
CHROMOSOME 8P23 AND HUMAN CANCERS
Inactivation of tumor suppressor genes due to gene alterations, notably loss of heterozygosity (LOH), plays a major role in the development of common human adult cancers [1, 2], with breast cancer as a good example [3-8]. Chromosome 8p23 is one of the most frequent LOH regions in common human adult epithelial malignancies, including breast, liver, lung, and gastrointestinal cancers [9-26]. For example, up to 70% of hepatocellular carcinomas [9-14] and 60% of human gastric cancer [26] exhibit LOH at 8p23 near the maker D8S277. 8p23 is also a common integration site for hepatitis B virus (HBV), a well-known major risk factor in liver cancer [10, 27]. Similarly, LOH on 8p is found in up to 50% of breast carcinomas and is often associated with advanced tumor stage and aggressive histology [15-19]. Although several potential tumor suppressors have been mapped to this region, including Nkx3.1 at 8p21 and FEZ1/LZTS1 at 8p22 [21, 28-30], even the combined rates of loss of these genes could not account for the extensive alterations seen in human tumors [21], indicating that major tumor suppressor gene(s) remain to be identified.
Telomerase activation and cancer
Maintaining optimal telomere length is crucial for cells since excessive telomere loss leads to telomere fusions, anaphase bridges, aberrant mitotic chromosome separation and aneuploidy [31-34]. Significantly, these telomere dysfunction phenotypes are very common in human cancer, especially epithelial cancers [34-38]. Moreover, long-term lack of telomerase in mice leads to telomere shortening and dysfunction, and promotes oncogenesis in a certain condition such as lack of tumor suppressors p53 [34, 39-43] or DNA mismatch repair protein MSH2 [44] or in the presence of telomeric repeat binding factor TRF2 overexpression [45], and also shifts the tumor spectrum of p53 mutant mice to include more epithelial cancers [42]. Thus, telomere dysfunction caused by excessive telomere loss has been proposed as a major cause of chromosome instability in epithelial carcinogenesis [35, 38, 42].
However, it has been well documented that telomere shortening due to knockout of telomerase activates DNA damage pathways, limits cell proliferation and inhibits tumorigenesis in a number of mouse models [46-50]. Moreover, although telomere loss in cancers has well documented, telomere elongation is quite common in a variety of epithelial cancers, e.g. in >40% of liver cancer [51], esophageal cancer [52] and brain tumors [53], and also correlates with advanced stages and/or poor survival in some cancers [51, 52, 54-57]. Even longer telomeres in blood cells are shown to be associated with higher cancer risk and poor survival in breast cancer [58]. However, little is known about the consequence of aberrant telomere elongation.
In mammalian cells, telomere surveillance is carried out in cis by shelterins, which include three telomeric DNA-binding proteins TRF1, TRF2 and POT1, and their associating proteins [59]. While TRF2 [32, 60] and POT1 [60-62] protect telomere integrity, TRF1 maintains telomeres at the optimal length [32, 59, 63]. Indeed, many TRF1-interacting or associated proteins influence telomere length [63-67]. We identified TRF1 as Pin2 in the same screen for Pin1 [68, 69]. Both Pin1 and Pin2/TRF1 are important for mitotic regulation [69-73] and Pin1 directly regulates TRF1 function in telomere maintenance and aging [74]. By identifying new TRF1/Pin2-interacting proteins, including PinX1-4, we have further identified important TRF1 function in telomere maintenance and mitosis [66, 74-76].
PINX1 IS A TELOMERASE INHIBITOR ESSENTIAL FOR MAINTAINING TELOMERASE ACTIVITY AND TELOMERE LENGTH
Unlike other TRF1-interacting proteins, PinX1 has a unique property to directly bind to TERT and TERC and inhibit telomerase activity at telomere [66, 77] . Furthermore, PinX1 inhibition in cancer cells activates telomerase and elongates telomeres, whereas PinX1 overexpression has the opposite effects [66]. The ability of PinX1 to regulate telomerase and telomere length is also conserved in yeast, rats and fish [78-80]. In fact, yeast PinX1 has been shown to inhibit telomerase by sequestering its protein catalytic subunit in an inactive complex lacking telomerase RNA in the nucleolus [78]. However, it was not clear why such telomerase inhibitor is needed until recently [81, 82].
By performing the extensive structure-function analysis to define the PinX1-TRF1 interaction and to dissect the biological function of the PinX1-TRF1 interaction in regulating telomere maintenance, we have demonstrated that the TRF1-PinX1 interaction is required not only for targeting PinX1 to telomeres, but also for PinX1 to prevent abnormal telomere elongation in cells [81]. Given the unique and potent ability of PinX1 to inhibit telomerase activity and telomere elongation in cells [66, 83], our results suggest that the TRF1-PinX1 interaction affects the loading of PinX1 onto telomeres to prevent telomere elongation [81]. This may provide an additional level of telomerase regulation, in conjunction to the physical occlusion from telomeres by Pot1 [67, 84], by supplying a link between TRF1 and telomerase inhibition that contributes toward maintaining telomere homeostasis (Fig. 1). TRF1 binds along the duplex part of the telomere and functions as a measuring device to assess telomere length [59, 63, 85, 86]. For telomere length homeostasis to be effective, the information about the length of the telomere needs to be relayed from TRF1 to telomerase via other proteins since TRF1 does not directly affect telomerase activity [59, 63]. Our data indicate that the telomerase inhibitor PinX1 might be recruited by TRF1 to a telomere to stop telomerase action as the telomere is being elongated and reaches a certain threshold [81].
This model (Fig. 1) has been recently supported by manipulating PinX1 expression in vitro and in vivo to demonstrate that PinX1 is a rate-limiting physiological regulator of telomerase and telomere length [82]. We have shown that reduced PinX1 expression activates telomerase and leads to telomere lengthening and chromosome instability, which can be fully rescued by TERT knockdown or knockout [82], confirming that PinX1 acts as a telomerase inhibitor to regulate telomere maintenance. The significance of these results is demonstrated by our observations that most PinX1+/- mice develop aggressive cancers that display telomere lengthening and chromosome instability [82]. These results provide the first evidence for an essential role of PinX1 in regulating telomerase activity and telomere length [82].
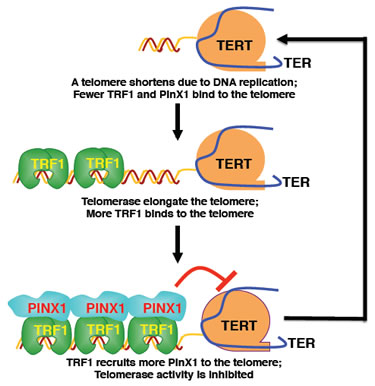
Figure 1: The role of PinX1 in maintaining telomeres at the optimal length. When telomerase extends a telomere to a certain length, the elongated telomere binds more TRF1, which might in turn recruit more PinX1 to the telomere. Once concentrated locally on telomeres at a high concentration, simple mass-action might allow PinX1 to more effectively stop telomerase from adding more repeats. Conversely, when a telomere is shortened following each cell division due to the end replication problem, the shortened telomere contains less TRF1, which might recruit fewer PinX1 or none at all to the telomere. Therefore, the telomere might have a greater chance of being elongated due to telomerase being inhibited less, or even not at all. This Pinx1-mediated negative feedback mechanism might help maintain telomeres at a similar median length. (Adapted from Soohoo,et al., 2011, J Biol Chem 286:3894-3906).
PINX1 IS A SOUGHT-AFTER MAJOR TUMOR SUPPRESSOR AT 8P23 ESSENTIAL FOR CHROMOSOME STABILITY
Notably, PinX1 gene localizes to human chromosome 8p23 [13, 66], which is one of the regions that experiences the most frequent loss of heterozygosity (LOH) in many common human cancers [9, 10, 12, 15, 19, 21, 25, 26]. For example, 8p23 LOH has been found in up to 70% of hepatocellular carcinomas [9, 10, 12, 13], 60% of gastric cancer [26], and 50% of breast cancer [15, 19]. This region is also a common integration site for hepatitis B virus, a well-known major risk factor in liver cancer [10] and PinX1 is reduced in ~ 40% hepatitis B virus-related liver cancer [13]. Moreover, PinX1 inhibition potently increases, whereas PinX1 overexpression suppresses, tumorigenicity of cancer cells [66]. Thus, PinX1 might be a putative tumor suppressor. Subsequent PinX1 studies on human cancer samples are inconclusive [26, 87]. Moreover, there is no genetic evidence for any involvement of PinX1 in cancer.
The recent results from my laboratory indicate that PinX1 is a major tumor suppressor at 8p23 [82]. PinX1 is downregulated in a large subset of human breast cancer tissues and in most breast cancer cell lines examined [82]. PinX1 expression is gene dosage-dependent; ablation of one allele reduces protein level by 60-70% in vitro and in vivo. Importantly, reducing PinX1 by gene knockout or knockdown not only increases telomerase activity and telomere length, but also leads to chromosome instability in cell models [82]. These telomere-related phenotypes are fully reversed by knockout or knockdown of telomerase. Moreover, telomere lengthening induced by PinX1 inhibition occurs at most chromosome ends, a feature of cancer cells [88]. The significance of these findings is further substantiated by the demonstrations that nearly all PinX1+/- mice develop a range of epithelial malignancies with evidence of telomere lengthening and chromosome instability.
Although most human cancers are epithelial carcinomas, common tumor suppressor mutant mice mainly develop lymphomas and soft tissue sarcomas, with a very few exceptions [89, 90]. Notably, most tumors in PinX1+/- mice are epithelial carcinomas arising in organs that are known to develop common cancers in humans, including lung, mammary, liver and gastrointestinal tract cancers [82], which are also known to have frequent LOH at 8p23 in humans [9, 10, 12, 20-22, 24, 25]. Most tumors showed features commonly seen in advanced human carcinomas such as nuclear atypia, desmoplasia, stromal invasion and/or lung metastasis [82]. Moreover, 20% of PinX1+/- mice developed two or three cancer types within the same animals. Even within one tumor type, there were diverse histopathologies in different mice, the same mice or even within the same tumors [82]. Thus, almost all PinX1+/- mice spontaneously develop a range of aggressive epithelial cancers [82], which are unusual in mice, even after deleting many other tumor suppressors [89, 90], but are known to have 8p23 LOH in humans. These results suggest that PinX1+/- cancers likely originate from multiple cells and behave aggressively. Given activation of telomerase in most human cancers and common downregulation of PinX1 in liver, gastric and breast cancers [13, 26, 82], these results indicate that PinX1 is a major tumor suppressor, whose downregulation activates telomerase, induces chromosome instability and eventually leads to tumorigenesis by [82].
HOW DOES PINX1 REGULATE CHROMOSOME STABILITY AND TUMORIGENESIS?
At the present time, we do not yet know the molecular mechanism by which reducing PinX1 function leads to chromosome instability and tumorigenesis. Notably, we have shown that the PinX1 knockout phenotypes including telomerase activation, telomere elongation, anaphase bridges, aneuploidy and chromosome instability are fully suppressed by knockdown or knockout of TERT or TERC, indicating that telomerase is essential for PinX1 reduction to induce chromosome instability [82]. Moreover, it takes time for PinX1-induced telomerase activation to induce telomere elongation and chromosome instability when PinX1 is knocked out or down [82]. Notably, the PinX1 and p53 double mutant mice have similar tumor spectrum [82] that are found in TERC and p53 double mutant mice due to telomere loss [42] or in TPP1/ACD and p53 double mutant mice due to telomere deprotection [91]. Furthermore, abnormal telomere elongation is common and also correlates with advanced stages and/or poor survival in some cancers [51, 52, 54, 57]. Moreover, TERC is required for the tumor-promoting effects of TERT overexpression in transgenic mice [92]. These results together suggest that abnormal telomerase activation and telomere lengthening due to loss of PinX1 might have similar effects on the development of epithelial cancers, as does telomere shortening or telomere deprotection. Given that PinX1 directly binds to and inhibits TERT [66] and is targeted by TRF1 to telomeres to prevent abnormal telomere elongation by telomerase [93], it is conceivable that when PinX1 is inhibited, telomerase is aberrantly activated without a proper brake and eventually leads to chromosome instability possibly via inducing aberrant telomere elongation to compromise telomere function [82]. Consistent with this idea is the previous findings that loss of p80/p95 in Tetrahymena induces telomere lengthening and chromosome instability [94]. However, telomerase has other telomere-independent function such as in DNA damage response [95, 96] and activating β-catenin [97]. Similarly, PinX1 might have non-telomeric functions such as in RNA maturation [98] and cell division [99]. Moreover, it has been shown that TERT overexpression extends life span in normal human cells in vitro [100-102] and that short-term telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice [103], although long-term TERT overexpression promotes tumorigenesis in vitro or in vivo [92, 104-110]. These results suggest that long-term loss of PinX1 function and/or telomerase overexpression might affect other cellular processes that contribute to chromosome instability and tumorigenesis. Therefore, further experiments are needed to define how PinX1 controls chromosome stability via telomere-dependent and/or -independent telomerase and/or other mechanisms unrelated to telomerase [82].
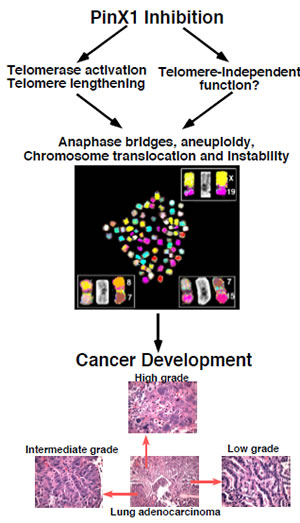
Figure 2: PinX1 as a sought-after major haploinsufficient tumor suppressor at 8p23 that is essential for chromosome stability. The human PinX1 gene is located at 8p23, a frequent LOH in common human epithelial cancers and its protein expression is often down-regulated in many human cancer tissues and cell lines. While PinX1 null mice or cells are lethal, reducing PinX1 expression via heterozygous knockout or knockdown increases telomerase activity and leads to telomere lengthening, anaphase bridges, chromosome translocation and instability, as shown in the picture. Moreover, PinX1 heterozygous knockout causes most mice to spontaneously develop a range of malignant tumors displaying evidence of telomere lengthening and chromosomal instability. Notably, the majority of cancers in the PinX1 mutant mice are carcinomas and share tissues of origin with human cancer types linked to 8p23 alterations. Furthermore, many PinX1 heterozygous knockout mice develop more than one type of tumor and diverse histopathologies are observed in the same types of tumors among different mice or even in the same tumors, as shown for lung cancer. (Adapted from Zhou,et al., 2011, J Clin Invest 121:1266-1282)
Although it is not clear how PinX1 downregulation leads to chromosome instability, we have demonstrated that PinX1-induced chromosome instability plays a major and novel role in tumorigenesis [82]. We show that PinX1 is reduced in most human breast cancer tissues and cells and that reducing PinX1 levels leads to telomerase activation, telomere elongation and chromosome instability [82]. Moreover, almost all PinX1 heterozygous knockout mice develop a range of epithelial malignancies, with multiple tumor types in the same mice, diverse cell morphologies/grades in one tumor type among mice, or even within individual tumors [82]. PinX1 heterozygous knockout also shifts the p53 mutant tumor spectrum to epithelial carcinomas [82]. Importantly, PinX1+/- cancer cells also display chromosome instability, similar to those in PinX1-inhibited cells [82]. Thus, PinX1 mutant tumors are likely derived from multiple epithelial cells, presumably due to chromosome instability. These results are consistently previous findings that PinX1 potently controls tumorigenicity of cancer cells [66], and that telomerase overexpression promotes tumorigenesis, but is not as oncogenic as PinX1 knockout. Notably, telomerase activation and telomere lengthening induced by due to lack of PinX1 [82] and telomere dysfunction due to telomere shortening and deprotection share some common features including anaphase bridges, aneuploidy, translocation and chromosome instability both in MEFs and tumors. However, unlike the latter, the former induces chromosome instability in the presence of functional p53 and the absence of TIF (telomere dysfunction-induced focus) [82]. These results suggest that downregulation of PinX1 induces chromosome instability and tumorigenesis without activating DNA damage pathways [82].
PINX1 IS AN ATTRACTIVE NEW TARGET IN CANCER THERAPY
An increasing body of evidence suggests that PinX1 is an attractive new target in cancer therapy. We have originally shown that whereas reducing PinX1 in human cancer cells increases their tumorigenicity, overexpression of PinX1, especially its small TID domain (telomerase-inhibitory domain) drives cancer cells into crisis and potently suppresses their ability to form tumors in mice [66]. These findings have now been confirmed and expanded to many different human cancer cells by transducing recombinant full length PinX1 protein or its protein fragments containing TID into cancer cells using HIV-Tat-mediated delivery [111] or mBAFF-targeted delivery [112]. Interestingly, overexpression of PinX1 also enhances the sensitivity of human cancer cells to chemotherapy drug 5-fluorouracil [113]. Furthermore, human PinX1 is located to 8p23 [13, 66], a frequent LOH region in common human epithelial cells [9, 10, 12, 15, 19, 21, 25, 26], and PinX1 protein is indeed downregulated commonly in many human epithelial cancers examined [13, 26, 66, 82]. Moreover, reducing PinX1 levels by heterozygous knockout or knockdown leads to chromosome instability and tumorigenesis in vitro and in vivo, with most tumors in PinX1+/- mice being carcinomas and sharing tissues of origin with human cancer types linked to 8p23 [82]. The therapeutic significance of these findings is further substantiated by the demonstration that the ability of PinX1 knockout or knockdown to induce chromosome instability is fully blocked by TERT knockout or knockdown [82]. Indeed, inhibition of telomerase by a wide range of approaches including chemical inhibitors or vaccines have been shown to potently inhibit cancer cell growth and even cancer stem cells in vitro, in vivo, with promising results in clinical trials [114, 115]. Therefore, these results together indicate that inhibition of telomerase by PinX1, especially its small telomerase inhibitory domain might be used to treat cancers that have telomerase activation.
Conclusion and future directions
Originally identified as a TRF1/Pin2-binding protein, PinX1 is a potent telomerase inhibitor. Recent results have not only demonstrated that PinX1 is essential for maintaining telomeres at the optimal length, but also discovered that Pinx1 is a sought-after major tumor suppressor at human chromosome 8p23 that is essential for maintaining chromosome stability in vitro and in vivo. Moreover, emerging evidence suggest that PinX1 and especially its small telomerase inhibitory domain might be a potential new drug target for treat cancers that have telomerase activation. However, many questions remained to be addressed including how PinX1 regulates chromosome stability and tumorigenesis, how PinX1 is regulated under physiological and pathological conditions, how to develop PinX1-based cancer therapy, and whether PinX1 has other new functions. Further studies on this relatively unknown protein would uncover novel insight into the regulation of chromosome stability and tumorigenesis and might eventually lead to new therapies for cancers.
CONFLICT OF INTEREST
The author has no conflict of interests to declare.
Acknowledgements
I am grateful to L. Cantley, P. P. Pandolfi, B. Neel, T. Hunter and K. P. Lu for constructive discussions during our study and the former and current members of the Zhou and Lu laboratories for their important contributions. The work was supported by NIH grant RO1CA122434 and Susan G. Komen for the Cure grant KG100958 to X.Z.Z.
References
1. Cavenee WK, Dryja TP, Phillips RA, Benedict WF, Godbout R, Gallie BL, Murphree AL, Strong LC, White RL: Expression of recessive alleles by chromosomal mechanisms in retinoblastoma. Nature 1983, 305(5937):779-784.
2. Foulkes WD: Inherited susceptibility to common cancers. N Engl J Med 2008, 359(20):2143-2153.
3. Nathanson KL, Wooster R, Weber BL: Breast cancer genetics: what we know and what we need. Nat Med 2001, 7(5):552-556.
4. Hodgson SV, Morrison PJ, Irving M: Breast cancer genetics: unsolved questions and open perspectives in an expanding clinical practice. Am J Med Genet C Semin Med Genet 2004, 129(1):56-64.
5. Hartman AR, Ford JM: BRCA1 and p53: compensatory roles in DNA repair. J Mol Med 2003, 81(11):700-707.
6. Keen JC, Davidson NE: The biology of breast carcinoma. Cancer 2003, 97(3 Suppl):825-833.
7. Moynahan ME: The cancer connection: BRCA1 and BRCA2 tumor suppression in mice and humans. Oncogene 2002, 21(58):8994-9007.
8. Buchholz TA, Weil MM, Story MD, Strom EA, Brock WA, McNeese MD: Tumor suppressor genes and breast cancer. Radiat Oncol Investig 1999, 7(2):55-65.
9. Emi M, Fujiwara Y, Nakajima T, Tsuchiya E, Tsuda H, Hirohashi S, Maeda Y, Tsuruta K, Miyaki M, Nakamura Y: Frequent loss of heterozygosity for loci on chromosome 8p in hepatocellular carcinoma, colorectal cancer, and lung cancer. Cancer Res 1992, 52(19):5368-5372.
10. Becker SA, Zhou YZ, Slagle BL: Frequent loss of chromosome 8p in hepatitis B virus-positive hepatocellular carcinomas from China. Cancer Res 1996, 56(21):5092-5097.
11. Nagai H, Pineau P, Tiollais P, Buendia MA, Dejean A: Comprehensive allelotyping of human hepatocellular carcinoma. Oncogene 1997, 14(24):2927-2933.
12. Pineau P, Nagai H, Prigent S, Wei Y, Gyapay G, Weissenbach J, Tiollais P, Buendia MA, Dejean A: Identification of three distinct regions of allelic deletions on the short arm of chromosome 8 in hepatocellular carcinoma. Oncogene 1999, 18(20):3127-3134.
13. Liao C, Zhao M, Song H, Uchida K, Yokoyama KK, Li T: Identification of the gene for a novel liver-related putative tumor suppressor at a high-frequency loss of heterozygosity region of chromosome 8p23 in human hepatocellular carcinoma. Hepatology 2000, 32(4 Pt 1):721-727.
14. Wong N, Lai P, Pang E, Fung LF, Sheng Z, Wong V, Wang W, Hayashi Y, Perlman E, Yuna S et al: Genomic aberrations in human hepatocellular carcinomas of differing etiologies. Clin Cancer Res 2000, 6(10):4000-4009.
15. Yokota T, Yoshimoto M, Akiyama F, Sakamoto G, Kasumi F, Nakamura Y, Emi M: Localization of a tumor suppressor gene associated with the progression of human breast carcinoma within a 1-cM interval of 8p22-p23.1. Cancer 1999, 85(2):447-452.
16. Wang JC, Radford DM, Holt MS, Helms C, Goate A, Brandt W, Parik M, Phillips NJ, DeSchryver K, Schuh ME et al: Sequence-ready contig for the 1.4-cM ductal carcinoma in situ loss of heterozygosity region on chromosome 8p22-p23. Genomics 1999, 60(1):1-11.
17. Loo LW, Grove DI, Williams EM, Neal CL, Cousens LA, Schubert EL, Holcomb IN, Massa HF, Glogovac J, Li CI et al: Array comparative genomic hybridization analysis of genomic alterations in breast cancer subtypes. Cancer Res 2004, 64(23):8541-8549.
18. Bhattacharya N, Chunder N, Basu D, Roy A, Mandal S, Majumder J, Roychowdhury S, Panda CK: Three discrete areas within the chromosomal 8p21.3-23 region are associated with the development of breast carcinoma of Indian patients. Exp Mol Pathol 2004, 76(3):264-271.
19. Varma G, Varma R, Huang H, Pryshchepava A, Groth J, Fleming D, Nowak NJ, McQuaid D, Conroy J, Mahoney M et al: Array comparative genomic hybridisation (aCGH) analysis of premenopausal breast cancers from a nuclear fallout area and matched cases from Western New York. Br J Cancer 2005, 93(6):699-708.
20. Kishimoto Y, Shiota G, Wada K, Kitano M, Nakamoto K, Kamisaki Y, Suou T, Itoh T, Kawasaki H: Frequent loss in chromosome 8p loci in liver cirrhosis accompanying hepatocellular carcinoma. J Cancer Res Clin Oncol 1996, 122(10):585-589.
21. Lai J, Flanagan J, Phillips WA, Chenevix-Trench G, Arnold J: Analysis of the candidate 8p21 tumour suppressor, BNIP3L, in breast and ovarian cancer. Br J Cancer 2003, 88(2):270-276.
22. Baffa R, Santoro R, Bullrich F, Mandes B, Ishii H, Croce CM: Definition and refinement of chromosome 8p regions of loss of heterozygosity in gastric cancer. Clin Cancer Res 2000, 6(4):1372-1377.
23. Gustafson CE, Wilson PJ, Lukeis R, Baker E, Woollatt E, Annab L, Hawke L, Barrett JC, Chenevix-Trench G: Functional evidence for a colorectal cancer tumor suppressor gene at chromosome 8p22-23 by monochromosome transfer. Cancer Res 1996, 56(22):5238-5245.
24. Sunwoo JB, Sun PC, Gupta VK, Schmidt AP, El-Mofty S, Scholnick SB: Localization of a putative tumor suppressor gene in the sub-telomeric region of chromosome 8p. Oncogene 1999, 18(16):2651-2655.
25. Wong MP, Lam WK, Wang E, Chiu SW, Lam CL, Chung LP: Primary adenocarcinomas of the lung in nonsmokers show a distinct pattern of allelic imbalance. Cancer Res 2002, 62(15):4464-4468.
26. Kondo T, Oue N, Mitani Y, Kuniyasu H, Noguchi T, Kuraoka K, Nakayama H, Yasui W: Loss of heterozygosity and histone hypoacetylation of the PINX1 gene are associated with reduced expression in gastric carcinoma. Oncogene 2005, 24(1):157-164.
27. Kahng YS, Lee YS, Kim BK, Park WS, Lee JY, Kang CS: Loss of heterozygosity of chromosome 8p and 11p in the dysplastic nodule and hepatocellular carcinoma. J Gastroenterol Hepatol 2003, 18(4):430-436.
28. Hamaguchi M, Meth JL, von Klitzing C, Wei W, Esposito D, Rodgers L, Walsh T, Welcsh P, King MC, Wigler MH: DBC2, a candidate for a tumor suppressor gene involved in breast cancer. Proc Natl Acad Sci U S A 2002, 99(21):13647-13652.
29. Ishii H, Vecchione A, Murakumo Y, Baldassarre G, Numata S, Trapasso F, Alder H, Baffa R, Croce CM: FEZ1/LZTS1 gene at 8p22 suppresses cancer cell growth and regulates mitosis. Proc Natl Acad Sci U S A 2001, 98(18):10374-10379.
30. Ishii H, Baffa R, Numata SI, Murakumo Y, Rattan S, Inoue H, Mori M, Fidanza V, Alder H, Croce CM: The FEZ1 gene at chromosome 8p22 encodes a leucine-zipper protein, and its expression is altered in multiple human tumors. Proc Natl Acad Sci U S A 1999, 96(7):3928-3933.
31. Kirk KE, Harmon BP, Reichardt IK, Sedat JW, Blackburn EH: Block in anaphase chromosome separation caused by a telomerase template mutation. Science 1997, 275:1478-1481.
32. van Steensel B, Smogorzewska A, de Lange T: TRF2 protects human telomeres from end-to-end fusions. Cell 1998, 92(3):401-413.
33. Samper E, Goytisolo FA, Slijepcevic P, van Buul PP, Blasco MA: Mammalian Ku86 protein prevents telomeric fusions independently of the length of TTAGGG repeats and the G-strand overhang. EMBO Rep 2000, 1(3):244-252.
34. Rudolph KL, Millard M, Bosenberg MW, DePinho RA: Telomere dysfunction and evolution of intestinal carcinoma in mice and humans. Nat Genet 2001, 28(2):155-159.
35. Gisselsson D, Jonson T, Petersen A, Strombeck B, Dal Cin P, Hoglund M, Mitelman F, Mertens F, Mandahl N: Telomere dysfunction triggers extensive DNA fragmentation and evolution of complex chromosome abnormalities in human malignant tumors. Proc Natl Acad Sci U S A 2001, 98(22):12683-12688.
36. Stewenius Y, Gorunova L, Jonson T, Larsson N, Hoglund M, Mandahl N, Mertens F, Mitelman F, Gisselsson D: Structural and numerical chromosome changes in colon cancer develop through telomere-mediated anaphase bridges, not through mitotic multipolarity. Proc Natl Acad Sci U S A 2005, 102(15):5541-5546.
37. Stewenius Y, Jin Y, Ora I, de Kraker J, Bras J, Frigyesi A, Alumets J, Sandstedt B, Meeker AK, Gisselsson D: Defective chromosome segregation and telomere dysfunction in aggressive Wilms’ tumors. Clin Cancer Res 2007, 13(22 Pt 1):6593-6602.
38. Artandi SE, DePinho RA: Telomeres and telomerase in cancer. Carcinogenesis 2010, 31(1):9-18.
39. Blasco MA, Lee HW, Hande MP, Samper E, Lansdorp PM, DePinho RA, Greider CW: Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell 1997, 91(1):25-34.
40. Chin L, Artandi SE, Shen Q, Tam A, Lee SL, Gottlieb GJ, Greider CW, DePinho RA: p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell 1999, 97(4):527-538.
41. Rudolph KL, Chang S, Lee HW, Blasco M, Gottlieb GJ, Greider C, DePinho RA: Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell 1999, 96(5):701-712.
42. Artandi SE, Chang S, Lee SL, Alson S, Gottlieb GJ, Chin L, DePinho RA: Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature 2000, 406(6796):641-645.
43. O’Hagan RC, Chang S, Maser RS, Mohan R, Artandi SE, Chin L, DePinho RA: Telomere dysfunction provokes regional amplification and deletion in cancer genomes. Cancer Cell 2002, 2(2):149-155.
44. Martinez P, Siegl-Cachedenier I, Flores JM, Blasco MA: MSH2 deficiency abolishes the anticancer and pro-aging activity of short telomeres. Aging Cell 2009, 8(1):2-17.
45. Blanco R, Munoz P, Flores JM, Klatt P, Blasco MA: Telomerase abrogation dramatically accelerates TRF2-induced epithelial carcinogenesis. Genes Dev 2007, 21(2):206-220.
46. Greenberg RA, Chin L, Femino A, Lee KH, Gottlieb GJ, Singer RH, Greider CW, DePinho RA: Short dysfunctional telomeres impair tumorigenesis in the INK4a(delta2/3) cancer-prone mouse. Cell 1999, 97(4):515-525.
47. Gonzalez-Suarez E, Samper E, Flores JM, Blasco MA: Telomerase-deficient mice with short telomeres are resistant to skin tumorigenesis. Nat Genet 2000, 26(1):114-117.
48. Feldser DM, Greider CW: Short telomeres limit tumor progression in vivo by inducing senescence. Cancer Cell 2007, 11(5):461-469.
49. Cosme-Blanco W, Shen MF, Lazar AJ, Pathak S, Lozano G, Multani AS, Chang S: Telomere dysfunction suppresses spontaneous tumorigenesis in vivo by initiating p53-dependent cellular senescence. EMBO Rep 2007, 8(5):497-503.
50. Jaskelioff M, Song W, Xia J, Liu C, Kramer J, Koido S, Gendler SJ, Calderwood SK, Gong J: Telomerase deficiency and telomere dysfunction inhibit mammary tumors induced by polyomavirus middle T oncogene. Oncogene 2009, 28(48):4225-4236.
51. Oh BK, Kim H, Park YN, Yoo JE, Choi J, Kim KS, Lee JJ, Park C: High telomerase activity and long telomeres in advanced hepatocellular carcinomas with poor prognosis. Lab Invest 2008, 88(2):144-152.
52. Gertler R, Doll D, Maak M, Feith M, Rosenberg R: Telomere length and telomerase subunits as diagnostic and prognostic biomarkers in Barrett carcinoma. Cancer 2008, 112(10):2173-2180.
53. Nurnberg P, Thiel G, Weber F, Epplen JT: Changes of telomere lengths in human intracranial tumours. Hum Genet 1993, 91(2):190-192.
54. Bisoffi M, Heaphy CM, Griffith JK: Telomeres: prognostic markers for solid tumors. Int J Cancer 2006, 119(10):2255-2260.
55. Garcia-Aranda C, de Juan C, Diaz-Lopez A, Sanchez-Pernaute A, Torres AJ, Diaz-Rubio E, Balibrea JL, Benito M, Iniesta P: Correlations of telomere length, telomerase activity, and telomeric-repeat binding factor 1 expression in colorectal carcinoma. Cancer 2006, 106(3):541-551.
56. Ohali A, Avigad S, Ash S, Goshen Y, Luria D, Feinmesser M, Zaizov R, Yaniv I: Telomere length is a prognostic factor in neuroblastoma. Cancer 2006, 107(6):1391-1399.
57. Gertler R, Rosenberg R, Stricker D, Friederichs J, Hoos A, Werner M, Ulm K, Holzmann B, Nekarda H, Siewert JR: Telomere length and human telomerase reverse transcriptase expression as markers for progression and prognosis of colorectal carcinoma. J Clin Oncol 2004, 22(10):1807-1814.
58. Svenson U, Nordfjall K, Stegmayr B, Manjer J, Nilsson P, Tavelin B, Henriksson R, Lenner P, Roos G: Breast cancer survival is associated with telomere length in peripheral blood cells. Cancer Res 2008, 68(10):3618-3623.
59. de Lange T: Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev 2005, 19(18):2100-2110.
60. Wu L, Multani AS, He H, Cosme-Blanco W, Deng Y, Deng JM, Bachilo O, Pathak S, Tahara H, Bailey SM et al: Pot1 deficiency initiates DNA damage checkpoint activation and aberrant homologous recombination at telomeres. Cell 2006, 126(1):49-62.
61. Baumann P, Cech TR: Pot1, the putative telomere end-binding protein in fission yeast and humans. Science 2001, 292(5519):1171-1175.
62. Hockemeyer D, Daniels JP, Takai H, de Lange T: Recent expansion of the telomeric complex in rodents: Two distinct POT1 proteins protect mouse telomeres. Cell 2006, 126(1):63-77.
63. van Steensel B, de Lange T: Control of telomere length by the human telomeric protein Trf1. Nature 1997, 385:740-743.
64. Smith S, Giriat I, Schmitt A, de Lange T: Tankyrase, a poly(ADP-ribose) polymerase at human telomeres. Science 1998, 282(5393):1484-1487.
65. Kim SH, Kaminker P, Campisi J: TIN2, a new regulator of telomere length in human cells. Nat Genet 1999, 23(4):405-412.
66. Zhou XZ, Lu KP: The Pin2/TRF1-interacting protein PinX1 is a potent telomerase inhibitor. Cell 2001, 107:347-359.
67. Loayza D, De Lange T: POT1 as a terminal transducer of TRF1 telomere length control. Nature 2003, 423(6943):1013-1018.
68. Lu KP, Hanes SD, Hunter T: A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature 1996, 380(6574):544-547.
69. Shen M, Haggblom C, Vogt M, Hunter T, Lu KP: Characterization and cell cycle regulation of related human telomeric proteins Pin2 and TRF1 suggest a role in mitosis. Proc Natl Acad Sci USA 1997, 94:13618-13623.
70. Lu KP, Zhou XZ: The prolyl isomerase Pin1: a pivotal new twist in phosphorylation signalling and human disease. Nat Rev Mol Cell Biol 2007, 8:904-916.
71. Kishi K, Lu KP: A critical role for Pin2/TRF1 in ATM-dependent regulation: Inhibition of Pin2/TRF1 function complements telomere shortening, the radiosensitivity and G2/M checkpoint defect of Ataxia-Telangiectasia cells. J Biol Chem 2002, 277:7420-7429.
72. Nakamura M, Zhou XZ, Kishi S, Kosugi I, Tsutsui Y, Lu KP: A specific interaction between the telomeric protein Pin2/TRF1 and the mitotic spindle. Curr Biol 2001, 11:1512-1516.
73. Zhu Q, Meng L, Hsu JK, Lin T, Teishima J, Tsai RY: GNL3L stabilizes the TRF1 complex and promotes mitotic transition. J Cell Biol 2009, 185(5):827-839.
74. Lee TH, Tun-Kyi A, Shi R, Lim J, Soohoo C, Finn G, Balastik M, Pastorino L, Wulf G, Zhou XZ et al: Essential role of Pin1 in the regulation of TRF1 stability and telomere maintenance. Nat Cell Biol 2009, 11(1):97-105.
75. Zhou XZ, Perrem K, Lu KP: Role of Pin2/TRF1 in telomere maintenance and cell cycle control. J Cell Biochem 2003, 89:19-37.
76. Lee TH, Perrem K, Harper JW, Lu KP, Zhou XZ: F-box protein Fbx4 targets Pin2/TRF1 for ubiquitin-mediated degradation and regulates telomere maintenance. J Biol Chem 2006, 281:759-768.
77. Banik SS, Counter CM: Characterization of interactions between PinX1 and human telomerase subunits hTERT and hTR. J Biol Chem 2004, 279(50):51745-51748.
78. Lin J, Blackburn EH: Nucleolar protein PinX1p regulates telomerase by sequestering its protein catalytic subunit in an inactive complex lacking telomerase RNA. Genes Dev 2004, 18(4):387-396.
79. Oh BK, Yoon SM, Lee CH, Park YN: Rat homolog of PinX1 is a nucleolar protein involved in the regulation of telomere length. Gene 2007, 400(1-2):35-43.
80. Sun C, Wu Z, Jia F, Wang Y, Li T, Zhao M: Identification of zebrafish LPTS: a gene with similarities to human LPTS/PinX1 that inhibits telomerase activity. Gene 2008, 420(1):90-98.
81. Soohoo CY, Shi R, Lee TH, Huang P, Lu KP, Zhou XZ: Telomerase inhibitor PINX1 provides a link between TRF1 and telomerase to prevent telomere elongation. J Biol Chem 2011, 286:3894-3906.
82. Zhou XZ, Huang P, Shi R, Lee TH, Lu G, Zhang Z, Bronson R, Lu KP: The telomerase inhibitor PinX1 is a major haploinsufficient tumor suppressor essential for chromosome stability. J Clin Invest 2011, 121:1266-1282.
83. Liao C, Zhao MJ, Zhao J, Jia D, Song H, Li ZP: Over-expression of LPTS-L in hepatocellular carcinoma cell line SMMC-7721 induces crisis. World J Gastroenterol 2002, 8(6):1050-1052.
84. Kelleher C, Kurth I, Lingner J: Human protection of telomeres 1 (POT1) is a negative regulator of telomerase activity in vitro. Mol Cell Biol 2005, 25(2):808-818.
85. Chong L, van SB, Broccoli D, Erdjument BH, Hanish J, Tempst P, de Lange T: A human telomeric protein. Science 1995, 270(5242):1663-1667.
86. Ancelin K, Brunori M, Bauwens S, Koering CE, Brun C, Ricoul M, Pommier JP, Sabatier L, Gilson E: Targeting assay to study the cis functions of human telomeric proteins: evidence for inhibition of telomerase by TRF1 and for activation of telomere degradation by TRF2. Mol Cell Biol 2002, 22(10):3474-3487.
87. Hawkins GA, Chang BL, Zheng SL, Isaacs SD, Wiley KE, Bleecker ER, Walsh PC, Meyers DA, Xu J, Isaacs WB: Mutational analysis of PINX1 in hereditary prostate cancer. Prostate 2004, 60(4):298-302.
88. Zhao Y, Sfeir AJ, Zou Y, Buseman CM, Chow TT, Shay JW, Wright WE: Telomere extension occurs at most chromosome ends and is uncoupled from fill-in in human cancer cells. Cell 2009, 138(3):463-475.
89. Wu X, Pandolfi PP: Mouse models for multistep tumorigenesis. Trends Cell Biol 2001, 11(11):S2-9.
90. Hakem R, Mak TW: Animal models of tumor-suppressor genes. Annnu Rev Genet 2001, 35:209-241.
91. Else T, Trovato A, Kim AC, Wu Y, Ferguson DO, Kuick RD, Lucas PC, Hammer GD: Genetic p53 deficiency partially rescues the adrenocortical dysplasia phenotype at the expense of increased tumorigenesis. Cancer Cell 2009, 15(6):465-476.
92. Cayuela ML, Flores JM, Blasco MA: The telomerase RNA component Terc is required for the tumour-promoting effects of Tert overexpression. EMBO Rep 2005, 6(3):268-274.
93. Soohoo CY, Shi R, Lee TH, Huang P, Lu KP, Zhou XZ: Telomerase inhibitor PINX1 provides a link between TRF1 and telomerase to prevent telomere elongation. J Biol Chem 2010:Nov 30. [Epub ahead of print].
94. Miller MC, Collins K: The Tetrahymena p80/p95 complex is required for proper telomere length maintenance and micronuclear genome stability. Mol Cell 2000, 6(4):827-837.
95. Masutomi K, Possemato R, Wong JM, Currier JL, Tothova Z, Manola JB, Ganesan S, Lansdorp PM, Collins K, Hahn WC: The telomerase reverse transcriptase regulates chromatin state and DNA damage responses. Proc Natl Acad Sci U S A 2005, 102(23):8222-8227.
96. Sharma GG, Gupta A, Wang H, Scherthan H, Dhar S, Gandhi V, Iliakis G, Shay JW, Young CS, Pandita TK: hTERT associates with human telomeres and enhances genomic stability and DNA repair. Oncogene 2003, 22(1):131-146.
97. Park JI, Venteicher AS, Hong JY, Choi J, Jun S, Shkreli M, Chang W, Meng Z, Cheung P, Ji H et al: Telomerase modulates Wnt signalling by association with target gene chromatin. Nature 2009, 460(7251):66-72.
98. Guglielmi B, Werner M: The yeast homolog of human PinX1 is involved in rRNA and small nucleolar RNA maturation, not in telomere elongation inhibition. J Biol Chem 2002, 277(38):35712-35719.
99. Yuan K, Li N, Jiang K, Zhu T, Huo Y, Wang C, Lu J, Shaw A, Thomas K, Zhang J et al: PinX1 is a novel microtubule-binding protein essential for accurate chromosome segregation. J Biol Chem 2009, 284(34):23072-23082.
100. Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE: Extension of life-span by introduction of telomerase into normal human cells. Science 1998, 279(5349):349-352.
101. Vaziri H, Benchimol S: Reconstitution of telomerase activity in normal human cells leads to elongation of telomeres and extended replicative life span. Curr Biol 1998, 8(5):279-282.
102. Kiyono T, Foster SA, Koop JI, McDougall JK, Galloway DA, Klingelhutz AJ: Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature 1998, 396(6706):84-88.
103. Jaskelioff M, Muller FL, Paik JH, Thomas E, Jiang S, Adams AC, Sahin E, Kost-Alimova M, Protopopov A, Cadinanos J et al: Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature 2011, 469(7328):102-106.
104. Gonzalez-Suarez E, Samper E, Ramirez A, Flores JM, Martin-Caballero J, Jorcano JL, Blasco MA: Increased epidermal tumors and increased skin wound healing in transgenic mice overexpressing the catalytic subunit of telomerase, mTERT, in basal keratinocytes. Embo J 2001, 20(11):2619-2630.
105. Artandi SE, Alson S, Tietze MK, Sharpless NE, Ye S, Greenberg RA, Castrillon DH, Horner JW, Weiler SR, Carrasco RD et al: Constitutive telomerase expression promotes mammary carcinomas in aging mice. Proc Natl Acad Sci U S A 2002, 99(12):8191-8196.
106. Stewart SA, Hahn WC, O’Connor BF, Banner EN, Lundberg AS, Modha P, Mizuno H, Brooks MW, Fleming M, Zimonjic DB et al: Telomerase contributes to tumorigenesis by a telomere length-independent mechanism. Proc Natl Acad Sci U S A 2002, 99(20):12606-12611.
107. Gonzalez-Suarez E, Flores JM, Blasco MA: Cooperation between p53 mutation and high telomerase transgenic expression in spontaneous cancer development. Mol Cell Biol 2002, 22(20):7291-7301.
108. Canela A, Martin-Caballero J, Flores JM, Blasco MA: Constitutive expression of tert in thymocytes leads to increased incidence and dissemination of T-cell lymphoma in Lck-Tert mice. Mol Cell Biol 2004, 24(10):4275-4293.
109. Gonzalez-Suarez E, Geserick C, Flores JM, Blasco MA: Antagonistic effects of telomerase on cancer and aging in K5-mTert transgenic mice. Oncogene 2005, 24(13):2256-2270.
110. Cristofari G, Lingner J: Telomere length homeostasis requires that telomerase levels are limiting. Embo J 2006, 25(3):565-574.
111. Chen G, Da L, Wang H, Xu Y, Sun C, Wang L, Zhao J, Zhang F, Feng J, Wang Y et al: HIV-Tat-mediated delivery of an LPTS functional fragment inhibits telomerase activity and tumorigenicity of hepatoma cells. Gastroenterology 2011, 140(1):332-343.
112. Zhang L, Jiang Y, Zheng Y, Zeng Y, Yang Z, Huang G, Liu D, Gao M, Shen X, Wu G et al: Selective killing of Burkitt’s lymphoma cells by mBAFF-targeted delivery of PinX1. Leukemia 2011, 25(2):331-340.
113. Wang HB, Wang WQ, Wang XW, Sun YG, Zhou G, Yang SM, Fan DC: PinX1 gene transfection enhances the sensitivity of gastric carcinoma cell line to 5-fluorouracil. Hepatogastroenterology 2011, 58(106):682-686.
114. Harley CB: Telomerase and cancer therapeutics. Nat Rev Cancer 2008, 8(3):167-179.
115. Joseph I, Tressler R, Bassett E, Harley C, Buseman CM, Pattamatta P, Wright WE, Shay JW, Go NF: The telomerase inhibitor imetelstat depletes cancer stem cells in breast and pancreatic cancer cell lines. Cancer Res 2010, 70(22):9494-9504.