bromodomains, functioning as acetyl-lysine binding domains, belong to a family of evolutionarily conserved protein modules originally found in proteins associated with chromatin and in nearly all nuclear HATs [10]. BRDs may contribute to highly specific histone acetylation by tethering transcriptional HATs to specific chromosomal sites, or to the activity of multi-protein complexes in chromatin remodeling [11]. Thus, BRDs modulate enzyme activities, protein assembly and protein-protein interactions (PPIs) via lysine acetylation, revealing broad implications for the mechanisms underlying a wide variety of cellular events, such as transcriptional activation and chromatin remodeling [12].
Human genome encodes 61 BRDs in 46 different proteins, in which legend specificity is imparted in the amino acid residue differences around the acetyl-lysine binding site [13]. BRD proteins mostly contain one or two bromodomains, while some proteins, such as nuclear scaffolding proteins (PB1), contain more than two BRDs [14]. Bromodomain and extra-terminal (BET), which taxonomically belongs to human BRD proteins family, shares a common domain architecture comprising two N-terminal bromodomains and an extra-C terminal domain. BET family consists of four mammalian members, including BRD containing 2 (BRD2), BRD3, BRD4 and BRDT, which both exhibit high levels of sequence conservation and a more divergent C-terminal recruitment [14]. Additionally, BET family proteins have been identified in oncogenic rearrangements, leading to highly oncogenic fusion proteins, and thus play key roles in development of several types of cancer. However, it is still unclear why only a subset of cells from diverse types of cancer responds to BET inhibitors [15].
Currently, there have been five registered active clinical trials investigating the targeting of BET family proteins, such as RVX-208, I-BET 762, OTX 015, CPI-0610 and TEN-010, in which OTX 015 has reported encouraging results in treating hematologic malignancies [16-19]. Meanwhile, multiple small-molecule inhibitors of BETs have also been developed and revealed great potential for clinical application, for instance, JQ1 and I-BET both exert the ability to interact with NF-κB and induce apoptosis in drug-resistant leukemia [20]. Hitherto, a number of landmark reports have revealed that the “reader” bromodomains are promising therapeutic targets in cancer. In this review, we summarize a series of small-molecule BET inhibitors and their molecular mechanisms in cancer, which may shed light on exploiting more novel BET inhibitors for future drug discovery.
STRUCTURE CHARACTERISTICS OF BET PROTEINS
BET family proteins all localize in the nucleus, and contain two tandems N-terminal BRDs, an extra-terminal (ET) domain and a more divergent C-terminal recruitment domain, all of which exhibit high levels of sequence conservation. Spanning 61 human BRDs, eight major BRD families are clustered by the derived phylogram analysis, in which BET family belongs to the sub-family II [21]. BRDs’ structural analysis of histones lysine-acetylated peptides recognition provides deep insights into characteristics and differences of biological ligand binding selectivity. Not only in BET family, but in all BRDs, it is likely that a hydrogen bond anchor Kac by a conserved asparagine residue their primary role is binding to Kac residues [22]. Besides, the conserved BRD fold contains a deep and largely hydrophobic acetyl lysine binding site, comprised of approximately 110 amino acids. BRDs contain 4 helices αZ, αA, αB, and αC, which form a characteristic antiparallel four-helix bundle, linked by two diverse loop regions, ZA and BC loops [23]. Helix αZ is flanked by a diverse sequence region, whose inserts are typically followed by a short helical segment in the ZA loop. The conserved motif follows the generic sequence Φ1X1X2(X3)Φ2X3X4X5(X6)Φ3, as Φi representing hydrophobic residues, N-terminal domain of BET family members specifically presents insertions at X3 [22]. At one end, the N and C termini come together, emphasizing the modular architecture of this domain and underscoring the idea that the BRD could act as a functional unit for PPIs. At the opposite end, the ZA loop packs against the BC loop, forming a central deep hydrophobic cavity that recognized as acetyl-lysine epitopes [24]. High-resolution co-crystal structures have shown that the first acetylated lysine mark of histone H4 docks directly onto the conserved asparagine. Conserved residue phenylalanine of BRDs deeply buried in helix αC stabilizes the C-terminus helical segment, while three conserved proline residues that ZA loop harbors may closely pack to hydrophobic residues in a C stabilizing the loop conformation. Simultaneously, similar as most BRD proteins, X-ray crystallography verifies BD1 of BRD2, 3 and 4 have an isoleucine at the position analogous to residue 162 in BRD2. This ‘gatekeeper’ residue, which varies in size across the BRD family, controls access to a lipophilic region comprising a tryptophan-proline-phenylalanine sequence (WPF shelf) which is present in a number of BRDs [25]. In BRD2, BRD3 and BRD4, affinity between H4 tail and different BRDs varied, for instance, BRD4 (1) seemed to specifically recognize multiple marks found on the H4 tail, while BRD4 (2) interacts with combinations of two and three acetylated lysines. However, studies indicate that BRDT requires two Kac residues for high–affinity binding. The importance of this shelf was also highlighted in BRDT, in which a diacetylated histone H4 tail to BRDT-BD1 and second AcK at position 8 lies across the WPF shelf leading to increased binding affinity [25]. BRDs distinguish different protein binding partners since they hold the sequence diversity in ZA and BC loop regions, binding to residues neighboring Kac in the target protein or peptide [26] (Figure 1).
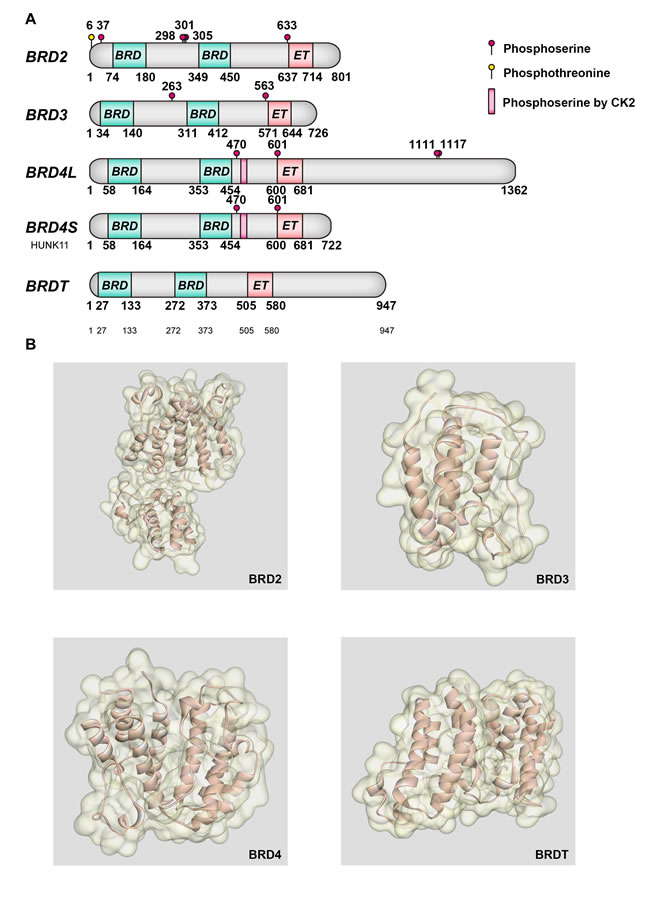
Figure 1: Molecular structures of BET bromodomains. Crucial post-translational modification sites and main features of BET proteins. (B) Structure of BET proteins.
BIOLOGICAL FUNCTION OF BET FAMILY
BET proteins are protein scaffolds, mitotic bookmarks and cell cycle regulators
Except BRDT specially locating in testis, BET proteins are widely distributed, and exert function to regulate an array of cellular processes. Firstly detected as protein scaffolds, BET family proteins recruit variety proteins to chromatin and transcription sites. During interphase, BRD4 recruits positive transcriptional elongation factor complex (PTEFb) to sites of active transcription, while another pool of BRD4 may be recruited by transcription mediator complexes independent of PTEFb [27]. In addition, the ET domain of BRD4 independently recruits transcription-modifying factors, including glioma tumor suppressor candidate region gene 1 (GLTSCR1); NSD3, a SET domain-containing histone methyltransferase; JMJD6, a histone arginine demethylase; and CHD4, a catalytic component of the NuRD nucleosome remodeling complex [28]. (Figure 2A) Different from BRD4, BRD2 binds to ε-aminoacetyl groups of nucleosomal histone lysines in a PTEFb-independent manner, and with related bromodomain proteins it provides a scaffold on chromatin to recruit E2F proteins , histone deacetylases (HDACs) , histone H4-specific acetyltransferase (HAT) and proteins involved in chromatin remodeling, thereby coupling histone acetylation to transcription [29] (Figure 2B). Similar with BRD2, BRD3 may regulate cell processes through E2F-RB pathway, moreover, it can directly recognized acetylated transcription factor, GATA1, which is essential for the targeting of GATA1 to chromatin [30] (Figure 2C).
BET family may also function as mitotic bookmark, identifying actively transcribed genes during mitosis by remaining associated chromatin when all the other factors dissociate. BRD4 remains associated with H4K5ac histones on chromatin during mitosis, leading to rapid de-compaction of the surrounding chromatin and to transcription post-mitotically [31]. BRD4 marks the start sites of many M/G1 genes, and accelerates expression of G1 genes and promotes cell cycle progression to S phase [32] BRD4 seems to be required for the G2 to M phase transition of the cell cycle because microinjection of BRD4-specific antibodies leads to cell cycle arrest [33] Then, BET family proteins function as cell cycle regulators, as mentioned before, that key transcriptional regulators genes of S phase, E2F1 and E2F2, are associated with BRD2 multi-protein complexes [33]. BRD3-dependent functional relationships with the cell cycle control machinery in normal cells are poorly understood, although forced expression of BRD3 down-regulates the RB–E2F pathway in nasopharyngeal carcinoma cells [33].
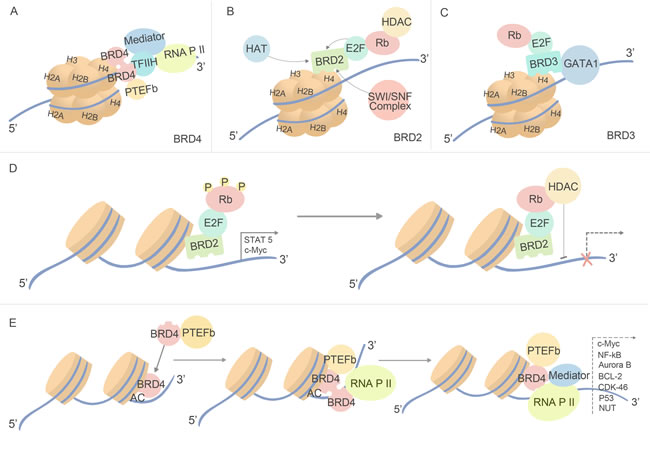
Figure 2: Biological function of BET bromodomains. Through sharing the highly similarity of amino acids sequences and all function as protein scaffolds, BRD2, BRD3 and BRD4 recruit different proteins. (A) BRD4 recruits proteins in a PTEFb-dependent manner, and it regulates the transcription process through coupling with RNA P II. (B) (C) BRD2 and BRD3 both exert function through E2F-RB pathway and in a PTEFb-independent manner, while BRD2 distinct from BRD3 for its ability to binding with SWI/SNF complex and regulating binding of ATP and histones. (D) The process how HDAC inhibits transcription through binding with BRD2 and E2F-Rb complex. (E) The process how BRD4 regulate transcription.
BET proteins function as transcription regulators
BET family has a pivotal role in regulating the transcription of growth-promoting genes [34], for instance, BRD4 and BRD2 are key mediators of transcriptional elongation by recruiting the PTEFb, which is composed of cyclin-dependent kinase 9 (CDK9) and its activator cyclin T. Productive transcription depends on the phosphorylation of the C-terminal repeat domain (CTD) of RNA polymerase II (RNAP II), and phosphorylation of the CTD residues Serine 5 (Ser5) and Serine 2 (Ser2) is necessary for the recruitment of RNA capping and splicing factors [35]. CTD Ser5 residues are phosphorylated primarily by the CDK7 kinase component of TFIIH, while subsequent Ser2 phosphorylation release RNAP II from an early elongation block and are necessary during productive elongation [36]. During elongation, Ser2 is phosphorylated by PTEFb, which depends on BRD4 for its nuclear localization and activation of its CDK9 kinase subunit [36]. BRD4 acts by affecting an acetylation and PTEFb dependent switch from basal transcription of immature unspliced transcripts to high levels of active mature mRNA. BRD4 is part of the transcription pre-initiation complex and remains associated with the RNAP II transcription complex until productive elongation, and it directly phosphorylates Ser2 while PTEFb only phosphorylates Ser2 if Ser5 has not been previously phosphorylated [25, 31]. Conversely, BRD4 is phosphorylated and activated by PTEFb. In addition, the same as TFIIH and PTEFb, BRD4 can directly interact with TAF7, a general transcription factor that regulates all three CTD kinases [36] (Figure 2C). BRD2 regulates transcription through E2F-RB pathway, while HDAC can phosphorylate RB to arrest the transcription process [37]. The involvement of BRD3 in cancer showed its role in the certain NMC translocations, and BRD3 can potentially associated with MLL fusion oncoproteins in leukemogenesis [38].Transcriptional inhibition is correlated with the prevention of signal-induced BRD-2, -3 and -4 protein recruitment to affected gene promoters. The inhibitor of BET selectively attenuated the induction of secondary response genes characterized by low CpG content, low basal H3 and H4 acetylation levels, low H3K4me3 methylation and low RNA polymerase II occupancy, suggesting BET proteins are involved in the recognition of gene promoters containing a combination of post-translational histone marks characteristic of poised but inactive secondary response genes [39]. BRD4 regulates NF-κB -dependent genes by binding acetylated RelA subunit of NF-kB [40]. The only specially expressed protein, BRDT, named as bromodomain testis-specific protein, has been reported in several stages of spermatogenesis. BRDT directly regulates activation of transcriptional repressors and activators in spermatocytes, and interacts with hyper-acetylated histone H4 tails to induce condensation of acetylated chromatin of haploid spermatids. In addition, it possesses similar functions of BRD4 in the recruitment of PTEFb to form PTEFb complex [38, 100]. Rapid development of both biological function and structural basis of BET protein has made it a newly emerging agent for therapeutic strategy of cancer. Development of small-molecule compounds targeting BET bromodomains has won increasingly attentions by biomedical researchers into this field.
BET proteins function in cancer
Histone acetylation levels have been associated with an open chromatin architecture and transcriptional activation, but specific marks have been linked to chromatin condensation [41], regulation of metabolism [42] and DNA repair [43]. Therefore, inappropriate acetylation levels have been associated with an aberrant transcription of disease-promoting genes, including cancer-related genes [44]. BET family proteins have been reported to be involved in a variety of malignant tumors, between which NUT-NMC have been reported to closely related to BRD proteins, in which the BRD-NUT blocks cellular differentiation, and depletion of this oncogene in squamous differentiation and cell cycle arrest. NUT midline carcinoma (NMC), an aggressive squamous cell carcinoma, is accordance with acquired chromosomal rearrangements involving NUT, creating chimeric genes that encode fusion proteins. Usually BRD4-NUT fusion genes are been detected, and less commonly NUT-variant fusion genes involving BRD3 also exists, leading to the expression of BRD-NUT fusion proteins [45].
BET family proteins have reported to able to promote aberrant gene expression in leukemia. MYC-family transcription factors are key regulators of cell growth and survival, whose gene amplification is a common copy-number alteration in cancer, while over-expression or translocation of the MYC locus contributes to Myc activity deregulation. In hematologic cancer models, such as MLL-fusion leukemia [46], acute myeloid leukemia (AML) [47], Burkitt’s lymphoma [48], multiple myeloma [49], and B-cell acute lymphoblastic (BLL) leukemia [50], amplification of onco-protein Myc drives distinct transcription programs, and leads to a consequence of cell proliferation. BET family directly regulate the expression of MYC genes, and directly silencing MYC gene expression via disruption of BET protein binding at the MYC locus may largely reduce cell proliferation [51]. Amplification or over-expression of MYC is frequently observed in lung cancer, ovarian cancer and breast cancer [52]. By recruiting a histone H3K36 methytransferase, WHSC1, BET protein BRD3/4 plays a key role in tamoxifen resistance to the ESR1 gene. WHSC1 is critical for maintaining estrogen signaling in ER-positive cells via forming a positive feedback regulatory loop with ERα, which is overexpressed in breast cancer. BRD3/4 interacts with WHSC1 in BRD regions, recognizes acetylated lysines on histone tails of the ESR1 promoter and recruits WHSC1, promoting ESR1 transcription [53]. Recent studies have unraveled a possible mechanism about how BRD4 proteins are involved in the transcription of active genes in cancers, especially associated with a subset of these genes. As previous mentioned, BRD4 and Mediator form a complex in transcription process, this complex may related to super-enhancers, which are span large genomic regions and contain exceptional amounts of Mediator and BRD4. In addition, important tumor genes are also associated with super-enhancers, so far has been identified in myeloma, small-cell lung cancer and glioblastoma. Therefore, BRD4 may regulate ongenetic drivers, such as MYC, through occupying super-enhancers, while inhibition of BRD4 also leads to preferential disruption of super-enhancers and selective loss of oncogene expression [70].
Cancers of neural origin may be related to the distinct expression of BET proteins including glioblastoma, medulloblastoma, and neuroblastoma. For instance, neuroblastoma is a pediatric solid tumor associated with a high frequency of MYCN amplifications, and inhibition of BET proteins in neuroblastoma leads to cell arrest [54]. BET proteins are also required for glioblastoma cell proliferation, mRNA of BRD2 and BRD4 are significantly overexpressed in glioblastoma, while disruption of BRD4 expression reduced glioblastoma cell cycle progression [55]. In melanoma, tumor progression may contribute to epigenetic changes, thus epigenetic and/or transcriptional regulation of certain target genes may support melanoma tumor-genesis. NF-κB regulates cytokine and chemokine production in melanoma, and is believed to contribute to progression of the disease by up-regulation of cell cycle and anti-apoptotic genes [56]. BRD2 and BRD4 are overexpressed in human primary and metastatic melanomas, whose inhibition resulted in down-regulating production of cytokines such as IL-6 and IL-8 [57].
The largely hydrophobic nature of the central acetylated lysine binding pocket and special extra-terminal domain of BET are necessary to accommodate the charge-neutralized acetylated lysine and recruits related proteins, making these modules particularly attractive for the development of inhibitors. Many proteins that use BRDs for their recruitment to specific regulatory complexes have been implicated in the development of cancer [58, 45]. The key principle that identification of small-molecule inhibitors with micromolar affinities has been identified, and recently two selective nanomolar inhibitors of BET proteins, JQ1 and I-BET, have been reported [59, 60]. Increased studies have focus on exploring new small-molecule compounds to selectively inhibit BET bromodomains for cancer therapy, and to date, several compounds have been found to exert their remarkable anti-cancer capacities (Table 1).
Table1: BET bromodomain inhibitors in cancer
Name |
Cancer type |
Target |
Mechanism/pathway |
Reference |
JQ1 |
Tam-R breast cancer |
BRD3/4 |
Suppresses the classic estrogen receptor-α signaling pathway and the growth of Tam-R breast cancer cells in culture |
[53] |
NUT midline carcinoma (NMC) |
BRD3/4 |
Suppresses different BRD4-NUT translocations |
[73] |
|
AML cells |
BRD4 |
Reduce binding of BRD4 and RNA polymerase II to the DNA of c-MYC and BCL2 |
[62] |
|
OCI-AML3 cell line |
BRD4 |
Trigger caspase 3/7-mediated apoptosis and DNA damage response. |
[63] |
|
Erythroleukemia cell line UT7 |
BRD4 |
Inhibit Epo-induced UT7 proliferation and restoring terminal erythroid differentiation |
[95] |
|
B-cell non-Hodgkin lymphoma |
BRD4 |
Induce cell death through MYC-CYCLON pathway |
[96] |
|
Neuroblastoma |
BRD4 |
Induce cell death through targeting MYCN |
[67] |
|
Primary glioblastoma xenograft lines |
BRD4 |
Induced marked G1 cell-cycle arrest and apoptosis through Bcl-xL and p21(CIP1/WAF1) . |
[97] |
|
Osteosarcoma cells |
BRD4 |
Trigger transcriptional silencing of MYC and RUNX2, resulting from the depletion of BRD4 |
[98] |
|
|
BRD2 |
Decrease STAT5-dependent transcription of both heterologous reporter genes and endogenous STAT5 target genes |
[64] |
|
I-BET151 |
Myeloma cell |
BRD2/3/4 |
Induce apoptosis and exerts strong anti-proliferative effect associating with contrasting effects on oncogenic MYC and HEXIM1, and inhibit transcriptional activator PTEFb |
[77] |
AML |
BRD4 |
Suppress cell growth in a HOX gene independent manner, but relieving upon NPM1c mutation and cytosplasmic dislocation. |
[79] |
|
Erythroleukemic (HEL) cell |
BRD4 |
Suppress myeloproliferative neoplasms by constitutively active JAK2 kinase |
[78] |
|
I-BET762 |
Myeloma cell |
BRD2/3/4 |
Inhibit myeloma cell proliferation, resulting in survival advantage in a systemic myeloma xenograft model. |
[77] |
neuroblastoma tumor models |
BRD2/3/4 |
Suppress cell growth in apoptosis signaling, and N-Myc-driven pathways, including the direct suppression of BCL2 and MYCN. |
[76] |
|
CPI203 |
Mantle cell lymphoma (MCL) |
BRD2/3/4; |
Decreased tumor burden, involving simultaneous MYC and IRF4 downregulation and apoptosis induction. |
[81] |
RVX2135 |
Myc-induced murine lymphoma |
BRD2/3/4 |
Exhibit broad transcriptional effects in Myc-transgenic lymphoma cells affecting many transcription factor networks. |
[82] |
Dinaciclib |
leukemia |
BRDT,CDKs; |
Interact with the acetyl-lysine recognition site of the bromodomain |
[94] |
PFI-1 |
Leukemia |
BRD2/4 |
Induce G1 cell-cycle arrest, downregulation of MYC expression, downregulation of Aurora B kinase |
[84] |
RVX-208 |
; |
BRD3(BD2); |
Raise apoA-I and increase preβ-HDL particles. Displace BET proteins from chromatin modestly affecting BET-dependent gene transcription. |
[90] |
SMALL-MOLECULE INHIBITORS OF BET BROMODOMAINS IN CANCER
BRD2/4 Inhibitors
The close similarity, about 80% at the amino acid level in human, between the bromodomains of BRD2 and BRD4 implies that two proteins may share a highly substantial functional similarity. Identified functional studies have verified that both BRD2 and BRD4 play crucial roles in transcription regulation, chromatin remodeling and recruiting related-protein, although the specific proteins may vary. (Figure 3).
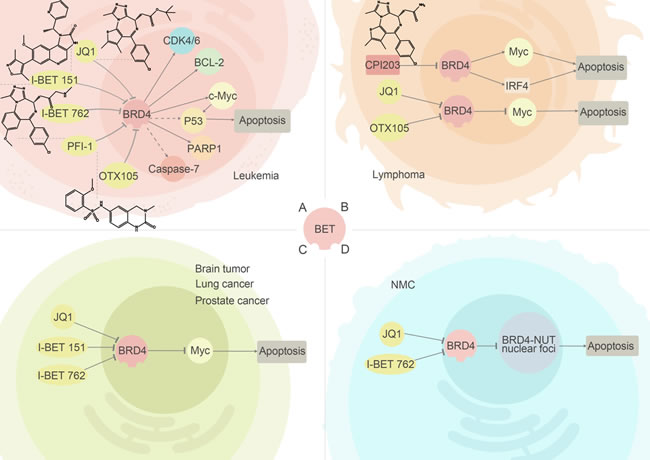
Figure 3: BRD2/4 inhibitors and their relevant anti-cancer pathways. Inhibitors of BRD2 and BRD4 have been detected to exert the anti-cancer ability in leukemia, lymphoma, NMC and other tumors. (A) In leukemia, inhibiting BRD4 to regulate transcription of c-Myc can effectively suppress the cell growth. (B) (C) In lymphoma and other solid tumors, inhibiting of BRD2/4 can lead to low expression of specific oncogenes, making BET inhibitors attractive anti-cancer agents. (D) In NMC, suppression of BRD4 can inhibit emerging of BRD4-NUT fusion proteins.
Due to the special structure interface of BRD4 protein, targeting bromodomain of BRD4 to inhibit the activity of BRD4-NUT fused protein become an attractive therapeutic strategy. With extensive researches, a small molecular compound, JQ1, exerts both strong inhibition efficiency and highly absorption rate towards NMC. JQ1 is a novel thieno-triazolo-1, 4-diazepine, whose competitive binding to acetyl-lysine recognition motifs enables it a successful anti-proliferative agent in BRD4 dependent cell lines. The bulky t-butyl ester functional group at C6 also allows it additional pendant group diversity and less binding to the central benazodiaepine receptor [59]. Additionally, results of screen of diverse inhibitors of BRD4-NUT and laboratory experiments confirmed JQ1 may process a better efficiency, comparing to the counterpart of other clinic drugs, suggesting its potential in future preclinical studies [61].
Since the crucial role BET proteins plays in leukemia, JQ1 also possesses significant anti-proliferative effect in leukemia, such as AML, T-cell ALL. In addition, JQ1 can inhibit growth and induce apoptosis of human AML cells, including those expressing FLT3-ITD(FMS-like tyrosine kinase 3- internal tandem duplication),a mutation of proto-oncogene. Recent studies also shows that co-treatment of JQ1 and FLT3-inhibitor, FLT3-TKI, can greatly attenuate the expression of c-MYC, BCL2, and CDK4/6, meanwhile synergistically induce apoptosis of cultured and primary CD34(+) human AML blast progenitor cells (BPC) expressing [62]. Activation of caspase 3/7, but not caspase 8, is found in JQ1-mediated apoptosis, indicating that the intrinsic apoptotic pathway is involved [63].
In other hematologic malignancies, JQ1 may exert anti-tumor ability through targeting BRD2, as BRD2 is the critical mediator for STAT5 activity. STAT5, a transcription factor signal, is constitutively activated and drives the expression of genes necessary for proliferation, survival, and self-renewal in leukemia. In experimental models of acute T-cell lymphoblastic leukemia, JQ1 decreases STAT5-dependent (but not STAT3-dependent) transcription of both heterologous reporter genes and endogenous STAT5 target genes, and shows strong synergy with tyrosine kinase inhibitors (TKI) in inducing apoptosis in leukemia cells [64].
BET family proteins may participate in the malignant brain tumor gene regulation, for instance, high-level BRD4 expression was detected in 99 of 115 pediatric primary medulloblastomas (75%) while it was only marginally (22%) expressed in normal cerebellar tissue. One significant clinical challenge for medulloblastoma in pediatric oncology stands that the overall survival currently remains under 70%, and patients with tumors overexpressing MYC or harboring MYC oncogene amplification have an extremely poor prognosis [65]. JQ1 treatment can significantly reduce cell proliferation and induce apoptosis and senescence in different human medulloblastoma cells. Those MYC-amplified cell lines, HD-MB3, ONS-76 and D-341, produced the strongest apoptotic response, while the expression of MYC-associated proteins, such as Cyclin D1 and E2F1, were reduced as well, suggesting JQ1 may exert its anti-tumor ability by targeting BRD4-Myc pathway [66]. Similarly in human neuroblastoma, JQ1 targeted BRD4 to regulate MYCN expression, and induced cell death [67]. In addition, JQ1 inhibited expression of BRD3 and BRD4, and suppressed the classic estrogen receptor-α signaling pathway, resulting in the growth suppression of Tamoxifen resistant breast cancer cells in culture [53].
Particularly in virus-induced tumors, inhibition of host-encoded BET regulates both transcriptional activation and transcriptional repression of virus promoters, making JQ1 a promising therapeutic agent as well. In etiology study of human T cell leukemia virus 1 (HTLV-1)-mediated adult T cell leukemia, JQ1 suppressed the proliferation of Tax-expressing rat fibroblasts, and inhibited transformation and tumor-genesis of Tax-positive HTLV-1-infected cells and Tax-mediated cell. Considering Tax can induce the acetylation of lysine 310 of RelA and promote BRD4 binding acetylated RelA, inhibition of BRD4 by JQ1 may suppress Tax-mediated transcriptional activation of NF-κB [68]. JQ1 treatment can reduce Epstein–Barr virus (EBV) promoter in lymphomas. EBV is the causative agent of lympho-proliferative diseases, whose nuclear antigen (EBNA) proteins providing a functional analogue of LANA1 (Latency-associated nuclear antigen). During the transcription elongation of EBV, inhibition of host-encoded BRD4 can suppress recruiting of PTEFb to viral C promoter, therefore negatively controlling the replication of EBV [69]. JQ1 treatment reduces BRD4 association with the promoter, providing a promising future of drug development in the treatment of latent virus infections by disrupting transcriptional co-regulator.
BET inhibitor, OTX 015, shows the clinically meaningful activity at nontoxic doses, in which inter results of an ongoing phase I trial in hematologic malignances are inspiring. Besides regulating the BRD4, OTX 015 can inhibit the growth of hematologic malignances through directly regulating MYC expression and activity [16]. Besides that, currently, OTX015 has been reported to exert anti-proliferative activity in diffuse large B-cell lymphoma (DLBCL). OTX 015 is highly sensitive to majority (9 of 13) of DLBCL cell lines, and suppression of MYC caused by OTX015 is reversible in DLBCL [71].
GSK525762A (I-BET762) is a specific and potent inhibitor of BET protein binding to acetylated bromodomains, and GSK has embarked it on Phase I clinical trial of NMC. It is derived from medicinal chemistry optimization of a hit derived from a phenotypic screen to identify small molecules able to enhance ApoA1 expression [72] The fusion between BRD4 (and to a lesser extent BRD3) with NUT gene leads to NMC [73], and this fusion is oncogenic due to the inability to sequester important regulatory molecules such as CBP/p300 into BRD4-NUT nuclear foci, which are formed in a bromodomain-dependent manner, and knockdown of BRD4-NUT with anti-NUT siRNAs has been shown to lead to cell differentiation and apoptosis [74]. Treatment of patient-derived samples with I-BET762 has been shown to lead to terminal differentiation and growth arrest of the malignant cells [74]. In addition, I-BET762 potently reduced MYC expression in LNCaP prostate cancer cell lines and a patient-derived tumor model with subsequent inhibition of cell growth and reduction of tumor burden in vivo [75]. In neuroblastoma tumor models, I-BET 762 can trigger apoptosis through BET inhibition of N-Myc-driven pathways, including the direct suppression of Bcl-2 and MYCN. And conversely, reversal of MYCN or BCL2 suppression reduces the potency of I-BET726-induced cytotoxicity in a cell line-specific manner [76]. Further, I-BET762 can influence myeloma cell proliferation, resulting in survival advantage in a systemic myeloma xenograft model [77].
Similar with JQ1, non-benzodiazepine I-BET151 is a pan-BET inhibitor, which can exhibit significant anti-tumor activity in murine models of NUT midline carcinoma, multiple myeloma, MLL and ALL, lung cancer, and malignant brain tumor. In cellular studies of myelo-proliferative neoplasms driven by mutant JAK2 (JAK2V617F), I-BET151 possessed growth inhibitory activity with concomitant down-regulation of LMO2, an important oncogenic regulator of hematopoietic stem cell development [78]. In acute myeloid leukemia (AML) with mutations of the nucleophosmin gene (NPM1), I-BET 151 treatment may down-regulate the core transcriptional program, which is HOX gene independent underlies sensitivity to I-BET treatment [79]. Similar with I-BET762, I-BET151 also inhibits myeloma cell proliferation through inducing apoptosis and exerting strong anti-proliferative effect in vitro and in vivo through transcriptional repression of MYC and MYC-dependent programs by abrogating recruitment to transcriptional activator PTEFb [77]. BRD2 is the main BET protein involved in regulation of NF-kB and that I-BET151 caused transcriptional downregulation of the NF-kB subunit p105/p50 [80].
CPI203, a BET bromodomain inhibitor, can affect the lymphoma cell growth. The development of Bortezomib resistance to proteasome inhibition in mantle cell lymphoma (MCL) may limit its efficacy of clinical activity. An increased tumorigenicity of bortezomib-resistant MCL cells, which is associated with plasmacytic differentiation features, like interferon regulatory factor 4 (IRF4) and Blimp-1 up-regulation. Repression of the IRF4 target gene MYC in bortezomib-resistant cells by gene knockdown or treatment with CPI203 synergistically induced cell death when combined with lenalidomide [81]. In mice, addition of CPI203 to lenalidomide therapy further decreased tumor burden, involving simultaneous MYC and IRF4 down-regulation and apoptosis induction [81]. RVX2135, a novel and orally bioavailable selective pan-BET inhibitor, presented anti-proliferative ability in Myc-induced lymphoma. What’s more, RVX2135 was reported that broad transcriptional changes are mediated, while these are genetically and functionally linked to histone deacetylase inhibitors [82].
PFI-1, a novel dihydroquinazolinone reported as a BET chemical probe, binds to BET bromodomain chemically distinct from previously reported BET inhibitors. Exposure of leukemia cells to PFI-1 results in induction of caspase-dependent apoptosis, differentiation and in down-regulation of the Aurora B kinase. Aurora kinases are highly expressed in diverse cancer types and are also frequently up-regulated in leukemia [83]. In the BET inhibitor sensitive cell line MV4, researchers observed strong induction of PARP1 and pro-caspase 7 cleavage after 24 h exposure with PFI-1 [84]. PFI-1 and JQ1 dissociate BRD4 from HOXA9 and promotes differentiation, as a marker of poor prognosis in patients with acute myeloid leukemia [85] and overexpression of HOXA9 leads to expansion of hematopoietic stem cells in bone marrow cells and development of leukemia in mice [84, 85].
Further, more efficient dual kinase-bromodomain inhibitors have been developed for rationally designed polypharmacology. For instance, two nanomolar activities on BRD4 inhibitors, BI-2536 and TG-101348, have been identified to inhibit bromodomains with therapeutically relevant potencies, particularly noteworthy as shedding light on independent oncogenic pathways [99].
BRD3 Inhibitors
Diverse from BRD2-dependent roles in regulating differentiation of adipose tissue and neurons, BRD3 mainly functions in recruitment of GATA1 in hematopoietic cells through regulating maturation of erythroid, megakaryocyte, and mast cell lineages [86, 87]. Inhibitors of BRD3 are less studied than their counterparts in BRD2 and BRD4, due to the lacking of specific mechanism of BRD3. However, pan-BET inhibitors, like JQ1 and I-BET-151, have been found to target BRD3 in NMC and leukemia [88], and inhibition with an I-BET762 analogue led to disruption of normal erythroid maturation.
Currently, a disappointing result of negative clinical finding of RVX-208 has been reported, which is acting as an ApoA1 modulator in phase I/II clinical trials for the treatment of cardiovascular diseases [89] The quinazolone RVX-208, a derivative of the plant polyphenol resveratrol, acts as interaction partner of ApoA1 and performs a preferentially binding ability to the BD2 of BRD3, exhibiting selectivity over BD1 of up to 23-fold [90]. However, previous studies of BRD3 that showed that its recruitment to acetylated sites on GATA1 is mediated by BD1 [91], suggesting the selective inhibition of RVX-208 may cause drugs nullity. Considering the important role ApoA1 played in hepatocellular carcinoma, and chemical inhibition of BDs has been associated with ApoA1 up-regulation, RVX-208 can be used as drugs of hepatocellular carcinoma. In addition, other potent BET inhibitor, JQ1 has strongly stimulated ApoA-I production in Hep-G2 cells in a post-translational regulation manner [92], making it appealing of developing multi-target inhibitors in hepatocellular carcinoma (Figure 4A).
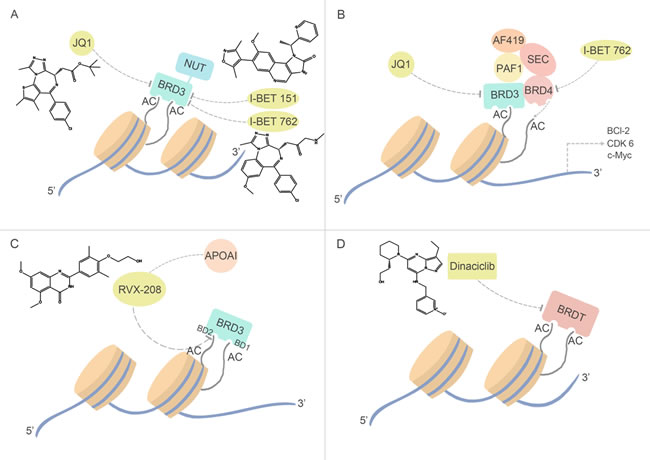
Figure 4: BRD3 and BRDT inhibitors and their relevant anti-cancer pathways. (A) Fused with NUT gene, BRD3 may encode BRD3-NUT fusion proteins in NMC, which is similar with BRD4. Specifically inhibiting BRD3-NUT fusion protein can block BRD3-NUT gene transcription, and inhibit NMC cell growth. (B) Pan-inhibitors I-BET762 and JQ1 inhibits BRD3. (C) As the only member of BET family can bind with transcription factor GATA1, inhibition of BRD3 through BRD3-GATA pathway by clinical experimental compound, RVX-208, may be a therapeutic strategy in hepatocellular carcinoma. (D) The least known BET family member, BRDT, is also been detected the anti-cancer capacity in leukemia by an unspecific-target drug Dinaciclib.
BRDT Inhibitors
BRDT, as a tesis-specific BET family member, have been observed the selective function of its two BDs, in which deletion of BD1 in BRDT may result in abnormal spermatids and sterility [93]. Furthermore, altered histone modifications in mice have been observed in the BRDT promoter region of sub-fertile patients. A potent inhibitor of cyclin-dependent kinases (CDKs), Dinaciclib can interact with BRDT and bind in the ZA channel of BRD [94], and reporting the potential of Dinaciclib to act as protein-protein inhibitors of bromodomains, and considering Dinaciclib has advanced to Phase III clinical trials for the treatment of leukemia [91], the new findings may provide a new structural framework for the design of next-generation bromodomain inhibitors using the vast chemical space (Figure 4B).
CONCLUSIONS
Bromodomain, as acetyl-lysine (Kac) “reader” domain, is part of the write−read−erase concept that has been linked with the transfer of epigenetic information. As part of the bromodomain-containing proteins, conserved BET family proteins processed dual-BD and an ET domain, suggesting a better structure feature for drug development. BET proteins mediate PPI network between diverse arrays of partners, and function as mitosis bookmark, protein scaffold and chromatin regulator in cellular processes. The information mentioned above has provided unique amino-acid BRDs signature and ability to regulating crucial cancer-regulation genes, revealing good potentials for drug targets in all subfamilies, thereby indicating that potent inhibitors targeting BET bromodomains would be developed.
Extensive studies have explored small-molecule inhibitors of BET family proteins for cancer therapy. Five small-molecule inhibitors have been used in clinical trials, such as I-BET762 in NMC and OTX-015 in hematologic cancers, inspiring researchers for further studies of BET inhibitors. Some hotspot inhibitors like JQ1 and I-BET151, I-BET 762 have been reported, which promote the clinical trial of these potential drugs, as well as gain more detailed information of the function and regulation activity of BET family. The experimental validation of pan-BET inhibitors have been further explored in different cancers, such as JQ1 and I-BET151, and experiments have validated that combination use of BET inhibitor and other drugs may reduce the drug resistance and raise the sensitivity towards other therapy. As a trend of current pharmaceutical design, developing multi-target inhibitors of BET family and combination use of BET inhibitors with other drugs would largely reduce the possibility of drugs resistance.
Hitherto, the limitation of BET proteins inhibitors also emerged, the negative finding of RVX-208 is disappointing for the potency and selectivity of this agent have not been disclosed in previous literatures. The selective inhibition of RVX-208 towards BD2 rather than BD1 in BRD3 suggests that specific targeting may be able to perturb and modulate BET function in a context-dependent manner. And notably, little study has been conducted about the diverse function of two BDs in one BET protein, except in BRDT that researchers confirmed that deletion of the first bromodomain in BRDT in mice is sufficient to confer sterility by blocking BRDT-dependent sperm maturation. Due to the alternative splicing, every BET family protein processes different isoforms, but the exact function and expression reason of these isoforms haven’t been fully studied. Therefore, further development of domain and isoform specific inhibitors need to be necessary for unravelling the exact roles of BET bromodomains in gene transcription. And, the alternative expression levels of BET proteins in various cancers still need to be further discovered. However, BET proteins mediated PPI network have not been fully unraveled; therefore, development of mechanism towards PPI of BET may be a promising studying flied, which may also benefit the drug development to BET inhibitors.
In conclusion, rapid success has been achieved with the BET family of bromodomains, and numbers of potent small-molecule inhibitors have been reported. Inspiring findings that various inhibitors bear their own anti-tumor abilities have been enlightening us for further designing and discovering more new potential BET inhibitors. Moreover, clinical studies and mechanism elucidation of such inhibitors have made it clear that they would be invaluable tools for dissecting the biological roles of bromodomains and being potentially of great value as therapeutic leads.
ACKNOWLEDGEMENTS
This work was supported by grants from the National 973 Basic Research Program of China (No. 2013CB911300), and National Natural Science Foundation of China (Nos. 81402496 and 81473091).
Conflicts of interest
None.
Author contribution
Conceived and designed the manuscript: B. Liu and J. Huang. Analyzed the data: L.L.Fu, J.J. Li, M. Tian and L. Ouyang. Wrote the paper: L.L. Fu, M. Tian, X Li and B. Liu. Revised the paper: Y.H. Zhang and B. Liu.
REFERENCES
1. Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV and Mann M. Lysine acetylationtargets protein complexes and co-regulates major cellular functions. Science. 2009; 325: 834-840.
2. Kuo MH and Allis CD. Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays. 1998; 20: 615-626.
3. Lombardi PM, Cole KE, Dowling DP and Christianson DW. Structure, mechanism, and inhibition of histone deacetylases and related metalloenzymes. Curr Opin Struct Biol. 2011; 21: 735-743.
4. Filippakopoulos P and Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov. 2014; 13: 337-356.
5. Barneda-Zahonero B and Parra M. Histone deacetylases and cancer. Mol Oncol. 2012;6:579-589.
6. Dhalluin C1, Carlson JE, Zeng L, He C, Aggarwal AK and Zhou MM. Structure and ligand of a histone acetyltransferase bromodomain. Nature. 1999; 6735: 491–496.
7. Seto E and Yoshida M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb Perspect Biol. 2014; 6:a018713.
8. Chen Y, Fu LL, Wen X, Wang XY, Liu J, Cheng Y and Huang J. Sirtuin-3 (SIRT3), a therapeutic target with oncogenic and tumor-suppressive function in cancer. Cell Death Dis. 2014; 5: e1047.
9. Anand P, Brown JD, Lin CY, Qi J, Zhang R, Artero PC, Alaiti MA, Bullard J, Alazem K, Margulies KB, Cappola TP, Lemieux M, Plutzky J, Bradner JE, Haldar SM. BET bromodomains mediate transcriptional pause release in heart failure. Cell. 2013; 154: 569-582.
10. Sanchez R, Meslamani J and Zhou MM. The bromodomain: from epigenome reader to druggable target. Biochim Biophys Acta. 2014; 8:676-685.
11. Barbieri I, Cannizzaro E and Dawson MA. Bromodomains as therapeutic targets in cancer. Brief Funct Genomics. 2013; 12(3):219–230.
12. Zhang G, Sanchez R and Zhou MM. Scaling the druggability landscape of human bromodomains, a new class of drug targets. J Med Chem. 2012; 55:7342–7345.
13. Filippakopoulos P, and Knapp, S. The bromodomain interaction module. FEBS Lett. 2012; 586: 2692–2704.
14. Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013; 153: 320-334.
15. Shi J and Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell. 2014; 5:728-736.
16. Herait, P.E. et al., BET-bromodomain inhibitor OTX015 shows clinically meaningful activity at nontoxic doses: interim results of an ongoing phase I trial in hematologic malignancies. In Proceedings of the 105th Annual Meeting of the American Association for Cancer Research, San Diego, CA. Philadelphia (PA): AACR; Abstract nr CT231. 2014; 5-9.
17. Ito T, Umehara T, Sasaki K, Nakamura Y, Nishino N, Terada T, Shirouzu M, Padmanabhan B, Yokoyama S, Ito A, Yoshida M. Real-time imaging of histone H4K12-specific acetylation determines the modes of action of histone deacetylase and bromodomain inhibitors. Chem Biol. 2011; 18: 495–507.
18. Chung CW , Coste H, White JH, Mirguet O, Wilde J, Gosmini RL, Delves C, Magny SM, Woodward R, Hughes SA, Boursier EV, Flynn H, Bouillot AM, Bamborough P, Brusq JM, Gellibert FJ et al. Discovery and characterization of small molecule inhibitors of the BET family bromodomains. J Med Chem. 2011; 54: 3827–3838.
19. Zeng L, Li J, Muller M, Yan S, Mujtaba S, Pan C, Wang Z and Zhou MM.Selective small molecules blocking HIV-1 Tat and coactivator PCAF association. J Am Chem Soc. 2005; 127: 2376–2377.
20. Ceribelli M, Kelly PN, Shaffer AL, Wright GW, Xiao W, Yang Y, Mathews Griner LA, Guha R, Shinn P, Keller JM, Liu D, Patel PR, Ferrer M, Joshi S et al. Blockade of oncogenic IκB kinase activity in diffuse large B-cell lymphoma by bromodomain and extraterminal domain protein inhibitors. Proc Natl Acad Sci USA. 2014; 31: 11365-11370.
21. Barr AJ, Ugochukwu E, Lee WH, King ON, Filippakopoulos P, Alfano I, Savitsky P, Burgess-Brown NA, Müller S and Knapp S. Large-scale structural analysis of the classical human protein tyrosine phosphatome. Cell. 2009; 136: 352–363.
22. Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, Felletar I, Volkmer R, Müller S, Pawson T, Gingras AC, Arrowsmith CH and Knapp S. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012; 149:214–231.
23. Calori G, Lattuada G, Piemonti L, Garancini MP, Ragogna F, Villa M, Mannino S, Crosignani P, Bosi E, Luzi L, Ruotolo G and Perseghin G. Prevalence, metabolic features, and prognosis of metabolically healthy obese Italian individuals: the Cremona Study. Diabetes Care. 2011; 34:210-215.
24. Zeng L and Zhou M. Bromodomain: an acetyl-lysine binding domain. FEBS Lett. 2002; 513:124−128.
25. Prinjha R K, Witherington J, Lee K. Place your BETs: the therapeutic potential of bromodomains[J]. Trends in pharmacological sciences, 2012, 33(3): 146-153.
26. Papavassiliou KA and Papavassiliou AG. Bromodomains: pockets with therapeutic potential. Trends Mol Med. 2014; 20:477-478.
27. Wu SY, and Chiang CM. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J Biol Chem. 2007; 13141-13145.
28. Rahman S, Sowa ME, Ottinger M, Smith JA, Shi Y, Harper JW and Howley PM. The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol Cell Biol. 2011; 31:2641–2652.
29. Houzelstein D, Bullock SL, Lynch DE, Grigorieva EF, Wilson VA and Beddington RS. Growth and early post implantation defects in mice deficient for the bromodomain-containing protein Brd4. Mol Cell Biol. 2002; 22:3794-3802.
30. Vollmuth F, Blankenfeldt W and Geyer M. Structures of the dual bromodomains of the P-TEFb-activating protein Brd4 at atomic resolution. J Biol Chem. 2009; 284:36547-36556.
31. Devaiah BN and Singer DS. Two faces of BRD4. Transcription. 2013;4(1): 13-17.
32. Zhao R, Nakamura T, Fu Y, Lazar Z and Spector DL. Gene bookmarking accelerates the kinetics of post mitotic transcriptional re-activation. Nat Cell Biol. 2011; 13:1295-1304.
33. Belkina AC and Denis GV. BET domain co-regulators in obesity, inflammation and cancer. Nature Reviews Cancer, 2012, 12(7): 465-477.
34. Houzelstein D, Bullock SL, Lynch DE, Grigorieva EF, Wilson VA and Beddington RS. Growth and early postimplantation defects in mice deficient for the bromodomain-containing protein Brd4. Mol Cell Biol. 2002; 22:3794–3802.
35. Devaiah BN, Lewis BA, Cherman N, Hewitt MC, Albrecht BK, Robey PG, Ozato K, Sims RJ 3rd and Singer DS. BRD4 is an atypical kinase that phosphorylates serine2 of the RNA polymerase II carboxy-terminal domain. Proc Natl Acad Sci U S A. 2012; 109:6927-6932.
36. Devaiah BN and Singer DS. Cross-talk among RNA polymerase II kinases modulates C-terminal domain phosphorylation. J Biol Chem. 2012; 287: 38755-38766.
37. Zou Z, Huang B, Wu X, Zhang H, Qi J, Bradner J, Nair S and Chen LF. Brd4 maintains constitutively active NF-κB in cancer cells by binding to acetylated RelA[J]. Oncogene, 2014, 33(18): 2395-2404.
38. Belkina AC, Nikolajczyk BS and Denis GV. BET protein function is required for inflammation: Brd2 genetic disruption and BET inhibitor JQ1 impair mouse macrophage inflammatory responses. The Journal of Immunology, 2013, 190(7): 3670-3678.
39. Gallenkamp D, Gelato KA, Haendler B and Weinmann H. Bromodomains and their pharmacological inhibitors. ChemMedChem. 2014; 9:438-464.
40. Shang E, Nickerson HD, Wen D, Wang X and Wolgemuth DJ. The first bromodomain of Brdt, a testis-specific member of the BET sub-family of double-bromodomain-containing proteins, is essential for male germ cell differentiation. Development. 2007; 134:3507-3515.
41. Bannister A J and Kouzarides T. Regulation of chromatin by histone modifications. Cell research, 2011, 21(3): 381-395.
42. Evertts AG, Zee BM, Dimaggio PA, Gonzales-Cope M, Coller HA and Garcia BA. Quantitative dynamics of the link between cellular metabolism and histone acetylation. Journal of Biological Chemistry, 2013, 288(17): 12142-12151.
43. Gong F and Miller K M. Mammalian DNA repair: HATs and HDACs make their mark through histone acetylation. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, 2013, 750(1): 23-30.
44. Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR and Peterson CL. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 2006; 311:844-847.
45. Muller S, Filippakopoulos P and Knapp S. Bromodomains as therapeutic targets. Expert Rev Mol Med. 2011; 13: e29.
46. Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, Robson SC, Chung CW, Hopf C, Savitski MM, Huthmacher C, Gudgin E, Lugo D, Beinke S, Chapman TD, Roberts EJ et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011; 478: 529-533.
47. Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, Taylor MJ, Johns C, Chicas A, Mulloy JC,Kogan SC, Brown P et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011; 478: 524-528.
48. Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl. Acad. Sci. USA 2011; 108: 16669-16674.
49. Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, Bergeron L, Sims RJ 3rd. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011; 146: 904-917.
50. Ott CJ, Kopp N, Bird L, Paranal RM, Qi J, Bowman T, Rodig SJ, Kung AL, Bradner JE, Weinstock DM. BET bromodomain inhibition targets both c-MYC and IL7R in high-risk acute lymphoblastic leukemia. Blood. 2012; 120: 2843–2852.
51. Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, Mc Henry KT, Pinchback RM, Ligon AH, Cho YJ, Haery L et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010; 463: 899-905.
52. Vita M, Henriksson M. The Myc oncoprotein as a therapeutic target for human cancer. Semin. Cancer Biol. 2006; 16: 318-330.
53. Feng Q, Zhang Z, Shea MJ, Creighton CJ, Coarfa C, Hilsenbeck SG, Lanz R, He B, Wang L, Fu X, Nardone A, Song Y, Bradner J,Mitsiades N, Mitsiades CS, Osborne CK et al. An epigenomic approach to therapy for tamoxifen-resistant breast cancer, Cell Res. 2014; 7: 809-819.
54. Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, Nekritz EA, Zeid R, Gustafson WC, Greninger P, Garnett MJ, McDermott U, Benes CH, Kung AL, Weiss WA et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov. 2013; 3: 308-323.
55. Pastori C, Daniel M, Penas C, Volmar CH, Johnstone AL, Brothers SP, Graham RM, Allen B, Sarkaria JN, Komotar RJ, Wahlestedt C, Ayad NG. BET bromodomain proteins are required for glioblastoma cell proliferation. Epigenetics. 2014; 9: 611-620.
56. Peng HH, Liang S, Henderson AJ, Dong C. Regulation of interleukin-8 expression in melanoma-stimulated neutrophil inflammatory response. Exp Cell Res. 2007; 313: 551-559.
57. Klein K, Kabala PA, Grabiec AM, Gay RE, Kolling C, Lin LL, Gay S, Tak PP, Prinjha RK, Ospelt C, Reedquist KA. The bromodomain protein inhibitor I-BET151 suppresses expression of inflammatory genes and matrix degrading enzymes in rheumatoid arthritis synovial fibroblasts. Ann Rheum Dis. 2014; pii: annrheumdis-2014-205809.
58. Filippakopoulos P and Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation[J]. Nature Reviews Drug Discovery, 2014, 13(5): 337-356.
59. Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, et al. Selective inhibition of BET bromodomains. Nature. 2010; 468:1067-1073.
60. Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, Chandwani R, Marazzi I, Wilson P, Coste H, White J, Kirilovsky J, Rice CM et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010; 468: 1119–1112
61. Beesley AH, Stirnweiss A, Ferrari E, Endersby R, Howlett M, Failes TW, Arndt GM, Charles AK, Cole CH and Kees UR. Comparative drug screening in NUT midline carcinoma. Br J Cancer. 2014; 110:1189-1198.
62. Fiskus W, Sharma S, Qi J, Shah B, Devaraj SG, Leveque C, Portier BP, Iyer S, Bradner JE and Bhalla KN. BET Protein Antagonist JQ1 Is Synergistically Lethal with FLT3 Tyrosine Kinase Inhibitor (TKI) and Overcomes Resistance to FLT3-TKI in AML Cells Expressing FLT-ITD. Mol Cancer Ther. 2014; 13:2315-2327.
63. Stewart HJ, Horne GA, Bastow S and Chevassut TJ. BRD4 associates with p53 in DNMT3A-mutated leukemia cells and is implicated in apoptosis by the bromodomain inhibitor JQ1. Cancer Med. 2013; 2:826-835.
64. Liu S, Walker SR, Nelson EA, Cerulli R, Xiang M, Toniolo PA, Qi J, Stone RM, Wadleigh M, Bradner JE and Frank DA. Targeting STAT5 in hematologic malignancies through inhibition of the bromodomain and extra-terminal (BET) bromodomain protein BRD2. Mol Cancer Ther. 2014; 13:1194-1205.
65. Henssen A, Thor T, Odersky A, Heukamp L, El-Hindy N, Beckers A, Speleman F, Althoff K, Schäfers S, Schramm A, Sure U, Fleischhack G, Eggert A and Schulte JH. BET bromodomain protein inhibition is a therapeutic option for medulloblastoma, Oncotarget. 2013; 4:2080-2095.
66. Bandopadhayay P, Bergthold G, Nguyen B, Schubert S, Gholamin S, Tang Y, Bolin S, Schumacher SE, Zeid R, Masoud S, Yu F, Vue N, Gibson WJ, Paolella BR, Mitra SS, Cheshier SH, et al. BET bromodomain inhibition of MYC-amplified medulloblastoma. Clin Cancer Res. 2014; 20:912-925.
67. Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, Nekritz EA, Zeid R, Gustafson WC, Greninger P, Garnett MJ, McDermott U, Benes CH, Kung AL, Weiss WA et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov. 2013; 3: 308-323.
68. Wu X, Qi J, Bradner JE, Xiao G and Chen LF. Bromodomain and Extraterminal (BET) Protein Inhibition Suppresses Human T Cell Leukemia Virus 1 (HTLV-1) Tax Protein-mediated Tumorigenesis by Inhibiting Nuclear Factor κB (NF-κB) Signaling[J]. Journal of Biological Chemistry, 2013, 288(50): 36094-36105.
69. Palermo RD, Webb HM and West MJ. RNA, Polymerase II stalling promotes nucleosome occlusion and pTEFb recruitment to drive immortalization by Epstein-Barr Virus. PLoS Pathog. 2011; 7:e1002334.
70. Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI and Young RA. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153(2): 320-334.
71. Bonetti P, Ponzoni M, Tibiletti MG, Stathis A, Heirat P, Zucca E, et al. The BRD-inhibitor OTX015 Shows Pre-clinical Activity in Diffuse Large B-cell Lymphoma (DLBCL). Best Pract Res Clin Haematol. 2012; 2:185-190.
72. Gosmini R, Nguyen VL, Toum J, Simon C, Brusq JM, Krysa G, Mirguet O, Riou-Eymard AM, Boursier EV, Trottet L, Bamborough P, Clark H, Chung CW, Cutler L, Demont EH, Kaur R, Lewis AJ et al. The Discovery of I-BET726 (GSK1324726A), a Potent Tetrahydroquinoline ApoA1 Up-Regulator and Selective BET Bromodomain Inhibitor[J]. Journal of medicinal chemistry, 2014, 57(19): 8111-8131.
73. French CA, Ramirez CL, Kolmakova J, Hickman TT, Cameron MJ, Thyne ME, Kutok JL, Toretsky JA, Tadavarthy AK, Kees UR, Fletcher JA and Aster JC. BRD-NUT oncoproteins: a family of closely related nuclear proteins that block epithelial differentiation and maintain the growth of carcinoma cells, Oncogene. 2007; 27:2237-2242.
74. Garnier J M, Sharp P P and Burns C J. BET bromodomain inhibitors: a patent review[J]. Expert opinion on therapeutic patents, 2014, 24(2): 185-199.
75. Wyce A, Degenhardt Y, Bai Y, Le B, Korenchuk S, Crouthame MC, McHugh CF, Vessella R, Creasy CL, Tummino PJ and Barbash O. Inhibition of BET bromodomain proteins as a therapeutic approach in prostate cancer. Oncotarget. 2013; 4:2419-2429.
76. Wyce A, Ganji G, Smitheman KN, Chung CW, Korenchuk S, Bai Y, Barbash O, Le B, Craggs PD, McCabe MT, Kennedy-Wilson KM, Sanchez LV, Gosmini RL, Parr N, McHugh CF, Dhanak D, et al. BET inhibition silences expression of MYCN and BCL2 and induces cytotoxicity in neuroblastoma tumor models. PLoS One. 2013; 8:e72967.
77. Chaidos A, Caputo V, Gouvedenou K, Liu B, Marigo I, Chaudhry MS, Rotolo A, Tough DF, Smithers NN, Bassil AK, Chapman TD, Harker NR, Barbash O, Tummino P, Al-Mahdi N, Haynes AC, et al. Potent antimyeloma activity of the novel bromodomain inhibitors I-BET151 and I-BET762. Blood. 2014; 123:697-705.
78. Wyspiańska BS, Bannister AJ, Barbieri I, Nangalia J, Godfrey A, Calero-Nieto FJ, Robson S, Rioja I, Li J, Wiese M, Cannizzaro E, Dawson MA et al. BET protein inhibition shows efficacy against JAK2V617F-driven neoplasms. Leukemia. 2014;1: 88-97.
79. Dawson MA, Gudgin EJ, Horton SJ, Giotopoulos G, Meduri E, Robson S, Cannizzaro E, Osaki H, Wiese M, Putwain S, Fong CY, Grove C, Craig J, Dittmann A, Lugo D, Jeffrey P, et al. Recurrent mutations, including NPM1c, activate a BRD4-dependent core transcriptional program in acute myeloid leukemia. Leukemia. 2014; 28:311-320.
80. Gallagher SJ, Mijatov B, Gunatilake D, Gowrishankar K, Tiffen J, James W, Jin L, Pupo G, Cullinane C, McArthur GA, Tummino PJ, Rizos H, Hersey P. Control of NF-kB activity in human melanoma by bromodomain and extra-terminal protein inhibitor I-BET151[J]. Pigment Cell Melanoma Res. 2014, 27(6): 1126-1137.
81. Moros A, Rodríguez V, Saborit-Villarroya I, Montraveta A, Balsas P, Sandy P, Martínez A, Wiestner A, Normant E, Campo E, Pérez-Galán P, Colomer D and Roué G. Synergistic antitumor activity of lenalidomide with the BET bromodomain inhibitor CPI203 in bortezomib-resistant mantle cell lymphoma. Leukemia. 2014; 18:2049-2059.
82. Bhadury J, Nilsson LM, Muralidharan SV, Green LC, Li Z, Gesner EM, Hansen HC, Keller UB, McLure KG, Nilsson JA. BET and HDAC inhibitors induce similar genes and biological effects and synergize to kill in Myc-induced murine lymphoma. Proc. Natl. Acad. Sci. USA. 2014; 111: E2721-2730.
83. de Paula Careta F, Gobessi S, Panepucci RA, Bojnik E, Morato de Oliveira F, Mazza Matos D, Falcão RP, Laurenti L, Zago MA and Efremov DG. The Aurora A and B kinases are upregulated in bone marrow-derived chronic lymphocytic leukemia cells and represent potential therapeutic targets. Haematologica. 2012; 97:1246–1254.
84. Picaud S, Da Costa D, Thanasopoulou A, Filippakopoulos P, Fish PV, Philpott M, Fedorov O, Brennan P, Bunnage ME, Owen DR, Bradner JE, Taniere P, O’Sullivan B, Müller S, Schwaller J, Stankovic T, et al. PFI-1 – A highly Selective Protein Interaction Inhibitor Targeting BET Bromodomains. Cancer Res. 2013; 73:3336–3346.
85. Thorsteinsdottir U, Mamo A, Kroon E, Jerome L, Bijl J, Lawrence HJ, Humphries K, and Sauvageau G. Overexpression of the myeloid leukemia–associatedHoxa9 gene in bone marrow cells induces stem cell expansion. Blood, 2002, 99(1): 121-129.
86. Gamsjaeger R, Webb SR, Lamonica JM, Billin A, Blobel GA and Mackay JP. Structural basis and specificity of acetylated transcription factor GATA1 recognition by BET family bromodomain protein Brd3. Mol Cell Biol. 2011; 31:2632–2640.
87. Lamonica JM, Deng W, Kadauke S, Campbell AE, Gamsjaeger R, Wang H, Cheng Y, Billin AN, Hardison RC, Mackay JP and Blobel GA. Bromodomain protein Brd3 associates with acetylated GATA1 to promote its chromatin occupancy at erythroid target genes. Proc Natl Acad Sci U S A. 2011; 108:E159–E168.
88. Oliver S S, Denu J M. Disrupting the reader of histone language. Angewandte Chemie International Edition, 2011, 50(26): 5801-5803.
89. Nicholls SJ, Gordon A, Johannson J, Ballantyne CM, Barter PJ, Brewer HB, Kastelein JJ, Wong NC, Borgman MR and Nissen SE. ApoA-I induction as a potential cardioprotective strategy: Rationale for the SUSTAIN and ASSURE studies. Cardiovasc Drugs Ther. 2012; 26:181–187.
90. Picaud S, Wells C, Felletar I, Brotherton D, Martin S, Savitsky P, Diez-Dacal B, Philpott M, Bountra C, Lingard H, Fedorov O, Müller S, Brennan PE,Knapp S, Filippakopoulos P. RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc. Natl. Acad. Sci. USA. 2013;110: 19754-19759.
91. Gamsjaeger R, Webb SR, Lamonica JM, Billin A, Blobel GA and Mackay JP. Structural basis and specificity of acetylated transcription factor GATA1 recognition by BET family bromodomain protein Brd3. Mol Cell Biol. 2011; 13:2632-2640.
92. Kempen HJ, Bellus D, Fedorov O, Nicklisch S, Filippakopoulos P, Picaud S, Knapp S. Stimulation of Hepatic Apolipoprotein A-I Production by Novel Thieno-Triazolodiazepines: Roles of the Classical Benzodiazepine Receptor, PAF Receptor, and Bromodomain Binding. Lipid Insights. 2013; 22: 47-54.
93. Morinière J, Rousseaux S, Steuerwald U, Soler-López M, Curtet S, Vitte AL, Govin J, Gaucher J, Sadoul K, Hart DJ, Krijgsveld J, Khochbin S, Müller CW, Petosa C. Cooperative binding of two acetylation marks on a histone tail by a single bromodomain. Nature. 2009; 461: 664-668.
94. Fabre C, Gobbi M, Ezzili C, Zoubir M, Sablin MP, Small K, Im E, Shinwari N, Zhang D, Zhou H, Le Tourneau C. Clinical study of the novel cyclin-dependent kinase inhibitor dinaciclib in combination with rituximab in relapsed/refractory chronic lymphocytic leukemia patients, Cancer Chemother Pharmacol. 2014; 74:1057-1064.
95. Goupille O, Penglong T, Lefèvre C, Granger M, Kadri Z, Fucharoen S, Maouche-Chrétien L, Leboulch P, Chrétien S. BET bromodomain inhibition rescues erythropoietin differentiation of human erythroleukemia cell line UT7. Biochem. Biophys. Res. Commun. 2012; 429: 1-5.
96. Emadali A, Rousseaux S, Bruder-Costa J, Rome C, Duley S, Hamaidia S, Betton P, Debernardi A, Leroux D, Bernay B, Kieffer-Jaquinod S, Combes F,Ferri E, McKenna CE, Petosa C, Bruley C et al. Identification of a novel BET bromodomain inhibitor-sensitive, gene regulatory circuit that controls Rituximab response and tumour growth in aggressive lymphoid cancers, EMBO Mol. Med. 2013; 8: 1180-1195.
97. Cheng Z, Gong Y, Ma Y, Lu K, Lu X, Pierce LA, Thompson RC, Muller S, Knapp S, Wang J. Inhibition of BET bromodomain targets genetically diverse glioblastoma. Clin. Cancer Res. 2013; 7: 1748-1759.
98. Lamoureux F, Baud’huin M, Rodriguez Calleja L, Jacques C, Berreur M, Rédini F, Lecanda F, Bradner JE, Heymann D, Ory B. Selective inhibition of BET bromodomain epigenetic signalling interferes with the bone-associated tumour vicious cycle. Nat. Commun. 2014; 5: 3511.
99. Ciceri P, Müller S, O’Mahony A, Fedorov O, Filippakopoulos P, Hunt JP, Lasater EA, Pallares G, Picaud S, Wells C, Martin S, Wodicka LM, Shah NP, Treiber DK and Knapp S. Dual kinase-bromodomain inhibitors for rationally designed polypharmacology. Nat Chem Biol. 201;10(4): 305-312.
100. Gaucher J, Boussouar F, Montellier E, Curtet S, Buchou T, Bertrand S, Hery P, Jounier S, Depaux A, Vitte AL, Guardiola P, Pernet K, Debernardi A, Lopez F, Holota H, Imbert J, et al. Bromodomain-dependent stage-specific male genome programming by Brdt. EMBO J. 2012; 31: 3809-3820.