
Introduction
The nuclear factor kappa B (NF-κB) is a dimeric transcription factor which plays versatile crucial roles in a plethora of normal cellular functions by controlling a panoply of downstream genes [1-4]. This pro-inflammatory transcription factor consists of rel family proteins, which are related through a highly conserved DNA-binding/dimerization domain called the Rel homology (RH) domain [5]. Currently, five mammalian NF-κB family members have been identified and studied. These include NF-κB1 (p50/p105), NF-κB2 (p52/p100), RelA (p65), RelB and c-Rel [6-8]. The C-terminal regions of RelA, RelB and c-Rel contain a transactivating domain that is important for NF-κB-mediated gene transactivation, while the C-termini of p105 and p100 contain multiple copies of the ankyrin repeats, a 33-residue sequence motif, which is also found in Inhibitor of κB family members [9-11].
In unstimulated state, NF-κB complexes are sequestered in the cytoplasm by Inhibitor of kappa-B (I-κB), which mask the nuclear localization signal (NLS) of NF-κB [12-14]. Upon activation of NF-κB, an upstream IB kinase phosphorylates IκBs at the critical amino acid residues (Ser-32 and Ser-36 of IκBα; Ser-19 and Ser-23 of IκBβ), which are subsequently ubiquitinated by β-transducin repeat-containing protein (βTrCP) and then degraded by the 26S proteasome, allowing freed NF-κB dimers to translocate to the nucleus and transactivate κB-responsive elements [3, 15-17].
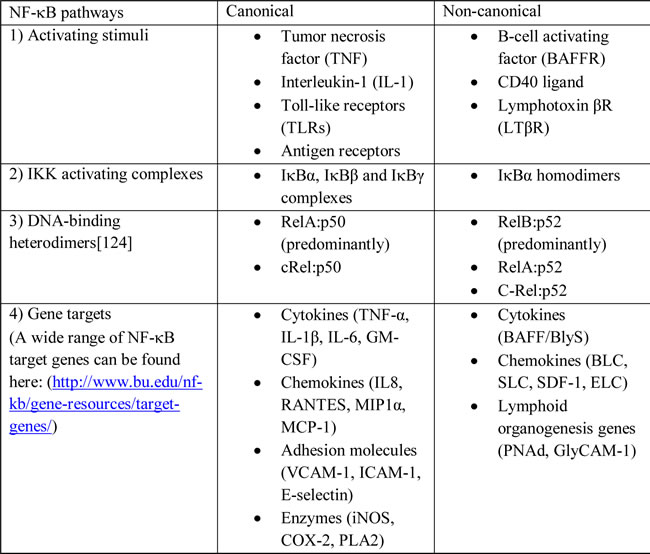
NF-κB signaling can occur through either the canonical or non-canonical pathways (Figure 1) [18-21]. These two pathways have different 1) activating stimuli, 2) IKK activating complexes, 3) DNA-binding heterodimers and 4) gene target [22]. The details of these two pathways were summarized in Table 1.
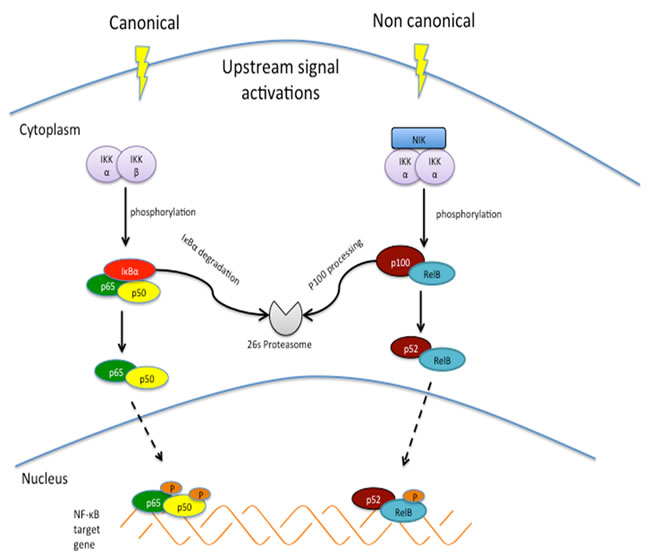
Figure 1: Canonical and non-canonical NF-κB signaling pathways. Canonical pathway involves activation of IκB kinase [125] complex, which subsequently phosphorylates IκBα for ubiquitin mediated proteolysis, enabling phosphorylation and transient nuclear translocation of p65/p50 NF-κB heterodimer. Non canonical pathway depends on NF-κB inducing kinase [126] and IKKα complex to achieve phosphorylation–induced p100 processing, leading to RelB/p52 complex.
NF-κB signaling pathway has been shown to regulate cell survival and apoptosis. Activation of inducible nitric oxide synthase (iNOS) to increase nitric oxide (NO) has been described as a pro-apoptotic function of NF-κB activation [23-25]. However, a study by Brandão et.al reported high iNOS expression in blood samples of AML patients in comparison to controls, which makes this apoptotic pathway questionable [26]. It is possible that acute production of NO triggers apoptosis, in contrast, the chronic production of NO by constitutively activate NF-κB signaling could inhibit the programmed suicide [26]. On the other hand, it is generally accepted that NF-κB activation is responsible for apoptosis resistance, cell proliferation and invasiveness [27-29]. Many tumours have been reported to show upregulation of a large number of NFκB target genes, for examples, FLICE-like inhibitory protein (FLIP), Inhibitors of Apoptosis (IAPs) and some members of anti-apoptotic Bcl-2 family to inhibit apoptosis; cyclin D1, c-myc and c-myb to enhance cell proliferation; and cell adhesion molecules (ICAM-1, E-selectin), matrix metalloproteinases and several angiogenic factors such as vascular endothelial growth factor (VEGF) to promote cancer cell invasion [27, 30-35].
It has been well known that Heme oxygenase-1 (HO-1) is an evolutionarily conserved key enzyme that catabolizes free heme [36]. Free heme is lipophilic, so it causes damage in lipid bilayers of cellular membrane, intracellular organelles [37]. Thus, HO-1 has function in protecting cells from apoptosis by escalating free heme catabolism. HO-1 promoter region contains NFκB responsive element and HO-1 expression is regulated by NFκB, in collaboration with other transcription factors [38]. In AML, induction of HO-1 expression has been reported as the mechanism by which AML cells evade tumour necrosis factor-α (TNF)-induced apoptosis [39], as well as chemotherapy-induced apoptosis [40]. Therefore, it appears an attractive approach by targeting both NFκB and HO-1 for anti-AML therapy [41].
Table 1: Overview of canonical and non-canonical NF-kB pathways
Molecular mechanisms of aberrant activation of NF-κB in AML
Constitutive activity of NF-κB is frequently observed in different types of cancer and has been correlated with resistance of cancer cells to radiation and chemotherapies [15, 16, 23, 42-46]. Causes of such aberrant activity could be due to alterations of genes that encode NF-κB and/or its inhibitors that promote NF-κB activation; constitutive activation of IKKs that accelerate IκB phosphorylation following degradation; or exposure to inflammatory stimuli in the tumour microenvironment that constantly trigger the signaling pathway. About 40% of patients with AML have shown increased activity of NF-κB [47]. Here we will discuss various mechanisms leading to aberrant activation of this pathway in AML.
ATM
Ataxia Telangiectasia Mutated (ATM) gene encodes a serine/threonine protein kinase, which is a master regulator of cell cycle checkpoint in response to DNA damage for the maintenance of genomic stability [48-50]. The development of AML involves multiple steps of genetic and epigenetic changes, including activation of oncogenes and inactivation of tumor suppressor genes [51]. These activated oncogenes in AML cells often induce oxidative stress (high production of reactive oxygen species, ROS) and replication stress, triggering DNA damage response (DDR) pathways, which, in turn, results in phosphorylation of ATM, CHK-1, CHK-2 and H2AX [52, 53]. In AML cells, phosphorylated (activated) ATM interacts with NFκB essential modulator (NEMO), a subunit of IκB kinase complex, and p53-induced death domain protein (PIDD) in the nucleus. Both NEMO and PIDD activate NFκB pathway [54]. Treatment of AML cells with pharmacological inhibitors of ATM or siRNA silencing ATM induces relocalization of NFκB from the nucleus to the cytoplasm, resulting in apoptosis of AML cells [54]. These results suggest constitutively active ATM leads to activate NFκB pathway in AML.
C/EBPα
CCAAT/enhancer-binding protein alpha (C/EBPα) consists of three transactivation domains (TAD1, TAD2 and TAD3) in the amino terminus (N-termal) and a basic leucine zipper domain (bZIP) at its carboxy terminus (C-termal) for DNA binding. C/EBPα is a bZIP transcription factor, which plays a critical role in myeloid development [55-57]. The expression of C/EBPα is tightly regulated during myeloid hematopoiesis. C/EBPα expresses at low level in the HSC and terminal differentiation stage, but high at the transition stage: common myeloid progenitor (CMP) and the granulocyte-monocyte progenitor (GMP) [55, 56]. Consistent with this expression pattern, the study of C/EBPα knock-out mice shows that deletion of C/EBPα selectively blocks myeloid differentiation at transition stage and reduces formation of neutrophils and monocytes [58]. Mutations in the C/EBPα gene have been detected in 10 - 15% of patients with AML [59]. Except for some rare types of mutations, C/EBPα mutations can be classified into two main categories: (1) N-terminal mutations that lead to premature termination of protein translation, resulting in translation of a dominant negative, short C/EBPα p30 isoform; (2) C-terminal mutations that disrupt the bZIP region, resulting loss of DNA binding capacity [60, 61]. The majority of AML patients with C/EBPα mutations have double mutations, i.e., two allele carrying different types of mutations. However, some patients harbor single mutation on one allele only. Of note, only double mutations, but not single mutation of C/EBPα, are associated with favorable prognosis [59, 62]. C/EBPα and its mutant forms, harboring with N-terminal mutations or C-terminal mutations, interact with NFκB components in AML cells [63]. Several lines of evidence indicate that C/EBPα, as well as its mutant variants, interacts with NFκB p50 and induces a subset of NFκB target genes, including pro-survival Bcl-2, FLIP, through promoter binding [63, 64]. Saturating mutagenesis analysis shows that some key residues in the basic region of bZIP domain of C/EBPα is critical for the interaction with NFκB p50 [65]. The expression of C/EBPα is 3-fold lower in NFκB p50 knockout cells and p50 binds to the promoter of C/EBPα αand regulates its expression [66]. On the other hand, C/EBPα and its mutant forms can replace histone deacetylase 1 to 3 on the p50 promoter, inducing p50 expression and activating NFκB activity in AML [67].
RUNX1
RUNX1 (runt-related transcription factor 1) is heterodimeric transcription factor belonging to RUNX gene family (RUNX1, 2, 3). RUNX1 plays a pivotal role in development of definitive hematopoiesis and primitive hematopoiesis [68-71]. Chromosomal abnormalities or point mutation involved in RUNX1 gene define a prognosis and biology distinct subset of AML patients [72, 73]. In mouse RUNX-1 deficient hematopoietic progenitor cells, both canonical and noncanonical pathways of NF-κB signaling are constitutively increased as evidenced by increased nuclear localization of p65 and p52 proteins [74]. Wild type RUNX1 binds to IKK complex in the cytoplasm and attenuates its kinase activity, thus repressing NFκB signaling. However, mutant RUNX1 forms lose the ability to inhibit IKK, leading to aberrant activation of NFκB pathway in AML cases with RUNX1 abnormalities [74].
TNF-α
Tumor necrosis factor-alpha (TNF-α) is a type II transmembrane protein and the soluble form of TNF-α is secreted by immune systems including macrophages, monocytes, neutrophils, T-cells, nature killer (NK)-cells in response to inflammatory stimulation [75, 76]. TNF binds two TNF receptors (TNFR1 and 2) and activates the canonical NFκB pathway [16]. In an AML mouse model, leukemia-initiating cells (LICs) or leukemia stem cells (LSCs) exhibit autocrine TNF-α secretion, which causes constitutive activation of NFκB activity in this unique cell population [77]. This finding is further supported by the positive correlation between NF-κB activity and autocrine TNF-α in human AML samples [77].
Oncogenic kinase activation
RAS protein family includes H-RAS, N-RAS and K-RAS, which are small GTPase proteins [78]. RAS proteins transmit signals from extracellular growth factors by cycling between inactive GDP-bound and active GTP-bound states. N-RAS or K-RAS mutations occur in approximate 20% of AML cases [79, 80]. However, aberrant RAS signaling has been detected in 40% of cases in addition to RAS mutation, primarily due to is somatic mutations in the other receptor tyrosine kinase like FLT3 and c-Kit [79, 80].
Birkenkamp and co-workers observed a significant association between constitutive NFκB DNA-binding activity and persistent RAS signaling in AML blasts [81]. In ex vivo experiments, AML blasts with high NFκB DNA-binding activity underwent less or no spontaneous apoptosis, compared to AML cases with no or low nuclear NFκB expression. By using small molecular inhibitor Ly294002 targeting PI3K/AKT pathway, L-744832 targeting RAS, PD98059 targeting ERK/MAPK signaling and AG1296 targeting FLT3, the authors found that NF-κB DNA-binding activity was inhibited only by RAS and PI3K/AKT inhibitors, thus concluded that increased NF-κB activity was regulated by RAS signaling, but not ERK and FLT3 pathways [81]. In contrast, several other studies clearly demonstrated that either FLT3 overexpression or FLT3 mutation increased NFκB activity in AML [82, 83]. Takahashi and colleagues showed that overexpression of FLT3 in BaF3 cells activated NFB reporter and increase level of IL-6, a NFκB target gene [82]. They also showed a modest positive correlation between FLT3 and IL-6 mRNA expression in AML samples [82]. Similarly, Grosjean-Raillard et al. reported that constitutive activation of FLT3 signaling resulted in activation of NF-κB, while inhibition of FLT3 signaling either by small molecule inhibitor or knockdown of Flt3 with RNAi reduced NF-κB activity and induced apoptosis in AML cell lines and CD34+ primary AML cells [83]. Furthermore, comprehensive biochemical experiments revealed the underlying mechanism of NF-κB activation in which FLT3 kinase physically bound and phosphorylates IKK2, an upstream regulator of canonical NF-κB pathway [83]. In addition, Internal tandem duplications of FLT3 (FLT3-ITD), one of the most common genetic abnormalities in AML [84, 85], induced expression of RelB and p52/NF-B heterodimers, thus promoting non-canonical NF-κB pathway [86]. In summary, activation of both canonical and non-canonical NF-κB pathways appears to be an important event contributing to the leukemic transformation initiated by some crucial oncogenic kinases.
NF-κB as a target for anti-AML therapy
Because a large body of evidence supports the important role of NFκB as a “hallmark of cancer”, there has been tremendous focus on the development of NFκB inhibitors for cancer treatment in both the academic community and the pharmaceutical industry [87-89]. Different NFκB inhibitors have been classified into 8 groups according to their chemical nature and have been reviewed in details [90-92]. Here, we focus on the proteasome inhibitor, Bortezomib (Velcade®, Millennium Pharmaceuticals) and other promising NFκB inhibitors in clinical trials for treatment of AML.
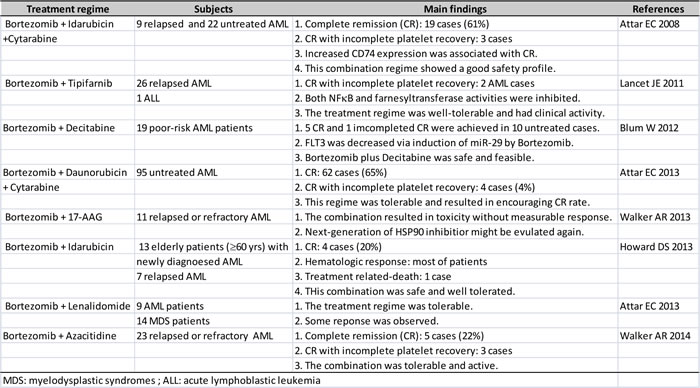
Bortezomib is the first-in-class proteasome inhibitor, which has been approved by FDA (USA) to treat multiple myeloma and now relapsed mantle cell lymphoma too [93-95]. Although the models of action by proteasome inhibition are not fully elucidated, one of the important mechanisms associated with the anti-myeloma functions of Bortezomib is its ability to suppress the NF-κB signaling pathway [96]. IκB, a cellular inhibitory protein of NFκB, is targeted by ubiquitin-proteasome pathway for degradation when it is phosphorylated at serine residue 32 and 36. Inhibition of the proteasome pathway by Bortezomib has been shown to block the degradation of IκBα, thus sequestering NFκB in the cytoplasm and preventing NFκB nuclear translocation and activation of NFκB target genes [97-99]. Because of its ability to inhibit NFκB activity, it provides a rationale to examine the effectiveness of Bortezomib either used alone or in combination with other drugs against AML in various clinical trials. Early phase I trial in AML evaluated Bortezomib as monotherapy in refractory or relapsed acute leukemias. As a monotherapy, the maximum tolerated dose (MTD) of Bortezomib was 1.25 mg/m2, and was shown to have transient hematological improvements in some patients [100]. In the subsequent trials in AML, Bortezomib was further investigated in combination with other agents. Eight clinical trials that enrolled majority of patients with AML aimed to evaluate the benefit of combination of Bortezomib with other drugs (Table 1) [101-108]. Two trials that combined the use of Bortezomib with Cytarabine and Anthracyclines (Idarubicin or Doxorubicin) showed a complete remission (CR) rate of 61% and 65%, respectively, and a good safety profile [101, 102]. One study of Bortezomib with Idarubicin in high risk of AML patients achieved a CR rate of 20% [106]. The other five trials that co-administrated Bortezomib with other targeted drugs or epigenetic drugs did not produced encouraging CR rate. But, one important note should be taken into consideration is that the subjects in these trials were high-risk patients with either refractory or relapsed AML or older than 60 years. These may be confounding factors that adversely impact on the clinical benefits of these combination therapies. In general, if these combination regimes are well tolerated, they should be further evaluated in standard risk patients. When new generation of inhibitors are developed, they might be tested in the combination regimes too.
Table 2: A Summary of Bortezomib in clinical trials for anti-AML therapy
Conclusions
Over the last decade, our understanding of NFκB signaling and our ability to target it has evolved significantly. Although there are now 8 different classes of more than 700 NFκB inhibitors, only a few of them have advanced into clinical trials for treatment of AML. At the same time, a growing body of evidence suggests the existence of leukemia stem cells (LSCs) in AML leading to the potential relapse of disease and treatment failure [109-111]. LSCs reside mostly in a quiescent cell cycle state, which is similar to their counterparts, the normal hematopoietic stem cells [112-114], thus escaping from the effects of standard chemotherapy drugs which usually target proliferative cells. NF-κB activity is aberrantly increased in primitive human leukemia cells compared to normal primitive bone marrow cells [47, 77, 115, 116], thus it provides a novel concept to treat AML by targeting the difference between HSCs and LSCs as exemplified by the different NF-κB activity between them. [117-120]. In fact, the small molecule NF-κB inhibitor dimethylaminoparthenolide (DMAPT /LC1) has been shown to selectively eradicate AML LSCs in the laboratory [92, 121-123] and is currently evaluated in a phase I-II ‘first in man’ clinical study at Cardiff University, UK (http://medicine.cf.ac.uk/person/dr-steven-knapper/research/). Hopefully, the result of this much anticipated trial will demonstrate effectiveness of NF-κB inhibitor in AML patients.
Acknowledgements
This research is supported by the National Research Foundation Singapore and the Singapore Ministry of Education under its Research Centres of Excellence initiative and NMRC Clinician-Scientist IRG Grant CNIG11nov38 (Zhou J). Chng WJ is also supported by NMRC Clinician Scientist Investigator award. The authors apologize to researchers and clinicians whose work could not be discussed due to space limitations.
Competing interests
The authors declare no competing interests.
Authors’ contributions
Zhou J, Ching YQ and Chng WJ all reviewed the literature and wrote the manuscript; all authors approved the final version of the manuscript.
References
1. Dey A, Tergaonkar V and Lane DP. Double-edged swords as cancer therapeutics: simultaneously targeting p53 and NF-kappaB pathways. Nature reviews Drug discovery. 2008; 7(12):1031-1040.
2. Hayden MS and Ghosh S. NF-kappaB, the first quarter-century: remarkable progress and outstanding questions. Genes & development. 2012; 26(3):203-234.
3. Hinz M and Scheidereit C. The IkappaB kinase complex in NF-kappaB regulation and beyond. EMBO reports. 2014; 15(1):46-61.
4. Wong ET and Tergaonkar V. Roles of NF-kappaB in health and disease: mechanisms and therapeutic potential. Clinical science. 2009; 116(6):451-465.
5. Hoffmann A, Natoli G and Ghosh G. Transcriptional regulation via the NF-kappaB signaling module. Oncogene. 2006; 25(51):6706-6716.
6. Huguet C, Crepieux P and Laudet V. Rel/NF-kappa B transcription factors and I kappa B inhibitors: evolution from a unique common ancestor. Oncogene. 1997; 15(24):2965-2974.
7. Ferreira V, Tarantino N and Korner M. Discrimination between RelA and RelB transcriptional regulation by a dominant negative mutant of IkappaBalpha. The Journal of biological chemistry. 1998; 273(1):592-599.
8. Chen FE, Huang DB, Chen YQ and Ghosh G. Crystal structure of p50/p65 heterodimer of transcription factor NF-kappaB bound to DNA. Nature. 1998; 391(6665):410-413.
9. Baldwin AS, Jr. The NF-kappa B and I kappa B proteins: new discoveries and insights. Annual review of immunology. 1996; 14:649-683.
10. Zandi E and Karin M. Bridging the gap: composition, regulation, and physiological function of the IkappaB kinase complex. Mol Cell Biol. 1999; 19(7):4547-4551.
11. Millet P, McCall C and Yoza B. RelB: an outlier in leukocyte biology. Journal of leukocyte biology. 2013; 94(5):941-951.
12. Gasparini C, Celeghini C, Monasta L and Zauli G. NF-kappaB pathways in hematological malignancies. Cellular and molecular life sciences : CMLS. 2014; 71(11):2083-2102.
13. Campbell KJ and Perkins ND. Regulation of NF-kappaB function. Biochemical Society symposium. 2006; (73):165-180.
14. Ghosh A, Saginc G, Leow SC, Khattar E, Shin EM, Yan TD, Wong M, Zhang Z, Li G, Sung WK, Zhou J, Chng WJ, Li S, Liu E and Tergaonkar V. Telomerase directly regulates NF-kappaB-dependent transcription. Nature cell biology. 2012; 14(12):1270-1281.
15. Hoesel B and Schmid JA. The complexity of NF-kappaB signaling in inflammation and cancer. Molecular cancer. 2013; 12:86.
16. Napetschnig J and Wu H. Molecular basis of NF-kappaB signaling. Annual review of biophysics. 2013; 42:443-468.
17. Verma IM, Stevenson JK, Schwarz EM, Van Antwerp D and Miyamoto S. Rel/NF-kappa B/I kappa B family: intimate tales of association and dissociation. Genes & development. 1995; 9(22):2723-2735.
18. Shih VF, Tsui R, Caldwell A and Hoffmann A. A single NFkappaB system for both canonical and non-canonical signaling. Cell research. 2011; 21(1):86-102.
19. Razani B, Reichardt AD and Cheng G. Non-canonical NF-kappaB signaling activation and regulation: principles and perspectives. Immunol Rev. 2011; 244(1):44-54.
20. Sun SC. Non-canonical NF-kappaB signaling pathway. Cell research. 2011; 21(1):71-85.
21. Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene. 2006; 25(51):6680-6684.
22. Ghosh S and Hayden MS. Celebrating 25 years of NF-kappaB research. Immunol Rev. 2012; 246(1):5-13.
23. Chandrasekar B, Friedrichs W, Donzis E, Silva J, Hidalgo M, Freeman J and Weiss G. NF-κB inhibition markedly enhances sensitivity of resistant breast cancer tumor cells to tamoxifen. Annals of oncology. 2004; 15(6):885-890.
24. Crowell JA, Steele VE, Sigman CC and Fay JR. Is Inducible Nitric Oxide Synthase a Target for Chemoprevention? Molecular Cancer Therapeutics. 2003; 2(8):815-823.
25. Pautz A, Art J, Hahn S, Nowag S, Voss C and Kleinert H. Regulation of the expression of inducible nitric oxide synthase. Nitric oxide : biology and chemistry / official journal of the Nitric Oxide Society. 2010; 23(2):75-93.
26. Brandão MM, Soares E, Salles TSI and Saad STO. Expression of Inducible Nitric Oxide Synthase Is Increased in Acute Myeloid Leukaemia. Acta Haematologica. 2001; 106(3):95-99.
27. Dolcet X, Llobet D, Pallares J and Matias-Guiu X. NF-kB in development and progression of human cancer. Virchows archiv. 2005; 446(5):475-482.
28. Ghosh S, Tergaonkar V, Rothlin CV, Correa RG, Bottero V, Bist P, Verma IM and Hunter T. Essential role of tuberous sclerosis genes TSC1 and TSC2 in NF-kappaB activation and cell survival. Cancer cell. 2006; 10(3):215-226.
29. Bai D, Ueno L and Vogt PK. Akt-mediated regulation of NFkappaB and the essentialness of NFkappaB for the oncogenicity of PI3K and Akt. International journal of cancer Journal international du cancer. 2009; 125(12):2863-2870.
30. Li X, Abdel-Mageed AB, Mondal D and Kandil E. The nuclear factor kappa-B signaling pathway as a therapeutic target against thyroid cancers. Thyroid : official journal of the American Thyroid Association. 2013; 23(2):209-218.
31. Carbone C and Melisi D. NF-kappaB as a target for pancreatic cancer therapy. Expert opinion on therapeutic targets. 2012; 16 Suppl 2:S1-10.
32. Ling J and Kumar R. Crosstalk between NFkB and glucocorticoid signaling: a potential target of breast cancer therapy. Cancer letters. 2012; 322(2):119-126.
33. Jain G, Cronauer MV, Schrader M, Moller P and Marienfeld RB. NF-kappaB signaling in prostate cancer: a promising therapeutic target? World journal of urology. 2012; 30(3):303-310.
34. Gyrd-Hansen M and Meier P. IAPs: from caspase inhibitors to modulators of NF-kappaB, inflammation and cancer. Nature reviews Cancer. 2010; 10(8):561-574.
35. Rushworth SA, Zaitseva L, Langa S, Bowles KM and MacEwan DJ. FLIP regulation of HO-1 and TNF signalling in human acute myeloid leukemia provides a unique secondary anti-apoptotic mechanism. Oncotarget. 2010; 1(5):359-366.
36. Hjortso MD and Andersen MH. The expression, function and targeting of haem oxygenase-1 in cancer. Current cancer drug targets. 2014; 14(4):337-347.
37. Jeney V, Balla J, Yachie A, Varga Z, Vercellotti GM, Eaton JW and Balla G. Pro-oxidant and cytotoxic effects of circulating heme. Blood. 2002; 100(3):879-887.
38. Kurata S, Matsumoto M, Tsuji Y and Nakajima H. Lipopolysaccharide activates transcription of the heme oxygenase gene in mouse M1 cells through oxidative activation of nuclear factor kappa B. European journal of biochemistry / FEBS. 1996; 239(3):566-571.
39. Rushworth SA and MacEwan DJ. HO-1 underlies resistance of AML cells to TNF-induced apoptosis. Blood. 2008; 111(7):3793-3801.
40. Heasman SA, Zaitseva L, Bowles KM, Rushworth SA and Macewan DJ. Protection of acute myeloid leukaemia cells from apoptosis induced by front-line chemotherapeutics is mediated by haem oxygenase-1. Oncotarget. 2011; 2(9):658-668.
41. Rushworth SA, Bowles KM, Raninga P and MacEwan DJ. NF-kappaB-inhibited acute myeloid leukemia cells are rescued from apoptosis by heme oxygenase-1 induction. Cancer research. 2010; 70(7):2973-2983.
42. Braunstein S, Formenti SC and Schneider RJ. Acquisition of stable inducible up-regulation of nuclear factor-κB by tumor necrosis factor exposure confers increased radiation resistance without increased transformation in breast cancer cells. Molecular Cancer Research. 2008; 6(1):78-88.
43. Voboril R and Weberova-Voborilova J. Constitutive NF-kappaB activity in colorectal cancer cells: impact on radiation-induced NF-kappaB activity, radiosensitivity, and apoptosis. Neoplasma. 2006; 53(6):518-523.
44. Natoli G. NF-kappaB and chromatin: ten years on the path from basic mechanisms to candidate drugs. Immunol Rev. 2012; 246(1):183-192.
45. Naugler WE and Karin M. NF-κB and cancer—identifying targets and mechanisms. Current opinion in genetics & development. 2008; 18(1):19-26.
46. Perkins ND. The diverse and complex roles of NF-kappaB subunits in cancer. Nature reviews Cancer. 2012; 12(2):121-132.
47. Guzman ML, Neering SJ, Upchurch D, Grimes B, Howard DS, Rizzieri DA, Luger SM and Jordan CT. Nuclear factor-kappaB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood. 2001; 98(8):2301-2307.
48. Boohaker RJ and Xu B. The versatile functions of ATM kinase. Biomedical journal. 2014; 37(1):3-9.
49. Shiloh Y and Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nature reviews Molecular cell biology. 2013; 14(4):197-210.
50. Chaudhary MW and Al-Baradie RS. Ataxia-telangiectasia: future prospects. The application of clinical genetics. 2014; 7:159-167.
51. Martelli AM, Evangelisti C, Chiarini F and McCubrey JA. The phosphatidylinositol 3-kinase/Akt/mTOR signaling network as a therapeutic target in acute myelogenous leukemia patients. Oncotarget. 2010; 1(2):89-103.
52. Esposito MT and So CW. DNA damage accumulation and repair defects in acute myeloid leukemia: implications for pathogenesis, disease progression, and chemotherapy resistance. Chromosoma. 2014; 123(6):545-561.
53. Boehrer S, Ades L, Tajeddine N, Hofmann WK, Kriener S, Bug G, Ottmann OG, Ruthardt M, Galluzzi L, Fouassier C, Tailler M, Olaussen KA, Gardin C, Eclache V, de Botton S, Thepot S, et al. Suppression of the DNA damage response in acute myeloid leukemia versus myelodysplastic syndrome. Oncogene. 2009; 28(22):2205-2218.
54. Grosjean-Raillard J, Tailler M, Ades L, Perfettini JL, Fabre C, Braun T, De Botton S, Fenaux P and Kroemer G. ATM mediates constitutive NF-kappaB activation in high-risk myelodysplastic syndrome and acute myeloid leukemia. Oncogene. 2009; 28(8):1099-1109.
55. Tenen DG. Abnormalities of the CEBP alpha transcription factor: a major target in acute myeloid leukemia. Leukemia. 2001; 15(4):688-689.
56. Paz-Priel I and Friedman A. C/EBPalpha dysregulation in AML and ALL. Critical reviews in oncogenesis. 2011; 16(1-2):93-102.
57. Roe JS and Vakoc CR. C/EBPalpha: critical at the origin of leukemic transformation. The Journal of experimental medicine. 2014; 211(1):1-4.
58. Zhang DE, Zhang P, Wang ND, Hetherington CJ, Darlington GJ and Tenen DG. Absence of granulocyte colony-stimulating factor signaling and neutrophil development in CCAAT enhancer binding protein alpha-deficient mice. Proceedings of the National Academy of Sciences of the United States of America. 1997; 94(2):569-574.
59. Preudhomme C, Sagot C, Boissel N, Cayuela JM, Tigaud I, de Botton S, Thomas X, Raffoux E, Lamandin C, Castaigne S, Fenaux P, Dombret H and Group A. Favorable prognostic significance of CEBPA mutations in patients with de novo acute myeloid leukemia: a study from the Acute Leukemia French Association (ALFA). Blood. 2002; 100(8):2717-2723.
60. Pabst T, Mueller BU, Zhang P, Radomska HS, Narravula S, Schnittger S, Behre G, Hiddemann W and Tenen DG. Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-alpha (C/EBPalpha), in acute myeloid leukemia. Nature genetics. 2001; 27(3):263-270.
61. Leroy H, Roumier C, Huyghe P, Biggio V, Fenaux P and Preudhomme C. CEBPA point mutations in hematological malignancies. Leukemia. 2005; 19(3):329-334.
62. Green CL, Koo KK, Hills RK, Burnett AK, Linch DC and Gale RE. Prognostic significance of CEBPA mutations in a large cohort of younger adult patients with acute myeloid leukemia: impact of double CEBPA mutations and the interaction with FLT3 and NPM1 mutations. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010; 28(16):2739-2747.
63. Paz-Priel I, Cai DH, Wang D, Kowalski J, Blackford A, Liu H, Heckman CA, Gombart AF, Koeffler HP, Boxer LM and Friedman AD. CCAAT/enhancer binding protein alpha (C/EBPalpha) and C/EBPalpha myeloid oncoproteins induce bcl-2 via interaction of their basic regions with nuclear factor-kappaB p50. Molecular cancer research : MCR. 2005; 3(10):585-596.
64. Paz-Priel I, Ghosal AK, Kowalski J and Friedman AD. C/EBPalpha or C/EBPalpha oncoproteins regulate the intrinsic and extrinsic apoptotic pathways by direct interaction with NF-kappaB p50 bound to the bcl-2 and FLIP gene promoters. Leukemia. 2009; 23(2):365-374.
65. Dooher JE, Paz-Priel I, Houng S, Baldwin AS, Jr. and Friedman AD. C/EBPalpha, C/EBPalpha oncoproteins, or C/EBPbeta preferentially bind NF-kappaB p50 compared with p65, focusing therapeutic targeting on the C/EBP:p50 interaction. Molecular cancer research : MCR. 2011; 9(10):1395-1405.
66. Wang D, Paz-Priel I and Friedman AD. NF-kappa B p50 regulates C/EBP alpha expression and inflammatory cytokine-induced neutrophil production. Journal of immunology. 2009; 182(9):5757-5762.
67. Paz-Priel I, Houng S, Dooher J and Friedman AD. C/EBPalpha and C/EBPalpha oncoproteins regulate nfkb1 and displace histone deacetylases from NF-kappaB p50 homodimers to induce NF-kappaB target genes. Blood. 2011; 117(15):4085-4094.
68. Wang CQ, Jacob B, Nah GS and Osato M. Runx family genes, niche, and stem cell quiescence. Blood cells, molecules & diseases. 2010; 44(4):275-286.
69. Koh CP, Wang CQ, Ng CE, Ito Y, Araki M, Tergaonkar V, Huang G and Osato M. RUNX1 meets MLL: epigenetic regulation of hematopoiesis by two leukemia genes. Leukemia. 2013; 27(9):1793-1802.
70. Ichikawa M, Yoshimi A, Nakagawa M, Nishimoto N, Watanabe-Okochi N and Kurokawa M. A role for RUNX1 in hematopoiesis and myeloid leukemia. International journal of hematology. 2013; 97(6):726-734.
71. Chuang LS, Ito K and Ito Y. RUNX family: Regulation and diversification of roles through interacting proteins. International journal of cancer Journal international du cancer. 2013; 132(6):1260-1271.
72. Lam K and Zhang DE. RUNX1 and RUNX1-ETO: roles in hematopoiesis and leukemogenesis. Frontiers in bioscience. 2012; 17:1120-1139.
73. Marcucci G, Caligiuri MA and Bloomfield CD. Molecular and clinical advances in core binding factor primary acute myeloid leukemia: a paradigm for translational research in malignant hematology. Cancer investigation. 2000; 18(8):768-780.
74. Nakagawa M, Shimabe M, Watanabe-Okochi N, Arai S, Yoshimi A, Shinohara A, Nishimoto N, Kataoka K, Sato T, Kumano K, Nannya Y, Ichikawa M, Imai Y and Kurokawa M. AML1/RUNX1 functions as a cytoplasmic attenuator of NF-kappaB signaling in the repression of myeloid tumors. Blood. 2011; 118(25):6626-6637.
75. Du W, Erden O and Pang Q. TNF-alpha signaling in Fanconi anemia. Blood cells, molecules & diseases. 2014; 52(1):2-11.
76. Hayden MS and Ghosh S. Regulation of NF-kappaB by TNF family cytokines. Seminars in immunology. 2014; 26(3):253-266.
77. Kagoya Y, Yoshimi A, Kataoka K, Nakagawa M, Kumano K, Arai S, Kobayashi H, Saito T, Iwakura Y and Kurokawa M. Positive feedback between NF-kappaB and TNF-alpha promotes leukemia-initiating cell capacity. The Journal of clinical investigation. 2014; 124(2):528-542.
78. Takashima A and Faller DV. Targeting the RAS oncogene. Expert opinion on therapeutic targets. 2013; 17(5):507-531.
79. Ward AF, Braun BS and Shannon KM. Targeting oncogenic Ras signaling in hematologic malignancies. Blood. 2012; 120(17):3397-3406.
80. Reuter CW, Morgan MA and Bergmann L. Targeting the Ras signaling pathway: a rational, mechanism-based treatment for hematologic malignancies? Blood. 2000; 96(5):1655-1669.
81. Birkenkamp KU, Geugien M, Schepers H, Westra J, Lemmink HH and Vellenga E. Constitutive NF-kappaB DNA-binding activity in AML is frequently mediated by a Ras/PI3-K/PKB-dependent pathway. Leukemia. 2004; 18(1):103-112.
82. Takahashi S, Harigae H, Ishii KK, Inomata M, Fujiwara T, Yokoyama H, Ishizawa K, Kameoka J, Licht JD, Sasaki T and Kaku M. Over-expression of Flt3 induces NF-kappaB pathway and increases the expression of IL-6. Leukemia research. 2005; 29(8):893-899.
83. Grosjean-Raillard J, Ades L, Boehrer S, Tailler M, Fabre C, Braun T, De Botton S, Israel A, Fenaux P and Kroemer G. Flt3 receptor inhibition reduces constitutive NFkappaB activation in high-risk myelodysplastic syndrome and acute myeloid leukemia. Apoptosis : an international journal on programmed cell death. 2008; 13(9):1148-1161.
84. Zhou J, Bi C, Janakakumara JV, Liu SC, Chng WJ, Tay KG, Poon LF, Xie Z, Palaniyandi S, Yu H, Glaser KB, Albert DH, Davidsen SK and Chen CS. Enhanced activation of STAT pathways and overexpression of survivin confer resistance to FLT3 inhibitors and could be therapeutic targets in AML. Blood. 2009; 113(17):4052-4062.
85. Zhou J, Goh BC, Albert DH and Chen CS. ABT-869, a promising multi-targeted tyrosine kinase inhibitor: from bench to bedside. Journal of hematology & oncology. 2009; 2:33.
86. Shanmugam R, Gade P, Wilson-Weekes A, Sayar H, Suvannasankha A, Goswami C, Li L, Gupta S, Cardoso AA, Al Baghdadi T, Sargent KJ, Cripe LD, Kalvakolanu DV and Boswell HS. A noncanonical Flt3ITD/NF-kappaB signaling pathway represses DAPK1 in acute myeloid leukemia. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012; 18(2):360-369.
87. Colotta F, Allavena P, Sica A, Garlanda C and Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009; 30(7):1073-1081.
88. Shen HM and Tergaonkar V. NFkappaB signaling in carcinogenesis and as a potential molecular target for cancer therapy. Apoptosis : an international journal on programmed cell death. 2009; 14(4):348-363.
89. Sethi G and Tergaonkar V. Potential pharmacological control of the NF-kappaB pathway. Trends in pharmacological sciences. 2009; 30(6):313-321.
90. Gilmore TD and Herscovitch M. Inhibitors of NF-kappaB signaling: 785 and counting. Oncogene. 2006; 25(51):6887-6899.
91. Reikvam H, Olsnes AM, Gjertsen BT, Ersvar E and Bruserud O. Nuclear factor-kappaB signaling: a contributor in leukemogenesis and a target for pharmacological intervention in human acute myelogenous leukemia. Critical reviews in oncogenesis. 2009; 15(1-2):1-41.
92. Fuchs O. Transcription factor NF-kappaB inhibitors as single therapeutic agents or in combination with classical chemotherapeutic agents for the treatment of hematologic malignancies. Current molecular pharmacology. 2010; 3(3):98-122.
93. Chng WJ, Lau LG, Yusof N and Mow BM. Targeted therapy in multiple myeloma. Cancer control : journal of the Moffitt Cancer Center. 2005; 12(2):91-104.
94. Blade J, Cibeira MT and Rosinol L. Bortezomib: a valuable new antineoplastic strategy in multiple myeloma. Acta oncologica. 2005; 44(5):440-448.
95. Demchenko YN and Kuehl WM. A critical role for the NFkB pathway in multiple myeloma. Oncotarget. 2010; 1(1):59-68.
96. Richardson PG, Mitsiades C, Schlossman R, Munshi N and Anderson K. New drugs for myeloma. The oncologist. 2007; 12(6):664-689.
97. Mani A and Gelmann EP. The ubiquitin-proteasome pathway and its role in cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2005; 23(21):4776-4789.
98. Shen M, Schmitt S, Buac D and Dou QP. Targeting the ubiquitin-proteasome system for cancer therapy. Expert opinion on therapeutic targets. 2013; 17(9):1091-1108.
99. Wu ZH and Shi Y. When ubiquitin meets NF-kappaB: a trove for anti-cancer drug development. Current pharmaceutical design. 2013; 19(18):3263-3275.
100. Cortes J, Thomas D, Koller C, Giles F, Estey E, Faderl S, Garcia-Manero G, McConkey D, Ruiz SL, Guerciolini R, Wright J and Kantarjian H. Phase I study of bortezomib in refractory or relapsed acute leukemias. Clinical cancer research : an official journal of the American Association for Cancer Research. 2004; 10(10):3371-3376.
101. Attar EC, De Angelo DJ, Supko JG, D’Amato F, Zahrieh D, Sirulnik A, Wadleigh M, Ballen KK, McAfee S, Miller KB, Levine J, Galinsky I, Trehu EG, Schenkein D, Neuberg D, Stone RM, et al. Phase I and pharmacokinetic study of bortezomib in combination with idarubicin and cytarabine in patients with acute myelogenous leukemia. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008; 14(5):1446-1454.
102. Attar EC, Johnson JL, Amrein PC, Lozanski G, Wadleigh M, DeAngelo DJ, Kolitz JE, Powell BL, Voorhees P, Wang ES, Blum W, Stone RM, Marcucci G, Bloomfield CD, Moser B and Larson RA. Bortezomib added to daunorubicin and cytarabine during induction therapy and to intermediate-dose cytarabine for consolidation in patients with previously untreated acute myeloid leukemia age 60 to 75 years: CALGB (Alliance) study 10502. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013; 31(7):923-929.
103. Lancet JE, Duong VH, Winton EF, Stuart RK, Burton M, Zhang S, Cubitt C, Blaskovich MA, Wright JJ, Sebti S and Sullivan DM. A phase I clinical-pharmacodynamic study of the farnesyltransferase inhibitor tipifarnib in combination with the proteasome inhibitor bortezomib in advanced acute leukemias. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011; 17(5):1140-1146.
104. Blum W, Schwind S, Tarighat SS, Geyer S, Eisfeld AK, Whitman S, Walker A, Klisovic R, Byrd JC, Santhanam R, Wang H, Curfman JP, Devine SM, Jacob S, Garr C, Kefauver C, et al. Clinical and pharmacodynamic activity of bortezomib and decitabine in acute myeloid leukemia. Blood. 2012; 119(25):6025-6031.
105. Walker AR, Klisovic RB, Garzon R, Schaaf LJ, Humphries K, Devine SM, Byrd JC, Grever MR, Marcucci G and Blum W. Phase I study of azacitidine and bortezomib in adults with relapsed or refractory acute myeloid leukemia. Leukemia & lymphoma. 2014; 55(6):1304-1308.
106. Howard DS, Liesveld J, Phillips GL, 2nd, Hayslip J, Weiss H, Jordan CT and Guzman ML. A phase I study using bortezomib with weekly idarubicin for treatment of elderly patients with acute myeloid leukemia. Leukemia research. 2013; 37(11):1502-1508.
107. Walker AR, Klisovic R, Johnston JS, Jiang Y, Geyer S, Kefauver C, Binkley P, Byrd JC, Grever MR, Garzon R, Phelps MA, Marcucci G, Blum KA and Blum W. Pharmacokinetics and dose escalation of the heat shock protein inhibitor 17-allyamino-17-demethoxygeldanamycin in combination with bortezomib in relapsed or refractory acute myeloid leukemia. Leukemia & lymphoma. 2013; 54(9):1996-2002.
108. Attar EC, Amrein PC, Fraser JW, Fathi AT, McAfee S, Wadleigh M, Deangelo DJ, Steensma DP, Stone RM, Foster J, Neuberg D and Ballen KK. Phase I dose escalation study of bortezomib in combination with lenalidomide in patients with myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML). Leukemia research. 2013; 37(9):1016-1020.
109. Kreso A and Dick JE. Evolution of the cancer stem cell model. Cell stem cell. 2014; 14(3):275-291.
110. Bonnet D and Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nature medicine. 1997; 3(7):730-737.
111. Terwijn M, Zeijlemaker W, Kelder A, Rutten AP, Snel AN, Scholten WJ, Pabst T, Verhoef G, Lowenberg B, Zweegman S, Ossenkoppele GJ and Schuurhuis GJ. Leukemic stem cell frequency: a strong biomarker for clinical outcome in acute myeloid leukemia. PloS one. 2014; 9(9):e107587.
112. Dick JE. Stem cell concepts renew cancer research. Blood. 2008; 112(13):4793-4807.
113. Lutz C, Hoang VT, Buss E and Ho AD. Identifying leukemia stem cells--is it feasible and does it matter? Cancer letters. 2013; 338(1):10-14.
114. Zhou J and Chng WJ. Identification and targeting leukemia stem cells: The path to the cure for acute myeloid leukemia. World journal of stem cells. 2014; 6(4):473-484.
115. Fang J, Barker B, Bolanos L, Liu X, Jerez A, Makishima H, Christie S, Chen X, Rao DS, Grimes HL, Komurov K, Weirauch MT, Cancelas JA, Maciejewski JP and Starczynowski DT. Myeloid malignancies with chromosome 5q deletions acquire a dependency on an intrachromosomal NF-kappaB gene network. Cell reports. 2014; 8(5):1328-1338.
116. Keutgens A, Robert I, Viatour P and Chariot A. Deregulated NF-kappaB activity in haematological malignancies. Biochemical pharmacology. 2006; 72(9):1069-1080.
117. Volk A, Li J, Xin J, You D, Zhang J, Liu X, Xiao Y, Breslin P, Li Z, Wei W, Schmidt R, Li X, Zhang Z, Kuo PC, Nand S, Zhang J, et al. Co-inhibition of NF-kappaB and JNK is synergistic in TNF-expressing human AML. The Journal of experimental medicine. 2014; 211(6):1093-1108.
118. Misaghian N, Ligresti G, Steelman LS, Bertrand FE, Basecke J, Libra M, Nicoletti F, Stivala F, Milella M, Tafuri A, Cervello M, Martelli AM and McCubrey JA. Targeting the leukemic stem cell: the Holy Grail of leukemia therapy. Leukemia. 2009; 23(1):25-42.
119. ten Cate B, de Bruyn M, Wei Y, Bremer E and Helfrich W. Targeted elimination of leukemia stem cells; a new therapeutic approach in hemato-oncology. Current drug targets. 2010; 11(1):95-110.
120. Ravandi F and Estrov Z. Eradication of leukemia stem cells as a new goal of therapy in leukemia. Clinical cancer research : an official journal of the American Association for Cancer Research. 2006; 12(2):340-344.
121. Guzman ML, Rossi RM, Neelakantan S, Li X, Corbett CA, Hassane DC, Becker MW, Bennett JM, Sullivan E, Lachowicz JL, Vaughan A, Sweeney CJ, Matthews W, Carroll M, Liesveld JL, Crooks PA, et al. (2007). An orally bioavailable parthenolide analog selectively eradicates acute myelogenous leukemia stem and progenitor cells.
122. Neelakantan S, Nasim S, Guzman ML, Jordan CT and Crooks PA. Aminoparthenolides as novel anti-leukemic agents: Discovery of the NF-κB inhibitor, DMAPT (LC-1). Bioorganic & medicinal chemistry letters. 2009; 19(15):4346-4349.
123. Ghantous A, Sinjab A, Herceg Z and Darwiche N. Parthenolide: from plant shoots to cancer roots. Drug discovery today. 2013; 18(17-18):894-905.
124. Hostager BS and Bishop GA. CD40-mediated activation of the NF-κB2 pathway. Frontiers in immunology. 2013; 4.
125. Stone RM, O’Donnell MR and Sekeres MA. Acute myeloid leukemia. ASH Education Program Book. 2004; 2004(1):98-117.
126. Boyerinas B, Park S-M, Hau A, Murmann AE and Peter ME. The role of let-7 in cell differentiation and cancer. Endocrine-Related Cancer. 2010; 17(1):F19-F36.