
INTRODUCTION
Breast cancer is a leading cause of neoplasia-associated mortality [1]. While studying intestinal cancer in mouse models, we observed that targeted orogastric infection with Helicobacter hepaticus, a murine bacterium frequently colonizing the gut of mice [2–4], causes mammary neoplasia with particularly increased frequency [5]. We hypothesized that unbalanced host immune responses to enteric bacteria may promote the development of cancer in epithelia distant from the gut [6]. Since that time, genetically-engineered and diet-associated mouse models of cancer, wound healing, and obesity have provided evidence to support our original hypothesis that gut bacteria have consequences in systemic immunity [7–9].
Several lines of evidence support the roles of GI tract microbial flora in promoting intestinal neoplasmatogenesis [3, 5, 10–14]. However, linking gut bacteria with systemic innate immune-related effects that enhance tumor formation throughout the body expands this paradigm [5, 6]. This notion is challenging, especially in the light of recent reports demonstrating that gut microbe dysbiosis undermines the outcome of both immune and non-immune chemotherapeutic cancer treatment modalities [15, 16]. In order to identify the key immune cell players, prior studies have used combinations of immunohistochemistry, targeted immune depletions, and adoptive cell transfers [5, 17–19]. In those studies, reciprocal relationships exist between innate immune neutrophils [11] and anti-inflammatory Interleukin (IL)-10 -dependent activities of CD4+ T regulatory (Treg) lymphocytes that bestow immune homeostasis [5, 10, 20]. Indeed, neutrophils have been identified as important contributors of cancer initiation and development [21]. Through the generation of reactive oxygen species (ROS), neutrophils induce genotoxic damage, thus contributing to the mutagenic events in the initial steps of carcinogenesis [22–24]. Also, cytokines and chemokines as well as serine proteases secreted by tumor-associated neutrophils shape the tumor microenvironment and promote tumor growth [25, 26].
In the present study, we used female genetically-engineered FVB-Tg(C3-1-TAg)cJeg/JegJ mice with a predilection to develop mammary cancer [27]. In this model system, the C3(1)/SV40 T antigen is over-expressed after exposure to sex steroid hormones during development of prostate and mammary cancer [28, 29]. This mouse model recapitulates all stages of human mammary carcinomas. These lesions have been previously classified as infiltrating ductal carcinomas, including ductal epithelial cell atypia and mammary intraepithelial neoplasia (MIN), which corresponds to the human ductal carcinoma in situ [29]. Here we record that targeted orogastric infection with H. hepaticus increases mammary tumor multiplicity recapitulating the classical C3-1-TAg mouse mammary tumorigenesis pattern. Further, systemic depletion of neutrophils, a key innate immune inflammatory cell, can block this extra-intestinal tumorigenic phenomenon. These data demonstrate that host inflammatory responses to environmental microbes significantly impact cancer progression in distant non-intestinal tissues by a neutrophil -mediated mechanism.
RESULTS
Orogastric gavage with Helicobacter hepaticus increases mammary tumor burden in genetically-prone C3-1-TAg mice
It was previously shown that infection with enteropathogenic H. hepaticus rapidly induced mammary tumor formation in genetically-susceptible ApcMin/+ [ApcMin] mice [5, 6, 30, 31]. However, the use of ApcMin mice as a model of mammary cancer has certain peculiarities, raising doubts about broader relevancy of roles of gut microbiota in mammary epithelial carcinogenesis. To examine this evident gut microbe-mammary linkage further, we first tested orogastric challenge with H. hepaticus in the FVB-Tg(C3-1-TAg)cJeg/JegJ mouse model [29]. Within three weeks of infection, we found numerous small palpable tumors arising in multiple mammary tissue sites of three-month-old C3-1-TAg mice infected with H. hepaticus (Figure 1A). By comparison, sham media-dosed matched control animals had significantly fewer palpable tumors (Figure 1B).
The unencapsulated expansile tumors in both H. hepaticus- and sham-treated mice had the typical C3-1-TAg mouse mammary adenocarcinoma histomorphology (Figures 2A–2C). In the non-tumoral mammary tissue, ducts and terminal duct lobular units (TDLU) showed a spectrum of hyperplastic, preneoplastic, and early neoplastic lesions depicting the well-characterized stages of the C3-1-TAg mouse mammary tumorigenesis progression (Figure 2D). In the non-tumoral areas the affected mammary epithelia of H. hepaticus-treated mice appeared to be in more advanced tumorigenesis stages by comparison with the uninfected controls. In order to confirm this observation we determined histomorphometrically the percentage of abnormal glands at each one of four critical histological stages of mammary tumorigenesis in H. hepaticus-infected and non-infected mice. We found that the classification of abnormal glands according to their histological stage differed significantly between experimental groups (P = 0.0307), with H. hepaticus-infected mice having a higher percentage of abnormal glands with mammary intraepithelial neoplasia (MIN) when compared to controls (Figure 2E).
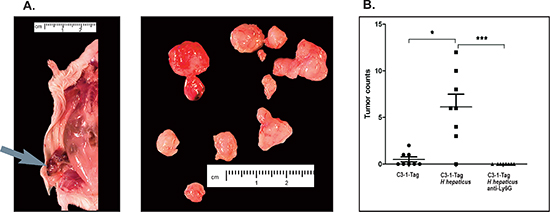
Figure 1: Tumor multiplicity assessment in experimental groups of 15-week-old C3-1-TAg mice. (A) Grossly visible tumors (arrow) found in each mouse were removed, placed on a petri dish and counted. (B) The statistical analysis of mammary tumor count data shows that infection with H. hepaticus accelerates tumorigenesis, whereas the depletion of neutrophils negates this effect. The y-axis depicts the mean ± SEM of mammary tumor counts. *p < 0.05. The points correspond to the mean of total tumors counted in each mouse.
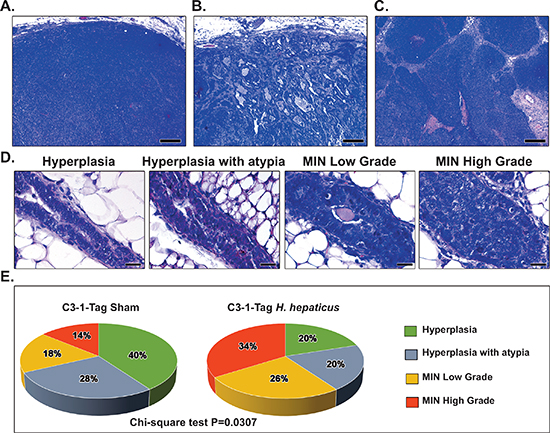
Figure 2: Effects of H. hepaticus on mammary gland carcinogenesis. Tumors of both H. hepaticus-infected and uninfected control C3-1-TAg mice shared similar histomorphological patterns. (A) Neoplastic cells arranged in solid sheets, cords or nests with minimal gland formation and small amounts of intervening stroma. (B) Glandular-like growth was seen in occasional areas at the periphery of the tumors. Note irregular glands in moderate to large amounts of desmoplastic stroma. (C) Large solid cord arrangement of neoplastic cells with variably sized areas of either diffuse or commedo type intratumoral necrosis. (D) The initial stages of mammary tumorigenesis. From left to the right there is progressively increased epithelial pseudostratification, cellular atypia, nuclear pleomorphism and mitotic figures. (E) Classification of abnormal glands in non-tumoral areas according to their histological stage. The mammary glands of H. hepaticus-infected mice are in more advanced stages of neoplastic progression compared to those of uninfected controls. Hematoxylin and Eosin (A, B, C and D); Scale bars: 250 μm (A, B and C) and 25 μm (D).
Gut microbial challenge leads to up-regulation of inflammatory cells in mammary tissue
Knowing that inflammatory cells and factors were pivotal in etiopathogenesis of microbe-induced mammary [5, 17, 32] and prostate [18] tumors, we next examined whether inflammatory cells were increased in C3-1-TAg mice undergoing infection with H. hepaticus. We found the tumor-associated inflammatory cell component residing at the periphery of well-defined tumors (Figure 3A) consisted of abundant macrophages, neutrophils, myeloid precursor cells with ring-shaped nuclei [33, 34], mononuclear cells, and mast cells. The same types of inflammatory cells were found in the connective tissue stroma within the tumor, with the exception of mast cells, which were sparse. Mast cells as well as neutrophils, however, were topographically associated with early neoplastic lesions such as MIN in the non-tumoral areas of the mammary glands.
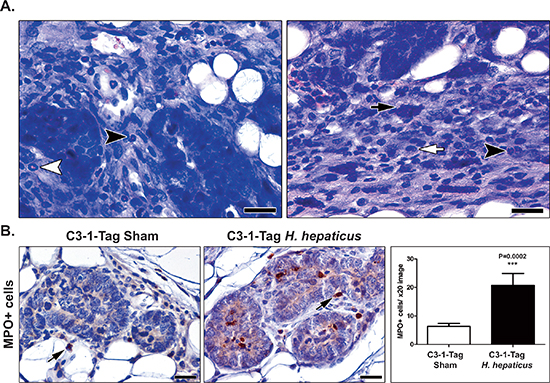
Figure 3: H. hepaticus infection up-regulates MIN-associated neutrophils. (A) The tumor-associated inflammation was comparable in large-sized neoplasms of both H. hepaticus-infected and uninfected control mice. Neutrophils (black arrow-heads), myeloid precursor cells (white arrow-head), mast cells (black arrow) and macrophages (white arrow) at the periphery of tumors are shown. (B) Morphometric counts of MPO-positive cells (arrows) in MIN lesions. The numbers of neutrophils are significantly higher in H-hepaticus infected mice compared to controls. Hematoxylin and Eosin (A) IHC; Diaminobenzidine chromogen, Hematoxylin counterstain (B) Scale bars: 25 μm (A) and 50 μm (B) Numbers on the y-axis of bar graph correspond to the mean ± SEM of MPO+cells. ***p < 0.0001.
Comparing histomorphologically similar MIN lesions in the two experimental groups, we noticed that neutrophils accumulated in higher numbers around the MIN lesions when mice were infected with H. hepaticus. Tumor associated neutrophils (TAN) were previously shown to enhance tumorigenesis in mouse models [35]. To quantify this result we performed counts of myeloperoxidase (MPO)-positive cells (neutrophils) in histologically comparable MIN lesions of both H. hepaticus-infected and uninfected mice. We found that the H. hepaticus infection status correlated with higher numbers of MIN-associated MPO-positive cells in statistically significant levels (P = 0.002) (Figure 3B).
Systemic depletion of neutrophils inhibits mammary tumor formation
Finally, based upon earlier findings showing that neutrophils are a consistent feature of H. hepaticus-induced inflammation [36] and cancer [10, 11, 20], and due to the emerging role of neutrophils in carcinogenesis [21, 37, 38], we tested whether Ly-6G+ neutrophils were required for mammary cancer in this mouse model. To test this we used thrice-weekly intraperitoneal injections with anti-Ly6G clone 18A previously shown to target mature neutrophils in mice [39]. While H. hepaticus-infected mice had multifocal well-sized mammary adenocarcinoma tumors (8/9, 88%), H. hepaticus-infected mice that were treated with anti-Ly6G antibody had no evidence of mammary adenocarcinoma tumors (0/9, 0%) (Figure 1B). The neutrophil depleted mice had only preneoplastic and early neoplastic lesions in their mammary gland epithelia (Figure 4). Specifically, the most advanced histopathological lesion found [7] was low and high grade MIN (carcinoma in situ) in three mice (3/9, 33%), hyperplasia with marked atypia in two (2/9, 22%), hyperplasia with atypia, intermediate grade in one (1/9, 0.09%) hyperplasia without atypia, low grade in three mice (3/9, 33%).
DISCUSSION
In the present study we use the well-characterized C3-1-TAg mouse model of mammary cancer to show that bacteria residing in the gut mucosa affect the evolution of neoplastic lesions in mammary glands. We find here that the neutrophil is an important cellular element of this mechanism, which links innate immune and neoplastic events occurring in topographically distant epithelia. This highlights the neutrophil as an important mediator of the remote effect of gut microbiota on mammary epithelial carcinogenesis. Further, these findings build upon the pivotal role of systemic immune balance in emerging pre-neoplastic lesions throughout the body not obviously linked with chronic inflammation.
Previously we have shown that H. hepaticus-infected C57BL/6 ApcMin Rag2-deficient female mice develop mammary tumors with increased frequency [5]. This was the first study providing compelling experimental evidence linking the effects of gastrointestinal bacteria with mammary carcinogenesis. Further, this provided evidence that innate immunity alone was sufficient for distant carcinogenesis triggered by gut bacterial infection. Subsequently we showed that male ApcMin mice infected with H. hepaticus develop prostate tumors transplantable to uninfected mice using purified lymph node cells obtained from H. hepaticus-infected mice alone [18], implicating systemic innate immune cell trafficking in distant carcinogenic events. Indeed, the abundance of morphologically-distinctive myeloid cells throughout target tissues in the ApcMin model supports the notion of gut bacteria-triggered systemic trafficking of immune precursor cells [18]. However, the use of ApcMin mice as a model of mammary or prostate carcinogenesis raises some questions. These mice exhibit a predilection for intestinal polypoid adenomas, due to their heterogygous adenomatosis polyposis coli gene status [30] and accelerated thymic involution [40]. Given microbe-dependent intestinal polyposis in this model [5, 41], it is difficult to assess whether the mammary cancer is due to H. hepaticus infection per se or to microbe-increased multiplicity of intestinal polyps. Taken together, these facts raise doubts about roles of gut microbiota in mammary epithelial carcinogenesis beyond ApcMin mice. The results of the present paper contribute towards showing that H. hepaticus accelerates mammary carcinogenesis in other animal systems, in this case the C3-1-TAg female mouse that is a widely utilized mouse model for hormonally-dependent cancer [28]. Thus the present findings build upon our previous observations and expand upon the ApcMin mouse mammary cancer paradigm.
Further, these data provide additional evidence that breast cancer is associated with microbial dysbiosis in the gut. Altering gut microbes can regulate the immune system and lower the risk of breast cancer; in particular, overgrowth or lack of certain types of bacteria in the gut have been associated with many diseases ranging from obesity, to digestive disorders, to cancers [42]. Differences in the bacterial populations in breast tumor tissue and healthy breast tissue have been reported [43]. Frequent use of antibiotics that may disrupt the microbiome is associated with breast cancer development and relapse [44–46]. Interestingly, immune dysregulation can be transferred in mice by fecal microbe transplant [47–50]. Additionally, the microbiome plays a key role in estrogen cycling in the body, and gut dysbiosis results in higher circulating estrogens which has been linked to postmenopausal breast cancer [51–54]. Estrogen and neutrophil dysregulation are hallmarks of breast cancer development [55], and a high neutrophil to lymphocyte ratio is associated with breast cancer relapse in patients. Both these cell populations are clearly modulated by the microbiome and inflammation [56–59].
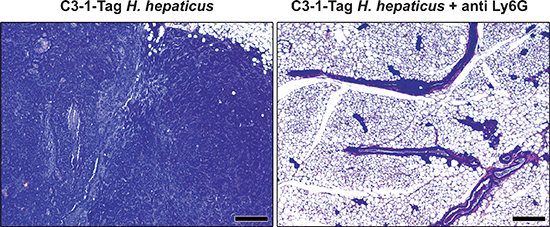
Figure 4: The depletion of neutrophils with anti-Ly-6G antibody blocks effect of H. hepaticus-promoted C3-1-TAg mouse mammary carcinogenesis. Fifteen-weeks-old H. hepaticus-infected C3-1-TAg mice had the typical for this mouse model mammary tumors. At the same timepoint their anti-Ly6G-treated counterparts had early neoplastic changes but no tumors at all. Hematoxylin and Eosin. Scale bars: 250 μm
Interestingly, our current results suggest that the remote effect of gut microbiota in mammary carcinogenesis does not specifically depend on the molecular pathway of mammary carcinogenesis involved. C3-1-TAg mice develop mammary cancer due to hormonally-dependent functional inactivation of the P53 and Rb tumor suppressor genes, which leads mammary epithelia to cell cycle regulation defects, uncontrolled proliferation and resistance to apoptosis [29]. The inactivation of these tumor suppressors and the dysregulation of the cell growth and apoptosis pathways they control are common in human breast cancer [29, 60]. Although the role of the APC gene, which is central in the β-catenin/WNT pathway of carcinogenesis in human breast cancer is emerging [61], neither APC mutant mice nor humans carrying germline mutations in APC develop spontaneously mammary cancer in a high percentage [62, 63]. By contrast C3-1-TAg mice inevitably develop mammary neoplasms [29]. This fact presumably reflects the more universal role of the P53 and Rb genes in the various types of neoplastic disease compared to the APC gene.
Our compiled results also show that the effect of intestinal H. hepaticus on mammary carcinogenesis does not connect specifically with the histological type of the mammary tumor. Previously we found H. hepaticus promotes formation of mammary adenosquamous carcinoma in ApcMin mice [5]. Here, we find in the C3-1-TAg mouse that it accelerates the growth of solid carcinomas, resembling infiltrating ductal carcinomas of humans [29, 64, 65].
Importantly, the C3-1-TAg mice on a FVB background develop neither IBD nor intestinal tumors [29]. This suggests that subclinical, rather than overt, alterations in the immune status are sufficient for the induction of phenotypically distinct effects on mammary carcinogenesis. Other proliferative and neoplastic pathologies reported to arise in different anatomical sites of female C3-1-TAg mice, such as the sweat glands of the foot pads, the salivary glands, and the vomeronasal organ [29] were not observed in the present study. This lack of tumors in other sites was predictable since these pathologies occur in older animals more than five-months-of-age, a time point that was not reached with young (less than four-months-of-age) mice in our present study. Unlike ApcMin, the C3-1-TAg mice are immunocompetent, which suggests that immune system defects are not necessary for the remote effect of GI tract bacteria on mammary carcinogenesis. Although T regulatory (Treg) cells function reciprocally with neutrophils [36], the roles for Treg cells in preventing mammary neoplasia were not specifically examined in this study. Knowing that C3-1-TAg mice exhibit hormone-dependent carcinogenesis [29], and that neutrophils robustly interact with estrogen and IL-10 [66], the relationships between microbes, hormones, immunity and cancer are a key topic for future studies.
It appears that H. hepaticus is not unique among gut bacteria exerting effects on mammary gland tissue. Our other recent findings show that the probiotic bacterium Lactobacillus reuteri, when introduced in the GI tract flora of mice, has the opposite effect of suppressing mammary tumor formation in genetically-susceptible HER2 mutant mice [7]. In both cases involving gut microbiota, whether the acceleration of mammary carcinogenesis by pathogenic H. hepaticus or suppression of mammary tumorigenesis by probiotic L. reuteri, we discover differences in the immune cell composition of the mammary gland microenvironment. An interesting aspect is that these gut-mammary connections may be covert; even in the absence of demonstrable inflammatory disease in the gut or elsewhere, we find that H. hepaticus-infected mice have not only higher risk for cancer but also more frequent inflammatory cells associating with mammary glands at early neoplastic stages.
In the present study we found that neutrophils associate with mammary lesions in higher numbers in the H. hepaticus-infected mice when compared to their uninfected controls. Neutrophils are a consistent feature of H. hepaticus-induced inflammation [36] and cancer [10, 11, 20]. Other evidence for the role of neutrophils in carcinogenesis and tumor evolution is emerging and the therapeutic approach of targeting the tumor-associated neutrophils has been recently introduced [21]. Studies in samples from many different types of human tumors suggest that a high neutrophil to lymphocyte ratios predicts a poor clinical outcome [38]. Also, the depletion of neutrophils has been shown to block cancer evolution in mouse models of fibrosarcoma [67], pancreatic islet cell [68], colonic [11], and pulmonary carcinoma [35].
Taken together with our earlier data these facts led us to test whether the depletion of neutrophils could affect mammary carcinogenesis in this mouse model, as well. Recognizing the etiopathogenic potential of myeloid immune precursor trafficking in lymph nodes, spleen, and target tissues after H. hepaticus infection [18], it's possible that depleting Ly-6G+ cells includes other relevant immune cells; although, clone 1A8 was previously shown to be specific for mature neutrophils [39]. We found that the H. hepaticus-infected C3-1-TAg mice depleted of Ly-6G+ neutrophils had significantly fewer mammary lesions, and in those cases only preneoplastic and early neoplastic lesions, in contrast to their non-depleted matching controls which exhibited palpable mammary tumors. The dependence of mammary tumorigenesis upon the presence of neutrophils highlights the neutrophil as an important mediator of the remote effect of gut microbiota on mammary epithelial carcinogenesis. Adoptive transfer experiments [18] using highly purified gut bacteria-stimulated myeloid and neutrophil cell populations will address this in future studies.
In conclusion, the results of the present study indicate that gut microbiota bacterial elements, in the absence of overt inflammatory disease, contribute to the character of the subclinical, systemic inflammatory tone of the mammalian organism. Based on these findings it appears that systemic inflammatory tone, which is mediated, at least in part, by neutrophils, affects the evolution of preneoplastic lesions to cancer in epithelia locating distantly from the gut, such as those of the mammary glands.
METHODS
Animals
Genetically-inbred FVB-Tg(C3-1-TAg)cJeg/JegJ female mice (Jackson Labs, Bar Harbor, ME), were housed and handled in Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC)-accredited facilities with diets, experimental methods, and housing as specifically approved by the Institutional Animal Care and Use Committee. Experimental design was to expose mice to Helicobacter hepaticus infection by gastric gavage at the age of twelve weeks, and then monitor for three weeks until euthanasia using carbon dioxide overdose. Control mice of the same age underwent gastric gavage with media alone. Subsets of infected mice were injected intraperitoneally with anti-Ly-6G antibody to deplete neutrophils, or with sham isotype-matched antibody alone, thrice weekly for 3.5–4 weeks starting three days before infection. Each experimental group included 5–10 animals per group with one replicate experiment with statistically similar outcomes to validate results. Mammary tissues were collected upon necropsy and then examined histologically.
Experimental infection with Helicobacter hepaticus
A total of 51 experimental mice were dosed at 12 weeks of age with H. hepaticus and then housed separately in a bio-containment area within the same animal facility. H. hepaticus (strain 3B1, ATCC #51449) [2] was grown under microaerobic conditions, prepared, and confirmed to be pure as described elsewhere [3]. Experimental mice received 0.2 ml of fresh inoculum by gastric gavage every other day for a total of three doses. Cecum and stool were collected at necropsy 3–4 weeks post-infection and analyzed by PCR using H. hepaticus-specific primers to confirm bowel colonization.
Systemic depletion of neutrophils
Mice were treated with anti-Ly6G antibody (clone 1A8; Bio- X-Cell, West Lebanon, NH) at 200 ug per mouse intraperitoneally 3X weekly for 3–4 weeks starting 3 days before infection with H. hepaticus. Treatment with antibody continued concurrent with H. hepaticus infection for a duration of 3 weeks, with euthanasia occurring at age 15 weeks. Treated mice were then compared with mice receiving a comparable dose of sham isotype antibody alone. Depletion of neutrophils was confirmed by undetectably low levels of MPO+ cells in spleens of mice treated with anti-Ly-6G antibody compared to sham-treated controls.
Statistical analyses
The Mann-Whitney U test was used for analyzing histomorphometry data. Mammary tumor multiplicity was evaluated using the Kruskal-Wallis analysis followed by the Dunn's multiple comparison test. Mammary gland lesion staging comparison between groups was done with the Chi-square test. Replicate experiments were not significantly different. A p-value < 0.05 was statistically significant.
Histopathology and immunohistochemistry
For histologic evaluation, formalin-fixed tissues were embedded in paraffin, cut at 5 μm, and stained with hematoxylin and eosin. Hyperplastic and preneoplastic lesions in the mammary gland were staged based on previously published consensus criteria [65] as previously described [7]. Rabbit polyclonal antibodies against Myeloperoxidase (MPO) (ThermoFisher Scientific/Lab Vision, Fremont, CA), were used for immunohistochemistry. Heat-induced antigen retrieval was performed with citrate buffer, pH 6. Rabbit primary antibody binding was detected with goat anti-rabbit polymer HRP (ZytoChem Plus, Berlin, Germany). Color was developed with DAB substrate-chromogen system (ThermoFisher Scientific/Lab Vision) and tissues were counterstained with hematoxylin.
For quantitative histomorphometry, MPO-positive cells were counted in 20 randomly-selected images of x20 representative high power fields using the ImageJ processing and analysis program (NIH, Bethesda, MD) as previously described [7] and results were recorded as number of cells per image. The relative percentage of abnormal glands belonging to different histological stages of mammary tumorigenesis in H. hepaticus-infected and non-infected mice was determined in 50 abnormal mammary duct profiles from each group. For that, histological sections from non-tumoral areas of 5 randomly selected mice per experimental group were used. Starting from the upper left corner of each histological section and moving towards its lower right, the first 10 abnormal glands (hyperplastic or with early neoplasia) found were staged and the result was recorded.
FUNDING
This work was supported by National Institutes of Health grants U01 CA164337 (S.E.E.) and T32 OD011141 (JGF and SEE).
CONFLICTS OF INTEREST
There are no competing financial or commercial interests.
Author contributions
Conceived and designed the experiments: JRL, TL, ECT, BF, NA, SEE. Performed the experiments: JRL, SM, BJV, TL, YMI, ECT, BF, NA, SG, SEE. Analyzed the data: JRL, TP, JMW, NP, SL, ZG, SEE. Contributed reagents/ materials/analysis tools: TP, JGF, ZG. Wrote the paper: JRL, TP, JMW, SEE.
REFERENCES
1. Boyle P, Levin B, International Agency for Research on Cancer. and World Health Organization. (2008). World cancer report 2008. (Lyon Geneva: International Agency for Research on Cancer; Distributed by WHO Press).
2. Fox JG, Dewhirst FE, Tully JG, Paster BJ, Yan L, Taylor NS, Collins MJ, Jr., Gorelick PL, Ward JM. Helicobacter hepaticus sp. nov., a microaerophilic bacterium isolated from livers and intestinal mucosal scrapings from mice. J Clin Microbiol. 1994; 32:1238–1245.
3. Erdman SE, Poutahidis T, Tomczak M, Rogers AB, Cormier K, Plank B, Horwitz BH, Fox JG. CD4+ CD25+ regulatory T lymphocytes inhibit microbially induced colon cancer in Rag2-deficient mice. Am J Pathol. 2003; 162:691–702.
4. Suerbaum S, Josenhans C, Sterzenbach T, Drescher B, Brandt P, Bell M, Droge M, Fartmann B, Fischer HP, Ge Z, Horster A, Holland R, Klein K, Konig J, Macko L, Mendz GL, et al. The complete genome sequence of the carcinogenic bacterium Helicobacter hepaticus. Proc Natl Acad Sci U S A. 2003; 100:7901–7906.
5. Rao VP, Poutahidis T, Ge Z, Nambiar PR, Boussahmain C, Wang YY, Horwitz BH, Fox JG, Erdman SE. Innate immune inflammatory response against enteric bacteria Helicobacter hepaticus induces mammary adenocarcinoma in mice. Cancer Res. 2006; 66:7395–7400.
6. Rao VP, Poutahidis T, Fox JG, Erdman SE. Breast cancer: should gastrointestinal bacteria be on our radar screen? Cancer Res. 2007; 67:847–850.
7. Lakritz JR, Poutahidis T, Levkovich T, Varian BJ, Ibrahim YM, Chatzigiagkos A, Mirabal S, Alm EJ, Erdman SE. Beneficial bacteria stimulate host immune cells to counteract dietary and genetic predisposition to mammary cancer in mice. Int J Cancer. 2014; 135:529–540.
8. Poutahidis T, Kleinewietfeld M, Smillie C, Levkovich T, Perrotta A, Bhela S, Varian BJ, Ibrahim YM, Lakritz JR, Kearney SM, Chatzigiagkos A, Hafler DA, Alm EJ, Erdman SE. Microbial reprogramming inhibits Western diet-associated obesity. PLoS One. 2013; 8:e68596.
9. Poutahidis T, Kearney SM, Levkovich T, Qi P, Varian BJ, Lakritz JR, Ibrahim YM, Chatzigiagkos A, Alm EJ, Erdman SE. Microbial Symbionts Accelerate Wound Healing via the Neuropeptide Hormone Oxytocin. PLoS One. 2013; 8:e78898.
10. Erdman SE, Rao VP, Poutahidis T, Ihrig MM, Ge Z, Feng Y, Tomczak M, Rogers AB, Horwitz BH, Fox JG. CD4+CD25+ regulatory lymphocytes require interleukin 10 to interrupt colon carcinogenesis in mice. Cancer Res. 2003; 63:6042–6050.
11. Erdman SE, Rao VP, Poutahidis T, Rogers AB, Taylor CL, Jackson EA, Ge Z, Lee CW, Schauer DB, Wogan GN, Tannenbaum SR, Fox JG. Nitric oxide and TNF-alpha trigger colonic inflammation and carcinogenesis in Helicobacter hepaticus-infected, Rag2-deficient mice. Proc Natl Acad Sci U S A. 2009; 106:1027–1032.
12. Li Y, Kundu P, Seow SW, de Matos CT, Aronsson L, Chin KC, Karre K, Pettersson S, Greicius G. Gut microbiota accelerate tumor growth via c-jun and STAT3 phosphorylation in APCMin/+ mice. Carcinogenesis. 2012; 33:1231–1238.
13. Nagamine CM, Rogers AB, Fox JG, Schauer DB. Helicobacter hepaticus promotes azoxymethane-initiated colon tumorigenesis in BALB/c-IL10-deficient mice. Int J Cancer. 2008; 122:832–838.
14. Arthur JC, Perez-Chanona E, Muhlbauer M, Tomkovich S, Uronis JM, Fan TJ, Campbell BJ, Abujamel T, Dogan B, Rogers AB, Rhodes JM, Stintzi A, Simpson KW, Hansen JJ, Keku TO, Fodor AA, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012; 338:120–123.
15. Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA, Molina DA, Salcedo R, Back T, Cramer S, Dai RM, Kiu H, Cardone M, Naik S, Patri AK, Wang E, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science. 2013; 342:967–970.
16. Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillere R, Hannani D, Enot DP, Pfirschke C, Engblom C, Pittet MJ, Schlitzer A, Ginhoux F, Apetoh L, Chachaty E, Woerther PL, Eberl G, et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science. 2013; 342:971–976.
17. Erdman SE, Rao VP, Olipitz W, Taylor CL, Jackson EA, Levkovich T, Lee CW, Horwitz BH, Fox JG, Ge Z, Poutahidis T. Unifying roles for regulatory T cells and inflammation in cancer. Int J Cancer. 2010; 126:1651–1665.
18. Poutahidis T, Cappelle K, Levkovich T, Lee CW, Doulberis M, Ge Z, Fox JG, Horwitz BH, Erdman SE. Pathogenic intestinal bacteria enhance prostate cancer development via systemic activation of immune cells in mice. PLoS One. 2013; 8:e73933.
19. Poutahidis T, Rao VP, Olipitz W, Taylor CL, Jackson EA, Levkovich T, Lee CW, Fox JG, Ge Z, Erdman SE. CD4+ lymphocytes modulate prostate cancer progression in mice. Int J Cancer. 2009; 125:868–878.
20. Poutahidis T, Haigis KM, Rao VP, Nambiar PR, Taylor CL, Ge Z, Watanabe K, Davidson A, Horwitz BH, Fox JG, Erdman SE. Rapid reversal of interleukin-6-dependent epithelial invasion in a mouse model of microbially induced colon carcinoma. Carcinogenesis. 2007; 28:2614–2623.
21. Gregory AD, Houghton AM. Tumor-associated neutrophils: new targets for cancer therapy. Cancer Res. 2011; 71:2411–2416.
22. Lonkar P, Dedon PC. Reactive species and DNA damage in chronic inflammation: reconciling chemical mechanisms and biological fates. Int J Cancer. 2011; 128:1999–2009.
23. Bouvard V, Baan R, Straif K, Grosse Y, Secretan B, El Ghissassi F, Benbrahim-Tallaa L, Guha N, Freeman C, Galichet L, Cogliano V, Group WHOIAfRoCMW. A review of human carcinogens—Part B: biological agents. Lancet Oncol. 2009; 10:321–322.
24. Shacter E, Weitzman SA. Chronic inflammation and cancer. Oncology (Williston Park). 2002; 16:217–226. 229, discussion 230–212.
25. Pham CT. Neutrophil serine proteases: specific regulators of inflammation. Nature reviews Immunology. 2006; 6:541–550.
26. Galdiero MR, Bonavita E, Barajon I, Garlanda C, Mantovani A, Jaillon S. Tumor associated macrophages and neutrophils in cancer. Immunobiology. 2013; 218:1402–1410.
27. Shibata MA, Jorcyk CL, Liu ML, Yoshidome K, Gold LG, Green JE. The C3(1)/SV40 T antigen transgenic mouse model of prostate and mammary cancer. Toxicologic pathology. 1998; 26:177–182.
28. Maroulakou IG, Anver M, Garrett L, Green JE. Prostate and mammary adenocarcinoma in transgenic mice carrying a rat C3(1) simian virus 40 large tumor antigen fusion gene. Proc Natl Acad Sci U S A. 1994; 91:11236–11240.
29. Green JE, Shibata MA, Yoshidome K, Liu ML, Jorcyk C, Anver MR, Wigginton J, Wiltrout R, Shibata E, Kaczmarczyk S, Wang W, Liu ZY, Calvo A, Couldrey C. The C3(1)/SV40 T-antigen transgenic mouse model of mammary cancer: ductal epithelial cell targeting with multistage progression to carcinoma. Oncogene. 2000; 19:1020–1027.
30. Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990; 247:322–324.
31. Moser AR, Luongo C, Gould KA, McNeley MK, Shoemaker AR, Dove WF. ApcMin: a mouse model for intestinal and mammary tumorigenesis. Eur J Cancer. 1995; 31A:1061–1064.
32. Rao VP, Poutahidis T, Ge Z, Nambiar PR, Horwitz BH, Fox JG, Erdman SE. Proinflammatory CD4+ CD45RB(hi) lymphocytes promote mammary and intestinal carcinogenesis in Apc(Min/+) mice. Cancer Res. 2006; 66:57–61.
33. Biermann H, Pietz B, Dreier R, Schmid KW, Sorg C, Sunderkotter C. Murine leukocytes with ring-shaped nuclei include granulocytes, monocytes, and their precursors. J Leukoc Biol. 1999; 65:217–231.
34. Rehg JE, Bush D, Ward JM. The utility of immunohistochemistry for the identification of hematopoietic and lymphoid cells in normal tissues and interpretation of proliferative and inflammatory lesions of mice and rats. Toxicologic pathology. 2012; 40:345–374.
35. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, Worthen GS, Albelda SM. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell. 2009; 16:183–194.
36. Maloy KJ, Salaun L, Cahill R, Dougan G, Saunders NJ, Powrie F. CD4+CD25+ T(R) cells suppress innate immune pathology through cytokine-dependent mechanisms. J Exp Med. 2003; 197:111–119.
37. Bromberg J, Wang TC. Inflammation and cancer: IL-6 and STAT3 complete the link. Cancer Cell. 2009; 15:79–80.
38. Dumitru CA, Lang S, Brandau S. Modulation of neutrophil granulocytes in the tumor microenvironment: mechanisms and consequences for tumor progression. Semin Cancer Biol. 2013; 23:141–148.
39. Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J Leukoc Biol. 2008; 83:64–70.
40. Coletta PL, Muller AM, Jones EA, Muhl B, Holwell S, Clarke D, Meade JL, Cook GP, Hawcroft G, Ponchel F, Lam WK, MacLennan KA, Hull MA, Bonifer C, Markham AF. Lymphodepletion in the ApcMin/+ mouse model of intestinal tumorigenesis. Blood. 2004; 103:1050–1058.
41. Newman JV, Kosaka T, Sheppard BJ, Fox JG, Schauer DB. Bacterial infection promotes colon tumorigenesis in Apc(Min/+) mice. J Infect Dis. 2001; 184:227–230.
42. Sheflin AM, Whitney AK, Weir TL. Cancer-promoting effects of microbial dysbiosis. Curr Oncol Rep. 2014; 16:406.
43. Xuan C, Shamonki JM, Chung A, Dinome ML, Chung M, Sieling PA, Lee DJ. Microbial dysbiosis is associated with human breast cancer. PLoS One. 2014; 9:e83744.
44. Koumarianou A, Tzeveleki I, Mekras D, Eleftheraki AG, Bobos M, Wirtz R, Fountzilas E, Valavanis C, Xanthakis I, Kalogeras KT, Basdanis G, Pentheroudakis G, Kotoula V, Fountzilas G. Prognostic markers in early-stage colorectal cancer: significance of TYMS mRNA expression. Anticancer Res. 2014; 34:4949–4962.
45. Velicer CM, Heckbert SR, Lampe JW, Potter JD, Robertson CA, Taplin SH. Antibiotic use in relation to the risk of breast cancer. JAMA. 2004; 291:827–835.
46. Tamim HM, Hanley JA, Hajeer AH, Boivin JF, Collet JP. Risk of breast cancer in relation to antibiotic use. Pharmacoepidemiology and drug safety. 2008; 17:144–150.
47. Geuking MB, Cahenzli J, Lawson MA, Ng DC, Slack E, Hapfelmeier S, McCoy KD, Macpherson AJ. Intestinal bacterial colonization induces mutualistic regulatory T cell responses. Immunity. 2011; 34:794–806.
48. Macpherson AJ, Geuking MB, McCoy KD. Innate and adaptive immunity in host-microbiota mutualism. Frontiers in bioscience. 2012; 4:685–698.
49. Chaudhry A, Samstein RM, Treuting P, Liang Y, Pils MC, Heinrich JM, Jack RS, Wunderlich FT, Bruning JC, Muller W, Rudensky AY. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity. 2011; 34:566–578.
50. Geuking MB, McCoy KD, Macpherson AJ. The continuum of intestinal CD4+ T cell adaptations in host-microbial mutualism. Gut Microbes. 2011; 2:353–357.
51. Roberts MS, Magnusson BM, Burczynski FJ, Weiss M. Enterohepatic circulation: physiological, pharmacokinetic and clinical implications. Clin Pharmacokinet. 2002; 41:751–790.
52. Plottel CS, Blaser MJ. Microbiome and malignancy. Cell Host Microbe. 2011; 10:324–335.
53. Woolcott CG, Shvetsov YB, Stanczyk FZ, Wilkens LR, White KK, Caberto C, Henderson BE, Le Marchand L, Kolonel LN, Goodman MT. Plasma sex hormone concentrations and breast cancer risk in an ethnically diverse population of postmenopausal women: the Multiethnic Cohort Study. Endocr Relat Cancer. 2010; 17:125–134.
54. Flores R, Shi J, Fuhrman B, Xu X, Veenstra TD, Gail MH, Gajer P, Ravel J, Goedert JJ. Fecal microbial determinants of fecal and systemic estrogens and estrogen metabolites: a cross-sectional study. J Transl Med. 2012; 10:253.
55. Clarke TB, Davis KM, Lysenko ES, Zhou AY, Yu Y, Weiser JN. Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nature medicine. 2010; 16:228–231.
56. Mercier BC, Ventre E, Fogeron ML, Debaud AL, Tomkowiak M, Marvel J, Bonnefoy N. NOD1 cooperates with TLR2 to enhance T cell receptor-mediated activation in CD8 T cells. PLoS One. 2012; 7:e42170.
57. Franchi L, Warner N, Viani K, Nunez G. Function of Nod-like receptors in microbial recognition and host defense. Immunol Rev. 2009; 227:106–128.
58. Keam B, Im SA, Koh Y, Han SW, Oh DY, Cho N, Kim JH, Han W, Kang KW, Moon WK, Kim TY, Park IA, Noh DY, Chung JK, Bang YJ. Predictive value of FDG PET/CT for pathologic axillary node involvement after neoadjuvant chemotherapy. Breast cancer. 2013; 20:167–173.
59. Azab B, Bhatt VR, Phookan J, Murukutla S, Kohn N, Terjanian T, Widmann WD. Usefulness of the neutrophil-to-lymphocyte ratio in predicting short- and long-term mortality in breast cancer patients. Ann Surg Oncol. 2012; 19:217–224.
60. Shackney SE, Silverman JF. Molecular evolutionary patterns in breast cancer. Advances in anatomic pathology. 2003; 10:278–290.
61. Mohinta S, Wu H, Chaurasia P, Watabe K. Wnt pathway and breast cancer. Front Biosci. 2007; 12:4020–4033.
62. Kuraguchi M, Ohene-Baah NY, Sonkin D, Bronson RT, Kucherlapati R. Genetic mechanisms in Apc-mediated mammary tumorigenesis. PLoS Genet. 2009; 5:e1000367.
63. Allen BA, Terdiman JP. Hereditary polyposis syndromes and hereditary non-polyposis colorectal cancer. Best practice & research Clinical gastroenterology. 2003; 17:237–258.
64. Shibata MA, Maroulakou IG, Jorcyk CL, Gold LG, Ward JM, Green JE. p53-independent apoptosis during mammary tumor progression in C3(1)/SV40 large T antigen transgenic mice: suppression of apoptosis during the transition from preneoplasia to carcinoma. Cancer Res. 1996; 56:2998–3003.
65. Cardiff RD, Anver MR, Gusterson BA, Hennighausen L, Jensen RA, Merino MJ, Rehm S, Russo J, Tavassoli FA, Wakefield LM, Ward JM, Green JE. The mammary pathology of genetically engineered mice: the consensus report and recommendations from the Annapolis meeting. Oncogene. 2000; 19:968–988.
66. Mjosberg J, Svensson J, Johansson E, Hellstrom L, Casas R, Jenmalm MC, Boij R, Matthiesen L, Jonsson JI, Berg G, Ernerudh J. Systemic reduction of functionally suppressive CD4dimCD25 highFoxp3+ Tregs in human second trimester pregnancy is induced by progesterone and 17beta-estradiol. J Immunol. 2009; 183:759–769.
67. Tazawa H, Okada F, Kobayashi T, Tada M, Mori Y, Une Y, Sendo F, Kobayashi M, Hosokawa M. Infiltration of neutrophils is required for acquisition of metastatic phenotype of benign murine fibrosarcoma cells: implication of inflammation-associated carcinogenesis and tumor progression. Am J Pathol. 2003; 163:2221–2232.
68. Nozawa H, Chiu C, Hanahan D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc Natl Acad Sci U S A. 2006; 103:12493–12498.