
Introduction
In the WHO 2008 classification, acute myeloid leukemia (AML) with myelodysplasia-related changes (AML-MRC) is defined as a distinct entity by the presence of multilineage dysplasia (MLD), and/or myelodysplastic syndrome (MDS)-related cytogenetics, and/or previously diagnosed MDS or MDS/Myeloproliferative neoplasm (MDS/MPN) [1]. The prognostic value of these three criteria is not established. The independent prognostic value of MLD is controversial and varies among different subsets of AML [2-7]. AML with MDS-related cytogenetics or previously diagnosed MDS or MDS/MPN have often unfavorable cytogenetics, and are associated with poorer outcome than AML without criteria of AML-MRC [2, 8, 9].
Although gene mutations are now a major tool for AML classification into distinct entities with specific prognosis [10-12], no molecular pattern is currently associated with AML-MRC. We hypothesized that the presence of mutations in targeted genes of interest could help identify subgroups of AML-MRC with distinct biological features and specific outcome. We had previously reported that AML-MRC have a specific mutation pattern sharing mutations found in both AML and high risk MDS and a particularly high frequency of ASXL1 mutation [13]. We report here a cohort of patients with AML-MRC for whom we analyzed the presence of mutational events according to AML-MRC criteria (MLD, cytogenetics and patient history) and identified mutation-based subgroups with specific poor outcome.
Results
Patient, disease and treatment characteristics
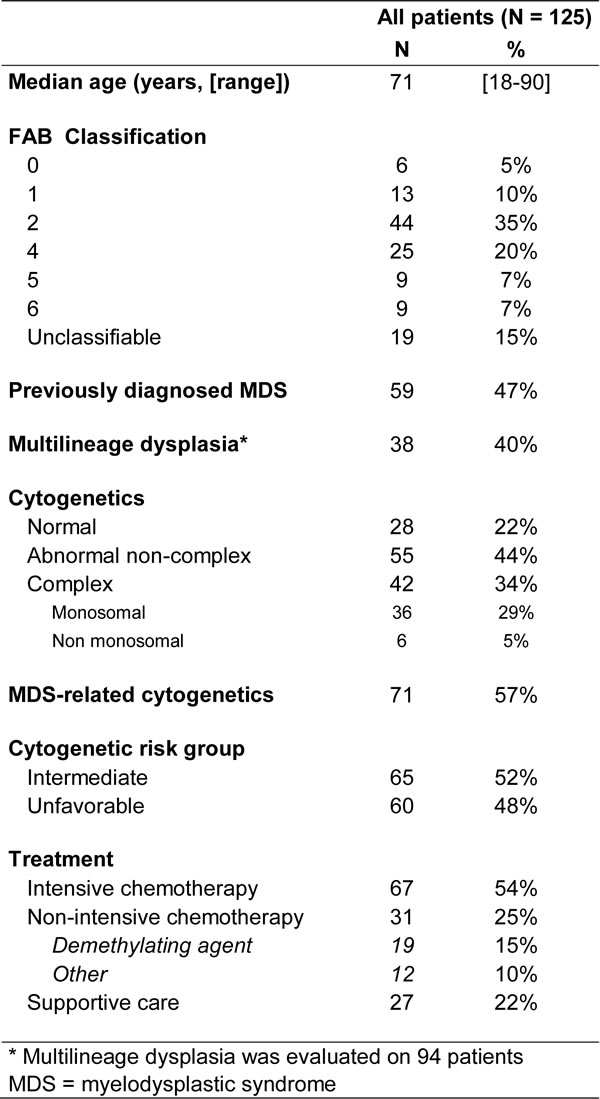
We studied 149 patients. After morphological review, 24 patients were excluded from the main analysis because of not enough dysplasia to reach the MLD criteria. These patients were analyzed separately (Supplemental Table 1). The remaining 125 fitting the AML-MRC criteria were considered for the main analysis and their characteristics are reported in Table 1. Median age was 71 years (range: 18-90). Fifty-nine patients (47%) had a previously diagnosed MDS. Seventy-one patients (57%) had MDS-related cytogenetics, including 42 patients (34%) with complex karyotype AML (CK-AML). Ninety-four patients were evaluable for morphological dysplasia by the double centralized review. Multilineage dysplasia (MLD) was found in 38/94 patients (40%). Sixty-seven patients (54%) received intensive induction chemotherapy. Mutations were found in ASXL1 (n = 26, 21%), RUNX1 (n = 15, 12%), DNMT3A (n = 11, 9%), NPM1 (n = 4, 3%), FLT3 (n = 9, 7%) and TP53 (n = 28, 22%). No mutation was found in 47 (38%) patients (31 with non-complex karyotype NCK-AML and 16 with CK-AML).
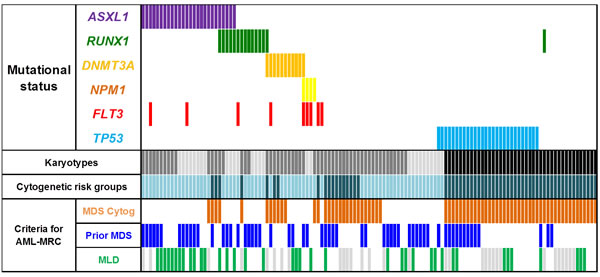
Figure 1: Co mutation profile of ASXL1, RUNX1, DNMT3A, NPM1, FLT3-ITD and TP53 genes in the 125 patients with AML-MRC: Karyotypes: normal (light grey), abnormal non-complex (dark grey) and complex (black). Cytogenetics risk group: intermediate (light blue) and unfavorable (dark blue). MLD: green bars indicate the presence of criteria for MLD and grey bars are samples that were not evaluable for MLD. These latter had at least one other criteria for AML-MRC to enrolled them in this study. MDS = myelodysplastic syndrome; MLD = multilineage dysplasia.
Table 1: Characteristics of the 125 patients with criteria for AML-MRC
Mutation profiles according to previous history of MDS, cytogenetics and MLD
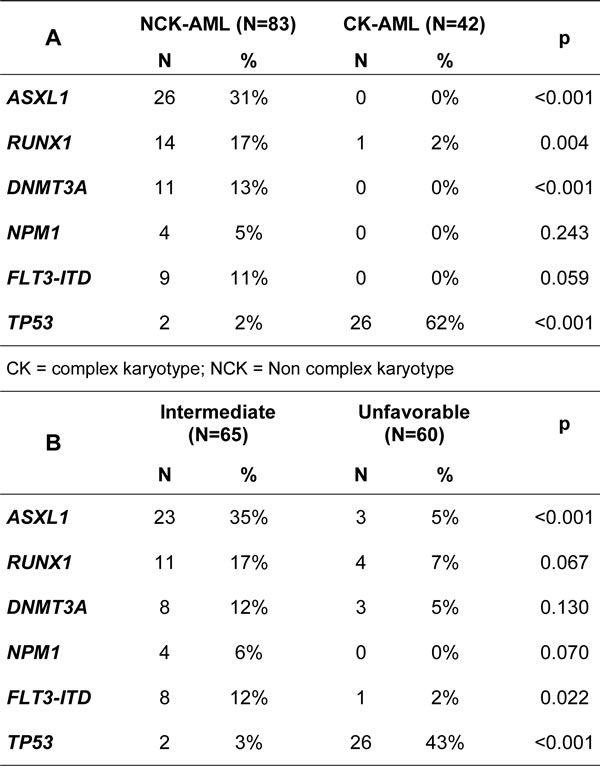
TP53 mutations were exclusive from all other mutations and all but two (26/28, 93%) were found in CK-AML. Mutations in the other genes were almost exclusively found in NCK-AMLs (Table 2A, Figure 1). In NCK-AMLs, ASXL1 was the most frequently mutated gene (26/83, 31%). These mutations were associated with the absence of MDS-related cytogenetics (MDS-related cytogenetics: yes vs. no: 4/29, 14% vs. 22/54, 41%, p = 0.013). Other mutations were equally distributed whether MDS-related cytogenetics was present or not (Figure 1).
We analyzed the presence of mutations according to the cytogenetic risk group (Table 2B). In the 94 patients evaluable for MLD, patients with criteria for MLD had more ASXL1 mutations (MLD: 15/38, 39% vs. no MLD: 8/48, 14%, p = 0.007). We did not find any correlation between the presence of MLD and other mutations. Median percentages of bone marrow DGP, DEP and DMP in the 94 patients were 64%, 24% and 45% respectively. ASXL1 mutations were associated with higher DGP (ASXL1-mut: 75% vs. ASXL1-wt: 55%, p = 0.030) but similar DEP (ASXL1-mut: 20% vs. ASXL1-wt: 25%, p = 0.933) and DMP (ASXL1-mut: 53% vs. ASXL1-wt: 40%, p = 0.139). There was no difference in percent of morphologic dysplasia according to any other genes (Supplemental Table 2). In a linear regression analysis including the mutation status of the 6 genes, mutation of ASXL1 remained associated with higher DGP (Coefficient beta = 20, p = 0.023, Supplemental Table 3). The frequencies of these mutations were not different according to the presence or not of prior MDS or MDS/MPN.
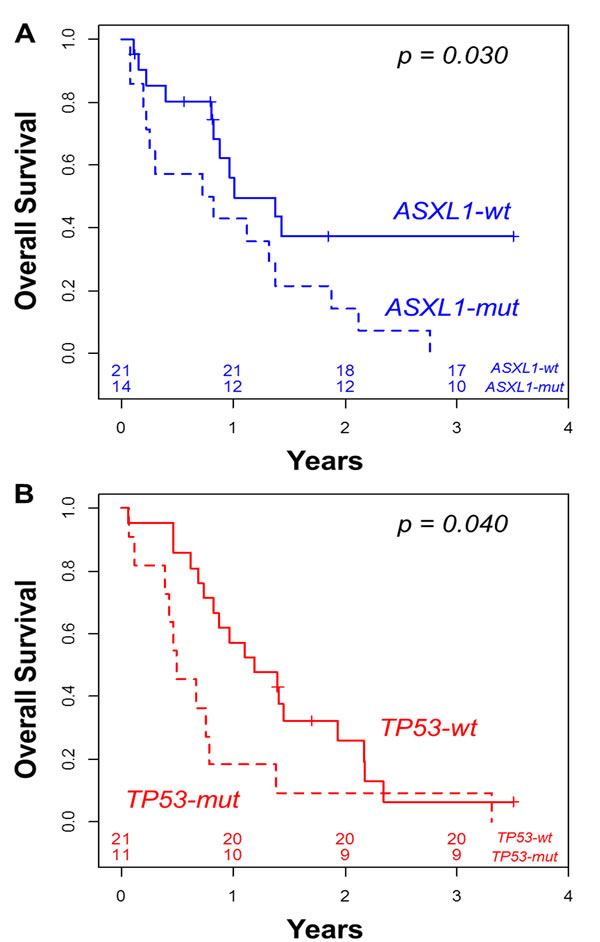
Figure 2: Overall survival according to the presence ASXL1 mutation in the intermediate cytogenetic patients (A) and according to the presence of TP53 mutation in the unfavorable cytogenetic patients (B)
Table 2: Frequencies of mutation in ASXL1, RUNX1, DNMT3A, NPM1, FLT3-ITD and TP53 in the 125 AML-MRC patients and according to the karyotypes (A) and to the cytogenetic risk groups (B)
Outcome after intensive chemotherapy in AML-MRC patients
Among the 125 AML-MRC patients, only the 67 patients who received intensive induction chemotherapy were considered for the outcome analyses. Among them, 42 achieved CR (63%). Cytogenetic risk group, the presence of MDS-related cytogenetics, previous history of MDS or MDS/MPN, and MLD did not influence the CR rate (data not shown). The presence of an ASXL1 mutation was associated with a lower CR rate (ASXL1-mut vs. ASXL1-wt: 40% vs. 69%, p = 0.039). Other gene mutations did not influence the CR rate.
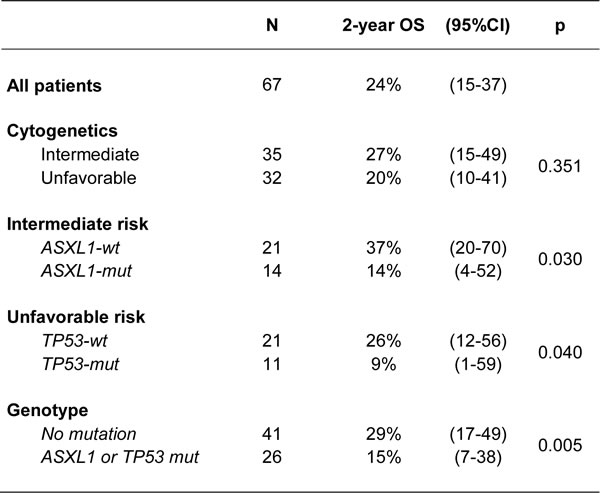
The 2-year OS was 24% in the 67 intensively treated patients. Cytogenetics did not predict outcome, with a 2-year OS of 27% and 20% in the intermediate and unfavorable groups, respectively (p=0.351, Table 3). Similarly, MDS-related cytogenetics, previous history of MDS or MDS/MPN, and MLD did not significantly influence OS (data not shown). Among the intermediate cytogenetic (IC-AML) patients, the presence of an ASXL1 mutation was associated with worse 2-year OS (14%) compared to patients without ASXL1 mutation (37%, p = 0.030, Figure 2A, Table 3).
In the unfavorable cytogenetic (UC-AML) group, TP53-mutated patients had a lower 2-year OS (9%) than TP53-wild type patients (26%, p = 0.040, Figure 2B, Table 3). In multivariate analyses adjusted for age and WBC, IC-AML with mutated ASXL1 (HR = 2.67, 95%CI = [1.15-6.24], p = 0.023) and UC-AML with mutated TP53 (HR = 5.44, 95%CI = [2.16-13.65], p < 0.001) had shorter OS than IC-AML with wild type ASXL1 (considered as reference, HR = 1). Of note, patients with UC-AML with wild type TP53 (HR = 1.14, 95%CI = [0.52-2.50], p = 0.743) had similar OS compared to those with intermediate cytogenetic and no ASXL1 mutation (considered as reference, HR = 1).
Using only the mutational status of ASXL1 and TP53 to stratify patient outcome, we found that patients who presented with either ASXL1 or TP53 mutation had worse 2-year OS (15%) than those with both ASXL1 and TP53 wild type (29%, p = 0.005, Table 3, Figure 3). In multivariate analysis including age, WBC and cytogenetics, the presence of either ASXL1 or TP53 mutation remained the only independent predictive factor associated with both lower CR rate (HR = 0.29, 95%CI = [0.09-0.89], p = 0.031) and shorter OS (HR = 2.53, 95%CI = [1.40-4.60], p = 0.002).
Table 3: Univariate analyses of overall survival
Patients with dysplasia but without criteria for MLD
These 24 patients were separately analyzed because they did not reach AML-MRC criteria after morphological review. Indeed, they had no previous history of MDS or MDS/MPN and no MDS-related cytogenetics. Morphological review showed dysplasia that did not reach criteria for MLD. All of them had intermediate cytogenetics and 15 (63%) had normal karyotype (Supplemental Table 1). Among these 24 patients, median percentage of DGP, DEP and DMP was 50% (range: 7-97%), 20% (range: 0-45%) et 26% (range: 0-83%), respectively. We found 4 mutations in ASXL1 (17%), 5 in RUNX1 (21%), 1 in DNMT3A (4%), 2 in NPM1 (8%), 4 in FLT3 (17%) and no TP53 mutation. ASXL1 mutated patients had a median percent of DGP of 62% (range: 35-90%) vs. 50% (range: 7-97%) in those with wild type ASXL1.
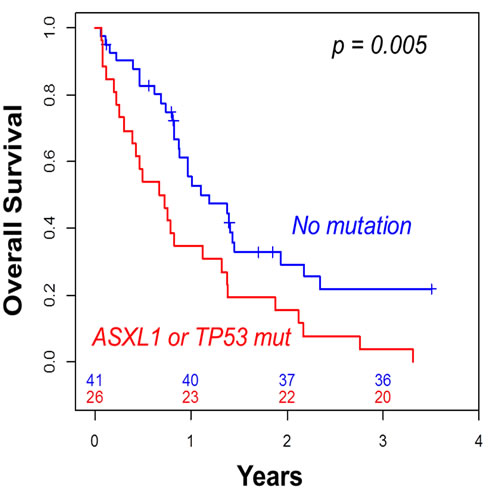
Figure 3: Overall survival according to the presence of ASXL1 or TP53 mutation
Discussion
We previously reported that AMLs with MRC harbor a specific mutational profile with a high proportion of ASXL1 and RUNX1 mutations and less DNMT3A, FLT3 and NPM1 mutations than AMLs without criteria of AML-MRC [13]. In this series including only patients with AML-MRC, we have evaluated the correlation between these mutations and the different criteria defining AML-MRC (previous history of MDS or MDS/MPN, MDS-related cytogenetics, and MLD). We found that none of the three MRC criteria could help identify a specific mutational profile within AML-MRC, suggesting that these criteria do not have a molecular basis, at least within the limit of the genes studied. This heterogeneity in terms of biological features as well as in prognostic significance suggests that AML-MRC is not a true distinct entity. Although they are defined as AML-MRC because of MDS-related cytogenetics, CK-AML should be considered separately because of the presence of a specific mutational profile consisting in a high frequency of TP53 mutations and the absence of other mutations. As expected, TP53 mutations predicted a worse outcome among patients with unfavorable cytogenetics, supporting that genetic stratification is useful to predict differential patient outcome although classified in the same cytogenetic risk group. This is in line with previous reports showing the close correlation between TP53 mutation, CK-AML and poor outcome [18, 19]. In NCK-AML, we found that ASXL1 was the most frequently mutated gene (31%). It was the only mutation associated with the presence of MLD and a higher proportion of DGP in bone marrow. This is in agreement with previous biological reports showing the role of ASXL1 in the appearance of dysplasia and myeloid transformation [20-23]. However, the overlap between ASXL1 mutations and morphological MLD was not complete, because 39% of patients with MLD had ASXL1 mutation and 14% patients without MLD had ASXL1 mutation. Thus, there is no strong association between morphological analyses and the presence of an ASXL1 mutation. Interestingly, ASXL1 mutations were associated with the absence of MDS-related cytogenetics among the NCK-AML, as if ASXL1 mutations and karyotype with MDS-related abnormalities were mutually exclusive. Finally, there was no correlation between ASXL1 mutation and previous history of MDS or MDS/MPN.
Taken together, these results suggest that the presence of an ASXL1 mutation could be considered as an independent molecular marker of dysplasia in AML that is not redundant with the criteria defining AML-MRC. This could be helpful in the perspective of developing a molecular classification of AML-MRC. This statement is also supported by the presence of a specific gene expression profile associated with ASXL1-mutated AML [24]. Like TP53 stratification of outcome in UC-AML, ASXL1 mutations were associated with a shorter OS in intermediate risk patients, with a 2-year OS (14%) close to that observed in unfavorable cytogenetic patients (20%). Different studies that did not specifically focus on AML-MRC also reported this poor outcome associated to ASXL1 mutations [25, 26]. Thus, stratification upon ASXL1 and TP53 mutation, which identified two distinct biological subgroups among AML-MRC, was the only significant predictor of outcome while usual cytogenetic risk classification or the different criteria defining AML-MRC (prior MDS or MDS/MPN, MDS-related cytogenetics and MLD) failed to predict patient outcome in our series. Because of the limited number of patients in some subgroups, these results need to be confirmed in larger studies.
Although ASXL1 and TP53 mutations could classify distinct subgroups of AML-MRC, patients without any of these mutations still represent a heterogeneous group of AML-MRC harboring different morphological, cytogenetic and molecular features. The mutations of DNMT3A and/or NPM1, which are usually found in de novo AML and are mutually exclusive with ASXL1 and TP53 mutations, could identify patients for whom the definition of AML-MRC should be questioned. Falini et al. reported that MLD did not identify distinct biological and clinical entities among NPM1-mutated AML, with overlapping gene expression profiling and similar outcome [4]. In contrast, some patients had morphological dysplasia but not enough to reach MLD criteria, leading to their exclusion from the AML-MRC category in the absence of other criteria. Among these patients, those who presented with ASXL1 mutations might be included in the same subgroup as ASXL1-mutated AML-MRC. This supports the need of redefining AML-MRC upon molecular abnormalities.
Finally, we did not find any mutation in the 6 genes studied in 47 patients, suggesting the need to identify other molecular markers to stratify these patients. Because ASXL1 is a key regulator of the polycomb repressive complex 2 (PRC2), it is possible that AML carrying alterations of other genes involved in methylation marks via PRC2 and myeloid transformation such as EZH2 [27, 28], JARID2 [29] or BAP1 [30, 31] could share characteristics of ASXL1-mutated AML. This could help identify AML-MRC through a common altered molecular pathway and could help develop future targeted treatments. An extensive mutational screening using whole genome or exome sequencing could be useful in this setting to improve the molecular characterization of these unmutated cases.
We conclude that the criteria defining AML-MRC do not identify distinct clinical and biological subgroups and do not predict outcome of patients with AML-MRC. In contrast, ASXL1 and TP53-mutated AML identify two distinct biological subgroups of AML-MRC with very poor outcome. This molecular characterization could be useful to redefine AML-MRC in a future classification aiming at merging biological characterization and specific prognostic value.
Patients and Methods
Selection criteria
We conducted a retrospective analysis of patients from two French centers. Selection criteria for analyses were: (i) Diagnosis of AML with criteria for AML-MRC according to the WHO classification[1]: previously diagnosed MDS or MPN/MDS; and/or multilineage dysplasia (MLD); and/or MDS-related cytogenetic abnormalities (complex karyotype (CK) defined by three or more chromosomal abnormalities, -7 or del(7q); -5 or del(5q); i(17q) or t(17p); -13 or del(13q); del(11q); del(12p) or t(12p); del(9q); idic(X)(q13); t(11;16)(q23;p13.3); t(3;21)(q26.2;q22.1); t(1;3)(p36.3;q21.1); t(2;11)(p21;q23); t(5;12)(q33;p12); t(5;7)(q33;q11.2); t(5;17)(q33;p13); t(5; 10)(q33;q21); t(3;5)(q25;q34); (ii) Genomic DNA available for mutational analyses.
AML with inv(3)/t(3;3), t(6;9) and t(v;11q23) or with favorable risk according to the ELN classification (inv(16)/t(16;16), t(8;21), t(15;17), normal karyotype AML with NPM1 or CEBPA mutations) were excluded as well as therapy-related AML [14].
Morphological analyses
Smears of bone marrow aspirates made at diagnosis were collected and stained by May-Grünwald-Giemsa. Each smear was retrospectively analyzed by two expert cytologists in both centers. We assessed the percentage of dysplastic cells in each lineage. MLD was assessed according to the WHO classification criteria: at least 50% of dysplastic cells in at least 2 lineages. We established the percentage of dysgranulopoiesis (DGP), dyserythropoiesis (DEP) and dysmegakaryopoiesis (DMP) for each smear.
Mutational analyses
Direct sequencing was done using the Sanger method as previously described [15]. We searched for mutation of ASXL1 (exon 12), RUNX1 (exon 1-8), DNMT3A (exon 15-23), NPM1 (exon 12), FLT3 (internal tandem duplication ITD, exon 14-15) and TP53 (exon 4-10).
Statistical analyses
Chi-square or Fischer tests were used to compare categorical variables. We used non-parametric test (U Mann Whitney) to compare the median percentage of dysplastic cells in each lineage according to mutational status for each gene. As multivariate model, linear regression was used to find correlation between morphologic dysplasia and the presence of mutation. Survivals were calculated using the Kaplan Meier estimator [16]. Time to event started from the date of diagnosis. We compared survivals using the Log-Rank test. Cox regression was used for multivariate of OS [17]. Statistics were computed using the R.3.1.0 software.
Acknowledgements
This work was supported by Inserm, Institut Paoli-Calmettes and grants from the Fondation ARC pour la Recherche sur le Cancer (DB) and INCa-DGOS-Inserm 6038.
Conflicts of interest
The authors have nothing to disclose.
References
1. Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, Harris NL, Le Beau MM, Hellström-Lindberg E, Tefferi A, Bloomfield CD. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5):937-51.
2. Haferlach T, Schoch C, Löffler H, Gassmann W, Kern W, Schnittger S, Fonatsch C, Ludwig W-D, Wuchter C, Schlegelberger B, Staib P, Reichle A, et al. Morphologic Dysplasia in De Novo Acute Myeloid Leukemia (AML) Is Related to Unfavorable Cytogenetics but Has No Independent Prognostic Relevance Under the Conditions of Intensive Induction Therapy: Results of a Multiparameter Analysis From the German AML Cooperative Group Studies. J Clin Oncol. 2003;21(2):256-65.
3. Miesner M, Haferlach C, Bacher U, Weiss T, Macijewski K, Kohlmann A, Klein H-U, Dugas M, Kern W, Schnittger S, Haferlach T. Multilineage dysplasia (MLD) in acute myeloid leukemia (AML) correlates with MDS-related cytogenetic abnormalities and a prior history of MDS or MDS/MPN but has no independent prognostic relevance: a comparison of 408 cases classified as « AML not otherwise specified » (AML-NOS) or « AML with myelodysplasia-related changes » (AML-MRC). Blood. 2010;116(15):2742-51.
4. Falini B, Macijewski K, Weiss T, Bacher U, Schnittger S, Kern W, Kohlmann A, Klein H-U, Vignetti M, Piciocchi A, Fazi P, Martelli MP, et al. Multilineage dysplasia has no impact on biologic, clinicopathologic, and prognostic features of AML with mutated nucleophosmin (NPM1). Blood. 2010;115(18):3776-86.
5. Bacher U, Schnittger S, Macijewski K, Grossmann V, Kohlmann A, Alpermann T, Kowarsch A, Nadarajah N, Kern W, Haferlach C, Haferlach T. Multilineage dysplasia does not influence prognosis in CEBPA-mutated AML, supporting the WHO proposal to classify these patients as a unique entity. Blood. 2012;119(20):4719-22.
6. Rozman M, Navarro J-T, Arenillas L, Aventín A, Giménez T, Alonso E, Perea G, Camós M, Navarrete M, Tuset E, Florensa L, Millá F, et al. Multilineage dysplasia is associated with a poorer prognosis in patients with de novo acute myeloid leukemia with intermediate-risk cytogenetics and wild-type NPM1. Ann Hematol. 2014;
7. Devillier R, Gelsi-Boyer V, Murati A, Prebet T, Rey J, Etienne A, D’Incan E, Charbonnier A, Blaise D, Mozziconacci M-J, Vey N. Prognostic significance of myelodysplasia-related changes according to the WHO classification among ELN-intermediate-risk AML patients. Am J Hematol. 2014;
8. Wandt H, Schäkel U, Kroschinsky F, Prange-Krex G, Mohr B, Thiede C, Pascheberg U, Soucek S, Schaich M, Ehninger G. MLD according to the WHO classification in AML has no correlation with age and no independent prognostic relevance as analyzed in 1766 patients. Blood. 2008;111(4):1855-61.
9. Xu X-Q, Wang J-M, Gao L, Qiu H-Y, Chen L, Jia L, Hu X-X, Yang J-M, Ni X, Chen J, Lü S-Q, Zhang W-P, et al. Characteristics of acute myeloid leukemia with myelodysplasia-related changes: A retrospective analysis in a cohort of Chinese patients. Am J Hematol. 2014;
10. Hou HA, Lin CC, Chou WC, Liu CY, Chen CY, Tang JL, Lai YJ, Tseng MH, Huang CF, Chiang YC, Lee FY, Kuo YY, et al. Integration of cytogenetic and molecular alterations in risk stratification of 318 patients with de novo non-M3 acute myeloid leukemia. Leukemia. 2014;28(1):50-8.
11. Patel JP, Gönen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, Van Vlierberghe P, Dolgalev I, Thomas S, Aminova O, Huberman K, Cheng J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366(12):1079-89.
12. Grossmann V, Schnittger S, Kohlmann A, Eder C, Roller A, Dicker F, Schmid C, Wendtner C-M, Staib P, Serve H, Kreuzer K-A, Kern W, et al. A novel hierarchical prognostic model of AML solely based on molecular mutations. Blood. 2012;120(15):2963-72.
13. Devillier R, Gelsi-Boyer V, Brecqueville M, Carbuccia N, Murati A, Vey N, Birnbaum D, Mozziconacci M-J. Acute myeloid leukemia with myelodysplasia-related changes are characterized by a specific molecular pattern with high frequency of ASXL1 mutations. Am J Hematol. 2012;87(7):659-62.
14. Döhner H, Estey EH, Amadori S, Appelbaum FR, Büchner T, Burnett AK, Dombret H, Fenaux P, Grimwade D, Larson RA, Lo-Coco F, Naoe T, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115(3):453-74.
15. Gelsi-Boyer V, Trouplin V, Adélaïde J, Bonansea J, Cervera N, Carbuccia N, Lagarde A, Prebet T, Nezri M, Sainty D, Olschwang S, Xerri L, et al. Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br J Haematol. 2009;145(6):788-800.
16. Kaplan E, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53(282):457-81.
17. Cox D. Regression models and life tables. J R Stat Soc Ser B Methodol. 1972;34(2):187-220.
18. Rücker FG, Schlenk RF, Bullinger L, Kayser S, Teleanu V, Kett H, Habdank M, Kugler C-M, Holzmann K, Gaidzik VI, Paschka P, Held G, et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood. 2012;119(9):2114-21.
19. Bowen D, Groves MJ, Burnett AK, Patel Y, Allen C, Green C, Gale RE, Hills R, Linch DC. TP53 gene mutation is frequent in patients with acute myeloid leukemia and complex karyotype, and is associated with very poor prognosis. Leukemia. 2009;23(1):203-6.
20. Abdel-Wahab O, Adli M, LaFave LM, Gao J, Hricik T, Shih AH, Pandey S, Patel JP, Chung YR, Koche R, Perna F, Zhao X, et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell. 2012;22(2):180-93.
21. Abdel-Wahab O, Gao J, Adli M, Dey A, Trimarchi T, Chung YR, Kuscu C, Hricik T, Ndiaye-Lobry D, Lafave LM, Koche R, Shih AH, et al. Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo. J Exp Med. 2013;210(12):2641-59.
22. West RR, Hsu AP, Holland SM, Cuellar-Rodriguez J, Hickstein DD. Acquired ASXL1 mutations are common in patients with inherited GATA2 mutations and correlate with myeloid transformation. Haematologica. 2014;99(2):276-81.
23. Inoue D, Kitaura J, Togami K, Nishimura K, Enomoto Y, Uchida T, Kagiyama Y, Kawabata KC, Nakahara F, Izawa K, Oki T, Maehara A, et al. Myelodysplastic syndromes are induced by histone methylation–altering ASXL1 mutations. J Clin Invest. 2013;123(11):4627-40.
24. Metzeler KH, Becker H, Maharry K, Radmacher MD, Kohlschmidt J, Mrózek K, Nicolet D, Whitman SP, Wu Y-Z, Schwind S, Powell BL, Carter TH, et al. ASXL1 mutations identify a high-risk subgroup of older patients with primary cytogenetically normal AML within the ELN Favorable genetic category. Blood. 2011;118(26):6920-9.
25. Schnittger S, Eder C, Jeromin S, Alpermann T, Fasan A, Grossmann V, Kohlmann A, Illig T, Klopp N, Wichmann H-E, Kreuzer K-A, Schmid C, et al. ASXL1 exon 12 mutations are frequent in AML with intermediate risk karyotype and are independently associated with an adverse outcome. Leukemia. 2013;27(1):82-91.
26. Chou W-C, Huang H-H, Hou H-A, Chen C-Y, Tang J-L, Yao M, Tsay W, Ko B-S, Wu S-J, Huang S-Y, Hsu S-C, Chen Y-C, et al. Distinct clinical and biological features of de novo acute myeloid leukemia with additional sex comb-like 1 (ASXL1) mutations. Blood. 2010;116(20):4086-94.
27. Wang X, Dai H, Wang Q, Wang Q, Xu Y, Wang Y, Sun A, Ruan J, Chen S, Wu D. EZH2 Mutations Are Related to Low Blast Percentage in Bone Marrow and -7/del(7q) in De Novo Acute Myeloid Leukemia. Richards KL, éditeur. PLoS ONE. 2013;8(4):e61341.
28. Xu F, Li X, Wu L, Zhang Q, Yang R, Yang Y, Zhang Z, He Q, Chang C. Overexpression of the EZH2, RING1 and BMI1 genes is common in myelodysplastic syndromes: relation to adverse epigenetic alteration and poor prognostic scoring. Ann Hematol. 2011;90(6):643-53.
29. Puda A, Milosevic JD, Berg T, Klampfl T, Harutyunyan AS, Gisslinger B, Rumi E, Pietra D, Malcovati L, Elena C, Doubek M, Steurer M, et al. Frequent deletions of JARID2 in leukemic transformation of chronic myeloid malignancies. Am J Hematol. 2012;87(3):245-50.
30. Abdel-Wahab O, Dey A. The ASXL-BAP1 axis: new factors in myelopoiesis, cancer and epigenetics. Leukemia. 2013;27(1):10-5.
31. Dey A, Seshasayee D, Noubade R, French DM, Liu J, Chaurushiya MS, Kirkpatrick DS, Pham VC, Lill JR, Bakalarski CE, Wu J, Phu L, et al. Loss of the tumor suppressor BAP1 causes myeloid transformation. Science. 2012;337(6101):1541-6.