
INTRODUCTION
DACH1 is a human homolog of drosophila gene dac, a key member of the Retinal Determination Gene Network (RDGN). RDGN is required for drosophila eye specification [1]. Through gene-specific recruitment of coactivator or corepressor that control precursor cell proliferation and survival, mamalian homologues, DACH1/SIX1/EYA network also determines the development of many organs, including lung [2, 3]. In this respect, transgenic model demonstrates that Six1 and Eya are required for maintaining epithelial progenitor cell, regulating epithelial branching, mesenchymal development as well as alveolarization during the saccular phase of lung morphogenesis [3, 4]. Recently, more evidence indicates RDGN network is implicated in the development of human cancer, including breast and lung carcinoma [5-10]. Functional studies have identified DACH1 as a negative regulator of TGF-β and Wnt signaling to repress cancer cell migration and invasion [11, 12]. DACH1 inhibits cell cycle progression and oncogenic transformation, as well as blocks paracrine signaling [13-15]. DACH1 attenuates the transcriptional activity of FOXM1b by a competitively binding to homologic DNA sequence [16]. In addition, DACH1 associates with the estrogen and androgen receptors (ER and AR) to regulate signal transduction and proliferation of breast and prostate cancer cells [17, 18]. DACH1 inhibits EMT and tumor initiated cells in breast cancer and glioma [19-21]. DACH1 can directly associate with p53 and enhance its function in breast and lung cancer [10, 22]. Inactivation of DACH1 in human cancer tissues was observed by mechanisms of gene deletion, mutation, or promoter hypermethylation [21, 23]. Clinically, a decreased expression of DACH1 in breast and endometrial cancer correlates with tumor progression, poor differentiation [23] and predicts a short survival [13, 24, 25]. In human lung adenocarcinoma, the transcription factor SIX1 is upregulated during preinvasive to invasive transition and that results in EMT and conferring a more malignant phenotype [26]. In contrast, DACH1 expression was reduced in human NSCLC. Reexpression of DACH1 reduced colony formation and tumor growth in NSCLC cell lines via synergistic action with p53 [10]. However, the mechanism by which DACH1 regulates lung cancer growth is not fully understood. In this study, we analyzed the association between the DACH1 expression and clinic-pathological characteristics in NSCLC. Our result revealed low expressions of DACH1 predicted unfavorable prognosis for survival. CXCL5 was identified as a downstream target of DACH1-mediated repression of cell invasion and tumorigenesis. Conversely, high expressions of CXCL5 is associated with worse prognosis.
RESULTS
Decreased expression of DACH1 correlates with tumor progression and poor survival in NSCLC
To examine the expression of DACH1 in normal and cancer tissues of lung, immunohistochemical stain was performed on human lung cancer tissue arrays consisting of normal and different types of cancers. As previously reported, DACH1 was highly expressed in the nuclei of lung epithelial cells [10], but dramatically reduced in adenocarcinoma, squamous carcinoma, and large cell carcinoma (Fig. 1A, B). A semi-quantitive analysis revealed progressive loss of DACH1 expression in relation to tumor stage and histological grade (Fig. 1C, D). To determine whether loss of DACH1 occurred in the early stage, we analyzed a gene expression dataset of 226 primary lung adenocarcinoma with pathological stage I-II, the result showed that expression of DACH1 mRNA was reduced in tumor tissues (Fig. 1E) and inversely correlated with tumor stage (Fig. 1F). T2 tumor had much lower DACH1 expression than T1 tumor. In addition, tumor tissues with lymph node metastasis (N1) had much less DACH1 than those without metastasis (N0) (Fig. 1G). Furthermore survival analysis demonstrated that patients with high expression of DACH1 had 90% relapse-free survival (RFS) at 100 months follow-up, while RFS rate for patients in lower DACH1 expression group were only 60% (Fig.1H). Similarly, patients with high expression of DACH1 were also associated with favorable overall survival (OS) than those with low expression, 95% vs 70% respectively (Fig. 1I). This results suggest that DACH1 expression was reduced in NSCLC tissues and inversely correlated with tumor progression. Low expression of DACH1 predicted worse prognosis.
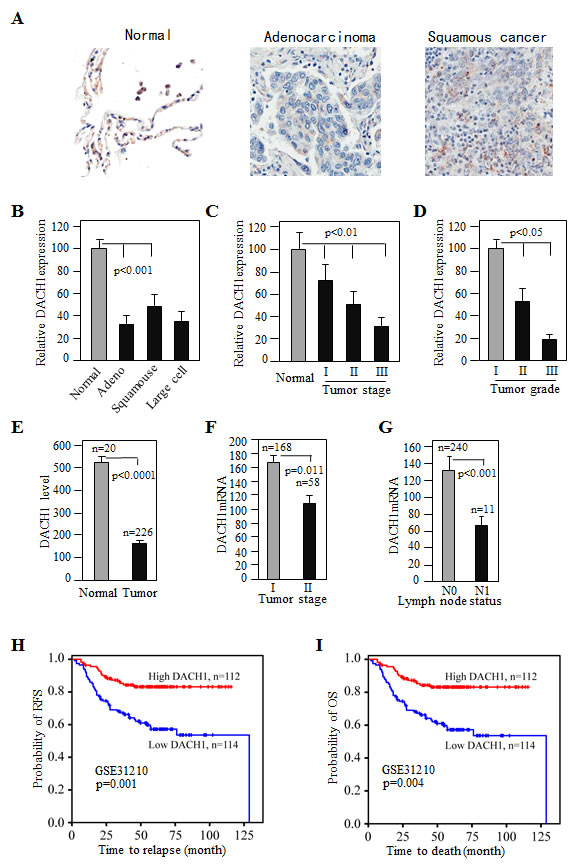
Figure 1: Decreased DACH1 expression in lung adenocarcinoma inversely correlated with tumor stage and related to survival. (A) Representative images of DACH1 immunohistochemical stain of normal and lung cancer tissues. (B-D) Relative intensity of DACH1 protein in cancer tissues in relation to histological type, stage and grade. Quantitate mRNA expression of DACH1 in normal and cancer tissues (E), and DACH1 abundance in relation to tumor size (F) and lymph node status (G). Impact of DACH1 mRNA expression on relapse-free survival (H) and overall survival (I) revealed by Kaplan-Meier curves from dataset GSE31210.
Restoration of DACH1 in NSCLC cells reduced cellular proliferation and tumor growth
In order to evaluate the role of DACH1 in NSCLC, we established DACH1 expressing lung adenocarcinoma cell line A-427 and A549 by lentivirus transduction. More than 95% of the cells were positive for ectopic expression of DACH1 as detected by fluorescent staining (Fig. 2A). Overexpression of DACH1 decreased A549 cellular proliferation assesssed by both MTT assay (Fig. 2B) and cell counting (Fig. 2C). The similar effects were observed in A-427 cell line (Fig. 2D-F). DS domain of DACH1 protein was required for inhibition of growth in breast and prostate cancer cells [20, 24, 26, 27]. To examine whether this domain also plays an essential role in NSCLC, sublines expressing DACH1 without DS domian (∆DS) were established. No significant effect on proliferation was observed by expression of ∆DS in both cell lines expressing. Cell cycle distribution analysis demonstrated that DACH1 decreased S phase ratio in comparision with vector control in both A-427 and A549 cells (Fig. 2G, H). Colony formation, especially contact-independent in soft agar, is a basic characteristic of transformed cells and partly represents the malignant potential and tumorigenicity. As we expected, ectopic expression of wild type DACH1 inhibited the clone number in both A-427 and A549 cells under contact-dependent growth. In contrast, equal expression of ∆DS had no repressive function (Fig. 3A). Colony formation in soft agar was also perfomed for both A-427 and A549 cells, the colony number was dramatically decreased in cells expressing wild type DACH1, whereas, there was no inhibitory effects by expressing ∆DS (Fig. 3B). To evaluate the role of DACH1 in tumorigenecity in vivo, A549 cells expressing either DACH1 or vector control were subcutaneously implanted into immunodeficient mice. As shown in (Fig. 3C), the dramatic decrease of tumor size was noticed in the animals inoculated with A549 cells expressing DACH1 compared with vector control. The average tumor weight decreased from 310mg in the control group to 40mg in the DACH1 group.
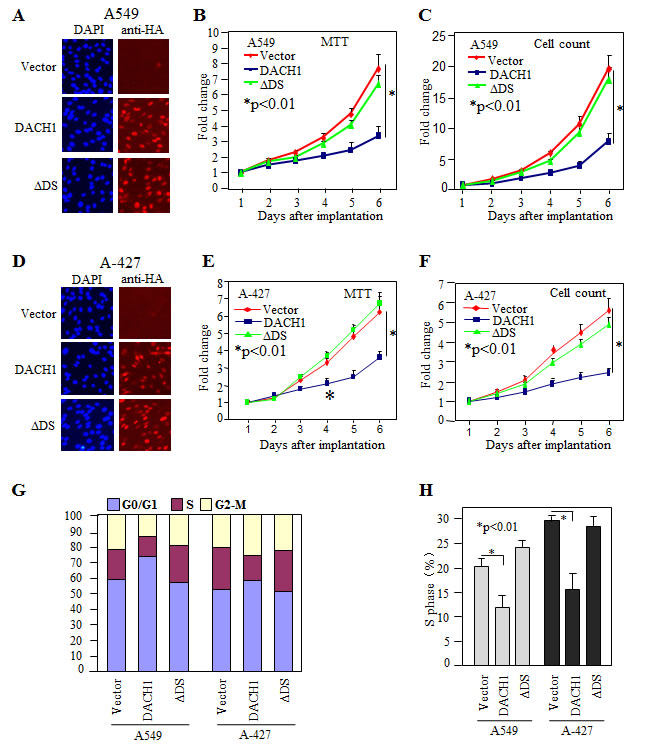
Figure 2: Ectopic expression of DACH1 inhibited cellular proliferation and cell cycle progression. Stable expression of HA-tagged DACH1 in lung cancer cell lines A549 (A-C) and A-427 by fluororescent staining (D-F). Proliferation was evaluated by MTT assay (B, E) and cell counting (C, F). Cell cycle distribution evaluated by FACS (G, H).
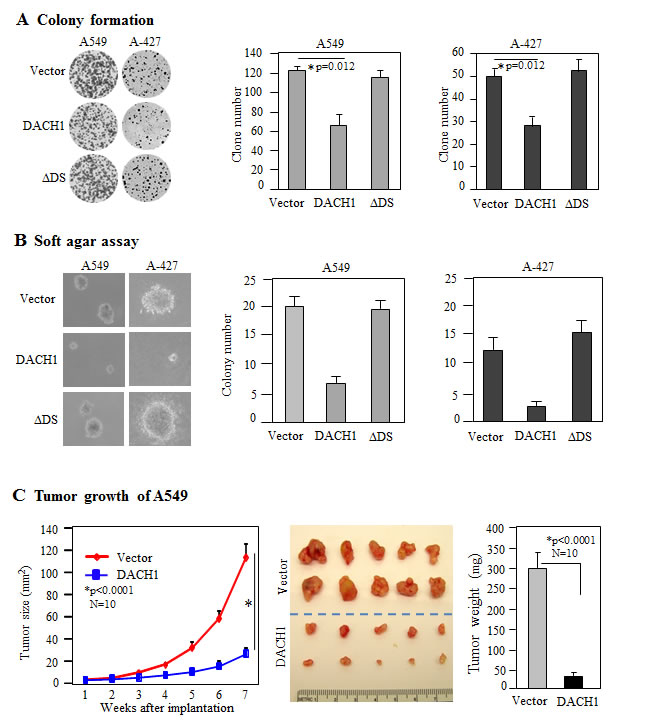
Figure 3: DACH1 inhibited clone formation and tumor growth in vivo. (A) Representative images of contact-dependent clone formation and relative efficiency of 3 experiments for A549 and A-427. (B) Representative images of clone formation in soft agar and relative efficiency of 3 experiments for A549 and A-427. (C) Tumor growth curves of subcutaneously implanted A549 cells with or without DACH1 expression, representative images of dissected tumors and tumor weight.
DACH1 repressed NSCLC migration and invasion accompanied with decreased CXCL5 secretion
To investigate the function of DACH1 in cell movement, wound healing assay was performed. In both A549 and A-427 cells, expression of DACH1 significantly inhibited wound-healing process, suggeting a decreased cell mobility induced by DACH1 (Fig. 4A, B). To determine the migratory capacity of single cell, we performed transwell assay. In comparision to cells with vector, migrative cell number was reduced by more than 50% by expression of wild type DACH1, but not DS deleted mutant (∆DS) (Fig. 4C). We further examined invasive capability using BD biocoated tumor invasion system. The inhibitory effect of DACH1 on invasion was even more dramatic compared to its effect on migration, decreased by more than 75% by DACH1 in comparision to vector. No significant effect was observed by ∆DS (Fig. 4D). We co-cultured A549 cells with conditional medium (CM) from either A549-vector or A549-DACH1, then observed migratory ability. The results indicated that incubation with supernatant from DACH1 expressing cells reduced cell motility, suggesting that CM in A549-DACH1 had less stimulative factors (Fig. 4E). Similar result was noticed in A-427 cells. To identify secreted factors, CM was employed to a cytokine antibody array spotted with 70 human chemokines/cytokines. Markedly reduced level of CXCL5 was noticed in CM from A549-DACH1 along with several other known cytokines, such as IL-8. To further confirm this results, we measured the CXCL5 levels by ELISA and found that there was more than 50% reduction of CXCL5 in both DACH1 expressing cell lines (Fig. 4F, G). To determine in vivo effect of DACH1 on CXCL5 expression, xenograft tumor tissues were immunohistochemically stained. The results showed that expression of CXCL5 was high in the cytoplasm of tumor cells expressing control vector, but very weak in tumor cells expressing DACH1 (Fig. 4H). Together, these results suggested that CXCL5 might be an important downstream target of DACH1 in repressing cell motility.
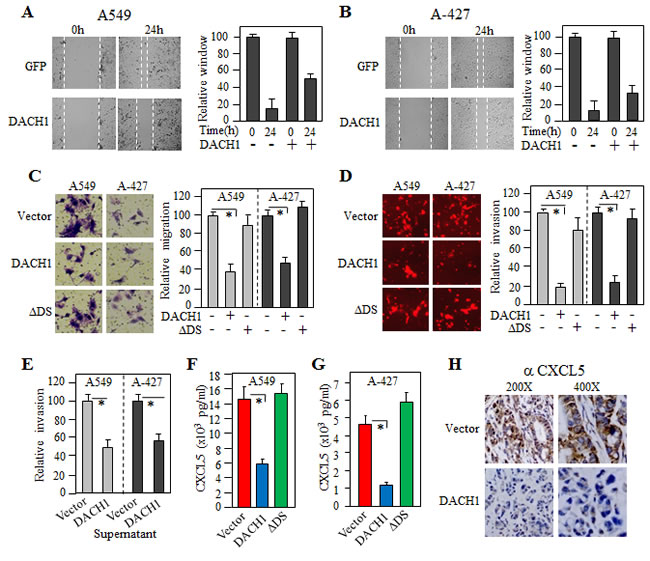
Figure 4: DACH1 inhibited lung cancer cell migration, invasion and CXCL5 secretion. Representative images of wound-healing assay of A549 (A) and A-427 (B) cells, relative wound space was calculated on the right panel. (C) Representative images and quantitatively analysis of migrated A549 and A-427 cells expressing DACH1 or mutant ∆DS versus vector control. (D) Representative images and quantitatively analysis of invaded A549 and A-427 cells expressing DACH1, mutant ∆DS or vector control. (E) Migratory promoting effect of supernatant collected from A549 cells expressing DACH1 versus empty vector. Quantitative measurement of CXCL5 in the condition medium collected from A549 (F) or A-427 (G) by ELISA. (H) Representative immunohistochemical images of CXCL5 in implanted tumor from A549 expressing DACH1 versus control vector.
CXCL5 rescued repression of cancer cell invasion and tumor formation by DACH1
To further explore the functional relationship between DACH1 and CXCL5, we searched lung cancer cell lines and tumor tissues database for mRNA expression. GEO dataset GSE7670 consisted of sixty-six affymetrix microarray information from 28 paired lung tumor and its adjacent normal tissues, 2 normal lung tissues and 8 cell lines, including immortalized human bronchial epithelial cell NL-20, adencarcinoma cell line A549, H1299, CL1-0 and large cell lung cancer cell line H661. The mRNA expression of DACH1 was inversely related to CXCL5 (R value = -0.77, p<0.025) (Fig. 5A) and such inverse relationship was also observed in normal and cancer tissues (Fig. 5B). To evaluate the role of CXCL5 in cell migration, purified CXCL5 was added into conditional medium, then migration and invasion was measured. Addition of CXCL5 to the medium enhanced cell migration and invasion in both A-427 and A549 cells (Fig. 5C, D). To further address the biological significance of CXCL5 in the CM, neutralized antibody to CXCL5 was added into the CM at final concentration of 1ng/ml before planting cells for motility assay. Antibody to CXCL5 significantly inhibited both migration and invasion in two cell lines at similar ratio (30%-40%) (Fig. 5E, F). To evaluate the role of CXCL5 in DACH1-mediated tumor growth, A549 cells stably expressing DACH1 (A549-DACH1) were co-transduced with lentivirus expressing CXCL5. In consist with (Fig. 3C), tumor derived from A549-DACH1 cells expressing vector control grown very slowly, but ectopic expresion of CXCL5 enhanced tumor growth to a rate similar to A549 parent cells (Fig. 5G). The average tumor weight were increased from 187mg to 735mg by CXCL5 (Fig. 5H, I). Immunohistochemical stain of xenograft tumor tissues confirmed strong ectopic expression of CXCL5 (Fig. 5J). Together, we provided an evidence that CXCL5 was a critical chemokine for DACH1-mediated repression of motility and tumor growth.
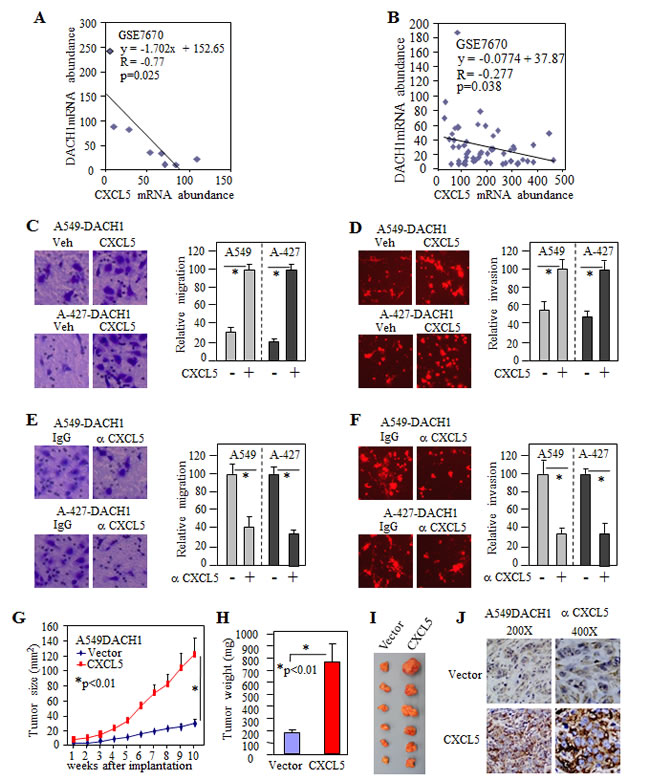
Figure 5: CXCL5 rescued invasion and antagonized tumor repression by DACH1. (A) The reciprocal mRNA expression between DACH1 and CXCL5 in normal lung epithelial cell line and 7 lung cancer cell lines from GSE7670. (B) The reciprocal mRNA expression between DACH1 and CXCL5 in NSCLC tissues from GSE7670. Addition of CXCL5 enhanced migration (C) and invasion (D) of A549 and A-427 cell expressing DACH1. Neutralized antibody to CXCL5 decreased migration (E) and invasion (F) of A549 and A-427 cells. Growth curve (G), weight (H) and representative images (I) of tumors from A549-DACH1 stable cells with ectopic CXCL5 expression versus vector control. (J) Representative immunohistochemical images of CXCL5 from A549-DACH1 cells with or without CXCL5 transduction.
CXCL5 expression correlated with tumor progression and might predict survival
To explore the potential value of CXCL5 expression as a biomarker in NSCLC, we analyzed GEO dataset GSE31210, which include 226 cases of pathological stage I-II lung adenocarcinomas with survival follow-up. CXCL5 mRNA abundance was positively related to tumor size with lowest expression in T1 and highest in T3-4 (Fig. 6A). Association of CXCL5 expression with histological differentiation status was also analyzed. Tumors with well differentiation expressed lowest level of CXCL5, whereas poorly differentiated tumors had highest expression of CXCL5 (Fig. 6B). In addition, we examined the prognostic value of CXCL5 mRNA for survival. 226 patients was divided as high versus low expression of CXCL5 based on mean value of CXCL5 mRNA. Kaplan-Meier survival curves showed that RFS rate at 100 months post operation was 70% versus 55% for patients with high expression of CXCL5 (Fig. 6C). The patients with low expression of CXCL5 had better OS, although not reaching statistical significance (Fig. 6D). We further analyzed mRNA profiles in another dataset with 439 lung adenocarcinomas, the patients were arbitrarily separated into three groups according to high, midium or low expression of CXCL5. The OS probabilities among the CXCL5 subgroups were statistically different (p=0.0093), indicating that CXCL5 expression was inversely correlated with overall survival (Fig. 6E).
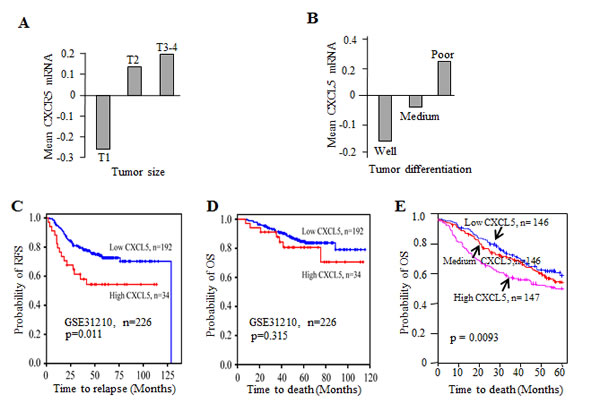
Figure 6: CXCL5 expression correlated with tumor stage and influenced survival in NSCLC. Association of CXCL5 mRNA abundance with tumor size (A) and differentiation (B). Kaplan-Meier curves for relapse-free survival (C) and overall survival (D) from dataset GSE31210. (E) Kaplan-Meiercurves for OS for lung adenocarcinoma patients with low, medium or high expressions of CXCL5 mRNA .
DISCUSSION
Recent findings supported the role of DACH1 as a novel tumor suppresor in several kinds of human cancers [5, 12-25]. The link of DACH1 to lung cancer was initially uncovered by whole-genome and transcriptome sequencing of NSCLC samples. Genomic mutations and copy number loss of DACH1 gene were identified along with decreased DACH1 mRNA expression in NSCLC [27]. A subsequent study demonstrated the p53-dependent effect of DACH1 on proliferation and tumor gowth [10]. The current study further supported DACH1 as lung tumor suppressor. We found in NSCLC cell lines that the conserved DS domain was required for DACH-mediated repression, indicating a p53-independent alternative mechanism might be involved. By unbiased chemokine screen analysis, CXCL5 was identified as a novel target of DACH1. Functional analysis showed that CXCL5 played an essential role in DACH1-induced inhibition of cell migration in vitro and tumorigenesis in vivo. Importantly, there was an inverse relationship between the expression of DACH1 and CXCL5 in human lung cancer cell lines and NSCLC tissues.
CXCL5, also known as epithelial-derived neutrophil-activationg protein 78 (ENA78), is originally discovered as a potent chemoattractant and activator of neutrophil function. Via its receptor CXCR2, CXCL5 has been shown to regulate cell proliferation, cell migration and invasion [28]. Down-regulation of CXCL5 resulted in significantly decreased cell proliferation and migration. The tumorigenic potential was ablated by knock down CXCL5 [29]. In additin, CXCL5 promoted metastasis by regulating tumor-associated angiogenesis [30]. In deed, over-expression of CXCL5 increased tumor metastatic capacity.
CXCL5 expression could be regulated by several signaling pathways. DACH1 represses CXCL5 expression might be through crosstalk with those pathways. For instance, it has been reported that CXCL5 expression can be up-regulated by CREB and NF-kappaB in lung carcinogenesis [31]. In breast cancer, DACH1 has been shown to repress CXCL8 through blocking activation from AP1 and NF- kappaB binding sites [15] and inhibit cyclin D1 transcription though AP-1/CREB [13]. Therefore, we surmise that DACH1 associates with transcriptional factors to block CXCL5 activation. CXCL5 is well known to be activated by Ras signaling and the genetic ablation of its receptor, mCXCR2, reduced oncogenic Ras-driven tumorigenesis in mice [32]. In malignant peripheral nerve sheath tumors, activation of RAS repressed DACH1expression and inbibition of RAS signaling normalized the expresion of DACH1 [33]. Interestingly, we previously reported that DACH1 attenuated Ras-induced migration, invasion and CXCL8 secretion in breast cancer [15]. These studies suggest the existence of mutual regulation between Ras and DACH1. Thus, activated Ras in NSCLC could repress the expression of DACH1, in turn, reduced expression of DACH1 might enhance Ras activity, and that could lead to increased production of CXCL5. In addition, the FOXM1 is abundantly expressed in human NSCLCs and it transcriptionally induces the expression of genes essential for proliferation of tumor cells. In FOXM1 transgene model, elevated tumor formation was associated with persistent pulmonary inflammation, macrophage infiltration and increased expressions of CXCL5, CXCL1 and CCL3 [34]. It has been demonstrated that DACH1 antagonizes FOXM1 signaling through competitively binding to the conserved forkhead specific DNA sequence in breast cancer [16]. Therefore, we speculate that functional inactivation of DACH1 might disrupt the balance of DACH1-FOXM1, leading to the activation of CXCL5.
Besides activation of CXCL5 by oncogenes, CXCL5 has been shown to be repressed by tumor suppressor p53 [35]. We previously demonstrated that DACH1 associated with p53 protein to enhance p53 function in NSCLC [10]. In this regard, it is worthy to validate whether repression of CXCL5 by DACH1 depends on p53.
As to the clinical-pathological relevance of DACH1 in NSCLC, we observed that DACH1 abundance was inversely corelated with tumor stage, grade and metastasis, which is consistent with previous reports [10, 12-14, 23]. Thus far, only a handful of reports have explored the value of DACH1 as a molecular marker for cancer prognosis. We first reported that breast cancer patients with low DACH1 expression have a 40-month survival disadvantage [13]. Recently, Dr. Powe’s study demonstrated that high expression of DACH1 predicted a better survival in luminal breast cancers subtype [24], further supporting the importance of DACH1 as a prognostic factor. Current analysis demonstrates that reduced expression of DACH1 is associated with unfavorable prognosis for RFS and OS. On the other hand, high expression of CXCL5 predicts worse prognosis for both RFS and OS. In deed, recent studies analyzed the effect of 18 out of the 23 differently expressed genes between the tumor and normal tissues on overall survival, only CXCL5 had significantly influence on overall and disease-free survival [36].
In summary, our study provides a functional link between DACH1 and CXCL5 in NSCLC. Considering the multiple functions of CXCL5 in terms of proliferation, invasion, inflamation and tumor-environment interaction, our results further enrich the understanding of the underlying mechanism by which DACH1 acts as a tumor suppressor.
METHODS
Patients and samples
The human lung cancer tissue arrays were purchased from Alenabio (Xi’an, China) and procedure was approved by the Ethical Committee of Tongji Hospital. There were 6 cases of normal lung tissues, 6 cases of cancer adjacent normal lung tissue, 57 cases of adenocarcinoma, 40 cases of squamous carcinoma, 31 cases of small cell carcinoma and 10 cases of large cell carcinoma. Tumor tissues were sub-grouped as grade I, well-differentiated; grade II, moderately differentiated; and grade III, poorly or undifferentiated. GSE31210 expression profiles consist of 226 cases with lung adenocarcinomas of pathological stage I-II [37].
Immunohistochemistry and immunofluorescence stain
Tissue immunohistochemical stains were performed using a streptavidin-biotin technique and semi-quantified [14, 20]. The polyclonal antibody to DACH1(Proteintech, 10914-1) and to CXCL5 (Santa Cruz, sc-73930) were used by the published method [14]. The staining intensity was graded as follows: no staining, 0; weakly positive, 1; moderately positive, 2; and strongly positive, 3. Immunofluorescence staining for transduced DACH1 was detected by HA probe (Santa Crux, sc-7392).
Cell culture and establishment of DACH1 stable cell lines
The human NSCLC cell lines A-427 and A549 were obtained from ATCC (Manassas, VA, USA) and were cultured in recommended medium supplemented with 10% fetal bovine serum. Human embryonic kidney 293T cells were maintained in DMEM containing 1% penicillin/streptomycin and supplemented with 10% FBS. Expressing vectors for wt DACH1 and ∆DS were described previously [13]. Lentivirus expression vector for human CXCL5 was purchased from DNASU plasmid repository. Lentivirus package and transduction were performed as manufacturer’s recommended protocol. Stably expressions of DACH1 in transduced cells were confirmed by immunofluorescence stain. For selection of CXCL5 stable cells, transduced cells were treated with blasticidin at 5ug/ml for 2 weeks.
Cell proliferation and colony formation assay
Cells expressing DACH1, ∆DS and vector control were seeded into 96-well plates in normal growth medium, and cell growth was measured by daily3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl-tetrazolium (MTT) assay as previously described [13, 14]. For growth curve assay, cells were seeded into 12-well plates and serially counted for 6 to 7 days. For contact-dependent growth, 4 × 103 cancer cells were plated in triplicate into 6cm dish and medium was changed every 3 days. The colonies were visualized by staining with 0.04% crystal violet in methanol for 1 hour [13]. For contact-independent growth, 4 × 103cells suspended in 0.3% soft agar were plated on the surface of 0.5% soft agar in 6cm dish. After 2-weeks growth, colony with more than 30 cells was counted under 200X microscope [13, 14, 17].
Cell cycle analysis
Cells were trypsinized and fixed in 70% ethanol, then processed by standard methods using propidium iodide staining of cellular DNA. Samples were analyzed on a FACScan flow cytometer (BD Biosciences). A minimum of 20,000 events for each sample were collected for statistical analysis of cell cycle distribution using ModFit version 2.0.
Migration and invasion assay
For the wound healing assay, cells in 90% confluent were streaked with a 10 µl sterile pipette tip, washed with PBS, and replenished with serum-free DMEM. Images were captured using an inverted microscope at 12 and 24 hours after wounding. Migration assays were performed in transwell inserts with 8-μm pore (Corning Inc.; Corning, NY, USA). Serum-starved cells were trypsynized and counted. 2 × 105 cells in 200 µl serum-free medium were placed into the upper chamber. The low chamber was filled with 500 µl of 10% FBS-DMEM. After 24 h, cells that migrated to the lower chamber were fixed and stained with hematoxylin. Invasion ability was quantitatively measured by BD coated invasion system.
Tumor implantation study
A549 cells (2 × 105) expressing either vector control or DACH1 were implanted subcutaneously to 6-week old ethymic male nude mice. The tumor growth was measured weekly for 4 to 5 weeks using a digital caliper. The isolated tumor mass was weighted after mice were sacrificed.
Statistical analysis
All data were expressed as the mean±standard error. Statistical analyses between groups were calculated by student’s t-test. P value <0.05 was considered stastically significant. Univariate cumulative survival analyses for relapse-free survival and overall survival were calculated by Kaplan-Meier curve using log rank tests.
Acknowledgements
This work was supported by China NSFC grants No. 81172422, 81072169, 81261120395 and 81301929, National Basic Research Program No. 2010CB912802 and 2012CB934002, and The Natural Science Foundation of Hubei Province No. 2014CFB218. We thank Dr. Ales Cvekl and Dr. Richard Pestell for providing plasmids. We thanks Dr. Guoan Chen for providing Kaplan-Meier analysis for patients with lung adenocarcinoma.
Competing interests
The authors declare that they have no competing interests.
Author’s contribution
KW designed the study and wrote the manuscript. NH, XY, HX and HW acquired and analyzed data; QC,YC, MG and SY help to design experiments and supervised study.
REFERENCES
1. Atkins M, Jiang Y, Sansores-Garcia L, Jusiak B, Halder G, Mardon G. Dynamic rewiring of the Drosophila retinal determination network switches its function from selector to differentiation. PLoS Genet. 2013; 9(8):e1003731.
2. Li X, Oghi KA, Zhang J, Krones A, Bush KT, Glass CK, Nigam SK, Aggarwal AK, Maas R, Rose DW, Rosenfeld MG. Eya protein phosphatase activity regulates Six1-Dach-Eya transcriptional effects in mammalian organogenesis. Nature. 2003; 426(6964):247-254.
3. Lu K, Reddy R, Berika M, Warburton D, El-Hashash AH. Abrogation of Eya1/Six1 disrupts the saccular phase of lung morphogenesis and causes remodeling. Dev Biol. 2013; 382(1):110-123.
4. El-Hashash AH, Al Alam D, Turcatel G, Rogers O, Li X, Bellusci S, Warburton D. Six1 transcription factor is critical for coordination of epithelial, mesenchymal and vascular morphogenesis in the mammalian lung. Dev Biol. 2011; 353(2):242-258.
5. Popov VM, Wu K, Zhou J, Powell MJ, Mardon G, Wang C, Pestell RG. The Dachshund gene in development and hormone-responsive tumorigenesis. Trends Endocrinol Metab. 2010; 21(1):41-49.
6. Wu W, Ren Z, Li P, Yu D, Chen J, Huang R, Liu H. Six1: A critical transcription factor in tumorigenesis. Int J Cancer. 2014; 136(6):1245-1253.
7. Christensen KL, Patrick AN, McCoy EL, Ford HL. The six family of homeobox genes in development and cancer. Adv Cancer Res. 2008; 101:93-126.
8. Tadjuidje E, Hegde RS. The Eyes Absent proteins in development and disease. Cell Mol Life Sci. 2013; 70(11):1897-1913.
9. Xia Y, Zhu Y, Ma T, Pan C, Wang J, He Z, Li Z, Qi X, Chen Y. miR-204 functions as a tumor suppressor by regulating SIX1 in NSCLC. FEBS Lett. 2014; 588(20):3703-3712.
10. Chen K, Wu K, Cai S, Zhang W, Zhou J, Wang J, Ertel A, Li Z, Rui H, Quong A, Lisanti MP, Tozeren A, Tanes C, Addya S, Gormley M, Wang C, McMahon SB, Pestell RG. Dachshund binds p53 to block the growth of lung adenocarcinoma cells. Cancer Res. 2013;73(11):3262-3274.
11. Wu K, Yang Y, Wang C, Davoli MA, D’Amico M, Li A, Cveklova K, Kozmik Z, Lisanti MP, Russell RG, Cvekl A, Pestell RG. DACH1 inhibits transforming growth factor-beta signaling through binding Smad4. J Biol Chem. 2003; 278(51):51673-51684.
12. Yan W, Wu K, Herman JG, Brock MV, Zhou Y, Lu Y, Zhang Z, Yang Y, Guo M. Epigenetic silencing of DACH1 induces the invasion and metastasis of gastric cancer by activating TGF-β signalling. J Cell Mol Med. 2014; 18(12):2499-2511.
13. Wu K, Li A, Rao M, Liu M, Dailey V, Yang Y, Di Vizio D, Wang C, Lisanti MP, Sauter G, Russell RG, Cvekl A, Pestell RG. DACH1 is a cell fate determination factor that inhibits cyclin D1 and breast tumor growth. Mol Cell Biol. 2006; 26(19):7116-29.
14. Chu Q, Han N, Yuan X, Nie X, Wu H, Guo M, Chen Y, Yu Y, Wu K. DACH1 inhibits cyclin D1 expression, cellular proliferation and tumor growth of renal cancer cell. J Hematol Oncol. 2014;7:73.
15. Wu K, Katiyar S, Li A, Liu M, Ju X, Popov VM, Jiao X, Lisanti MP, Casola A, Pestell RG. Dachshund inhibits oncogene-induced breast cancer cellular migration and invasion through suppression of interleukin-8. Proc Natl Acad Sci U S A. 2008;105(19):6924-6929.
16. Zhou J, Wang C, Wang Z, Dampier W, Wu K, Casimiro MC, Chepelev I, Popov VM, Quong A, Tozeren A, Zhao K, Lisanti MP, Pestell RG. Attenuation of Forkhead signaling by the retinal determination factor DACH1. Proc Natl Acad Sci U S A. 2010;107(15):6864-6869.
17. Wu K, Katiyar S, Witkiewicz A, Li A, McCue P, Song LN, Tian L, Jin M, Pestell RG. The cell fate determination factor dachshund inhibits androgen receptor signaling and prostate cancer cellular growth. Cancer Res. 2009; 69(8):3347-3355.
18. Popov VM, Zhou J, Shirley LA, Quong J, Yeow WS, Wright JA, Wu K, Rui H, Vadlamudi RK, Jiang J, Kumar R, Wang C, Pestell RG. The cell fate determination factor DACH1 is expressed in estrogen receptor-alpha-positive breast cancer and represses estrogen receptor-alpha signaling. Cancer Res. 2009; 69(14):5752-5760.
19. Wu K, Jiao X, Li Z, Katiyar S, Casimiro MC, Yang W, Zhang Q, Willmarth NE, Chepelev I, Crosariol M, Wei Z, Hu J, Zhao K, Pestell RG. Cell fate determination factor Dachshund reprograms breast cancer stem cell function. J Biol Chem. 2011; 286(3):2132-2142.
20. Wu K, Chen K, Wang C, Jiao X, Wang L, Zhou J, Wang J, Li Z, Addya S, Sorensen PH, Lisanti MP, Quong A, Ertel A, Pestell RG. Cell fate factor DACH1 represses YB-1-mediated oncogenic transcription and translation. Cancer Res. 2014; 74(3):829-839.
21. Watanabe A, Ogiwara H, Ehata S, Mukasa A, Ishikawa S, Maeda D, Ueki K, Ino Y, Todo T, Yamada Y, Fukayama M, Saito N, Miyazono K, Aburatani H. Homozygously deleted gene DACH1 regulates tumor-initiating activity of glioma cells. Proc Natl Acad Sci U S A. 2011; 108(30):12384-12389.
22. Chen K, Wu K, Gormley M, Ertel A, Wang J, Zhang W, Zhou J, Disante G, Li Z, Rui H, Quong AA, McMahon SB, Deng H, Lisanti MP, Wang C, Pestell RG. Acetylation of the cell-fate factor dachshund determines p53 binding and signaling modules in breast cancer. Oncotarget. 2013; 4(6):923-935.
23. Zhu H, Wu K, Yan W, Hu L, Yuan J, Dong Y, Li Y, Jing K, Yang Y, Guo M. Epigenetic silencing of DACH1 induces loss of transforming growth factor-β1 antiproliferative response in human hepatocellular carcinoma. Hepatology. 2013; 58(6):2012-2022.
24. Powe DG, Dhondalay GK, Lemetre C, Allen T, Habashy HO, Ellis IO, Rees R, Ball GR. DACH1: its role as a classifier of long term good prognosis in luminal breast cancer. PLoS One. 2014; 9(1):e84428.
25. Nan F, Lü Q, Zhou J, Cheng L, Popov VM, Wei S, Kong B, Pestell RG, Lisanti MP, Jiang J, Wang C. Altered expression of DACH1 and cyclin D1 in endometrial cancer. Cancer Biol Ther. 2009; 8(16):1534-1539.
26. Mimae T, Okada M, Hagiyama M, Miyata Y, Tsutani Y, Inoue T, Murakami Y, Ito A. Upregulation of notch2 and six1 is associated with progression of early-stage lung adenocarcinoma and a more aggressive phenotype at advanced stages. Clin Cancer Res. 2012; 18 (4):945-955.
27. Govindan R, Ding L, Griffith M, Subramanian J, Dees ND, Kanchi KL, Maher CA, Fulton R, Fulton L, Wallis J, Chen K, Walker J, McDonald S, Bose R, Ornitz D, Xiong D, You M, Dooling DJ, Watson M, Mardis ER, Wilson RK. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell. 2012;150(6):1121-1134.
28. Strieter RM, Burdick MD, Gomperts BN, Belperio JA, Keane MP. CXC chemokines in angiogenesis. Cytokine Growth Factor Rev. 2005;16(6):593-609.
29. Miyazaki H, Patel V, Wang H, Edmunds RK, Gutkind JS, Yeudall WA. Down-regulation of CXCL5 inhibits squamous carcinogenesis. Cancer Res. 2006;66(8):4279-4284.
30. Keeley EC, Mehrad B, Strieter RM. CXC chemokines in cancer angiogenesis and metastases. Adv Cancer Res. 2010;106:91-111.
31. Sun H, Chung WC, Ryu SH, Ju Z, Tran HT, Kim E, Kurie JM, Koo JS. Cyclic AMP-responsive element binding protein- and nuclear factor-kappaB-regulated CXC chemokine gene expression in lung carcinogenesis. Cancer Prev Res (Phila). 2008; 1(5):316-328.
32. O’Hayer KM, Brady DC, Counter CM. ELR+ CXC chemokines and oncogenic Ras-mediated tumorigenesis. Carcinogenesis. 2009; 30(11):1841-1847.
33. Miller SJ, Lan ZD, Hardiman A, Wu J, Kordich JJ, Patmore DM, Hegde RS, Cripe TP, Cancelas JA, Collins MH, Ratner N. Inhibition of Eyes Absent Homolog 4 expression induces malignant peripheral nerve sheath tumor necrosis. Oncogene. 2010; 29(3):368-379.
34. Wang IC, Meliton L, Tretiakova M, Costa RH, Kalinichenko VV, Kalin TV. Transgenic expression of the forkhead box M1 transcription factor induces formation of lung tumors. Oncogene. 2008; 27(30):4137-4149.
35. Yeudall WA, Vaughan CA, Miyazaki H, Ramamoorthy M, Choi MY, Chapman CG, Wang H, Black E, Bulysheva AA, Deb SP, Windle B, Deb S. Gain-of-function mutant p53 upregulates CXC chemokines and enhances cell migration. Carcinogenesis. 2012;33(2):442-451.
36. Kowalczuk O, Burzykowski T, Niklinska WE, Kozlowski M, Chyczewski L, Niklinski J. CXCL5 as a potential novel prognostic factor in early stage non-small cell lung cancer: results of a study of expression levels of 23 genes. Tumour Biol. 2014;35(5):4619-4628.
37. Okayama H, Kohno T, Ishii Y, Shimada Y, Shiraishi K, Iwakawa R, Furuta K, Tsuta K, Shibata T, Yamamoto S, Watanabe S, Sakamoto H, Kumamoto K, Takenoshita S, Gotoh N, Mizuno H, Sarai A, Kawano S, Yamaguchi R, Miyano S, Yokota J. Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer Res. 2012; 72(1):100-111.