
INTRODUCTION
Activation of the cyclin D1 oncogene, often by amplification or rearrangement, is a major driver of multiple types of human tumors including breast and squamous cell cancers, B-cell lymphoma, myeloma, nd parathyroid adenoma [1, 2]. The cyclin D1 gene is amplified or overexpressed in up to half of human breast cancers and its mammary-targeted overexpression induces mammary tumorigenesis in mice [3]. Cyclin D1 encodes the regulatory subunit of the cyclin-dependent kinase (CDK4/6) holoenzyme. Tumors overexpressing cyclin D1 tend to display normal levels of proliferation and expression of E2F target genes, which contrasts with tumors overexpressing cyclin E or an activator for pRb [4, 5]. Breast cancers overexpressing cyclin D1 that are wild type for pRb have relatively normal proliferation rates, in contrast to those caused by genetic inactivation of pRb, which show significantly increased proliferation rates [4–6]. Furthermore, the alternate splice form of cyclin D1, (cyclin D1b), has potent transforming ability, which does not correlate with the ability to phosphorylate the pRb protein [7, 8].
Much of the early work defined kinase-dependent functions of cyclin D1 (reviewed in [9]). Cyclin D1/CDK4/6 phosphorylates the retinoblastoma protein (pRb) to advance the G1S and phosphorylates NRF-1 to inhibit mitochondrial biogenesis thereby coordinating nuclear and mitochondrial functions [10–13]. Cyclin D1 regulates a pool of mammary progenitor cells (parity-identified mammary cells: PI-MEC) is kinase-dependent. The resistance of cyclin D1-/-/MMTV-ErbB2 mice to ErbB2 driven mammary tumors is thought to be dependent on a complete absence of PI mammary cells in cyclin D1-null mice [14]. Several other kinase-dependent properties of cyclin D1 have been identified including the induction of cellular migration, enhanced angiogenesis and mammary stem cell self-renewal [15–17].
In addition to the function of cyclin D1 as a regulatory subunit of a CDK holoenzyme, several CDK independent functions have been identified. Cyclin D1 also functions as a transcriptional regulator, usually in a CDK4-independent manner [8]. Cyclin D1 also mediates DNA-damage repair signaling in a CDK4-independent manner [18]. Chromatin immunoprecipitation studies identified cyclin D1 in the context of local chromatin, and the abundance of cyclin D1 determined the recruitment of transcription factors (TF) [19]. The recruitment of cyclin D1 to cis elements enriches for histone acetylases (p300/CBP), histone deacetylases, the methylase SUV39 and the heterochromatin protein HP1α in ChIP [20]. ChIP-ChIP demonstrated cyclin D1 and p300 together occupied genes in close proximity to the transcriptional start site [21], and whole genome ChIP-Seq demonstrated enrichment of cyclin D1 at genes that regulate mitosis and chromosomal stability [22]. In MEFs and in transgenic mice cyclin D1 induced chromosomal instability (CIN) gene expression. CIN occurs frequently in tumors [23] and is characterized by altered rates of loss or gain of whole chromosomes and/or structural chromosomal aberrations [24]. However, the contribution of CIN to the molecular mechanisms governing relatively early changes in tumor progression remains to be fully understood [25, 26], especially in an in vivo context. In view of recent findings that cyclin D1 is capable of inducing aneuploidy and prior findings that the cyclin D1 kinase function appears to be dispensable for several activities, and because of the crucial implications of this mechanism for cancer therapeutics, we determined the importance of cyclin D1 kinase function in the induction of CIN and mammary tumorigenesis in vivo.
RESULTS
Cyclin D1 induction of mitotic abnormalities is kinase-independent
Recent studies using SKY analysis and gene expression profiling have demonstrated that re-expression of cyclin D1WT in cyclin D1-deficient cells results in CIN [22]. In order to test the kinase-independent function of cyclin D1 in aneuploidy and tumorigenesis, we utilized a cyclin D1 point mutant, cyclin D1 K112E (cyclin D1KE), which contains a lysine to glutamine substitution at amino acid position 112 (Supplementary Figure S1A). The cyclin D1KE mutant was unable to bind CDKs in vitro [27]. Cyclin D1KE immunoprecipitated CDK4 and CDK6, and could efficiently bind p27Kip1, however in an in vitro kinase assay the cyclin D1KE complex showed dramatically reduced phosphorylation of pRb [28]. Cyclin D1KE in vivo binds CDK4 and p27Kip1 however the phosphorylation of pRb in vivo was reduced similar to levels seen in cyclin D1–/– mice [29]. In MEFs cyclin D1KE failed to bind CDK4 or p27Kip1 [16]. Collectively these studies demonstrate that the kinase function of cyclin D1KE is abrogated or substantially blunted.
Prior to engaging in studies to question whether the induction of aneuploidy by cyclin D1 is kinase-independent we verified the relative abundance and nuclear localization of cyclin D1KE. In cyclin D1–/– cells rescued with either cyclin D1WT or cyclin D1KE, the protein abundance was similar between the two cell lines (Supplementary Figure S1B). In addition there was no difference in the abundance within the nuclear compartment (Supplementary Figures S1C and S1D). Next we, determined the subcellular compartmentalization of cyclin D1KE and cyclin D1WT. We compared 3T3 wild type cells to 3T3 wild type cells transduced with MSCV-Cyclin D1KE and the localization of exogenous cyclin D1KE and endogenous cyclin D1WT protein monitored during aphidocoline block in G1 to release into S phase. Cyclin D1KE, like endogenous cyclin D1WT, was exported from the nucleus to the cytoplasm (Supplementary Figure S2). Next, to determine whether the alterations in mitotic abnormalities were induced by cyclin D1WT via its CDK-activating function, we performed immunofluorescence followed by high resolution confocal imaging of cyclin D1–/– 3T3 cells, rescued with either cyclin D1WT or cyclin D1KE (Figure 1A). The number of cells with multi-polar spindles was increased 28% in the cyclin D1–/–D1 Rescue cells and 31% in the cyclin D1–/–KE Rescue cells compared to control (p = 0.0051 and p = 0.0004 respectively) (Figures 1A and 1B). The generation of multi-polar spindle cells arising from abnormalities in centrosome number and distribution were quantitatively assessed using α-tubulin staining in conjunction with γ-tubulin. The cyclin D1-/-D1 Rescue and the cyclin D1–/–KE Rescue increased the percentage of prometaphase/metaphase cells with multiple centrosomes by 20% (p = 0.0021) and 28% (p = 0.0007) respectively compared to control cells (Figures 1A and 1C). The alteration of spindle architecture associated with metaphase plate disruption was measured by assessing metaphase plate length and width (ChL, Chw) and spindle length and width (SpL, SpW) (Figures 1D and 1E). Consistent with the increase in spindle/centrosome abnormalities, the ChW and SpL were significantly increased in cyclin D1-/-D1 Rescue and cyclin D1-/-KE Rescue cells compared with cyclin D1-/-Control cells.
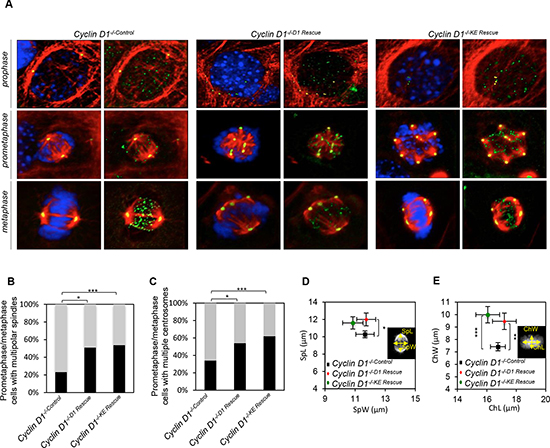
Figure 1: Cyclin D1 induction of centrosome amplification and mitotic spindle disorganization is independent of cyclin D1 kinase activity. (A) Representative confocal maximum Z projections of mitotic cells from cyclin D1-/-Control, cyclin D1-/-D1 Rescue and cyclin D1-/-KE Rescue. Cells were immunostained for α-tubulin (red), γ-tubulin (yellow), crest (green), and Hoechst (blue). Scalebar 5 μm. (B) Frequencies of mitotic cells with multiple polar spindles (**p = 0.0051, ***p = 0.0004; calculated by Fisher contingency test). (C) Frequency of cells with multiple chromosomes (*p = 0.021, ***p = 0.0007; calculated by Fisher contingency test). (D and E) Spindle measurements on maximum Z projections of metaphase cyclin D1-/-, cyclin D1-/-D1 Rescue and cyclin D1-/-KE Rescue cells. Measurement of metaphase plate dimensions (DAPI): ChL, chromatin length; ChW, chromatin width (**p = 0.0087, ***p < 0.001). Measurement of spindle dimensions (tubulin): SpW, spindle width; SpL, spindle length (*p = 0.0486; data are mean of ± SEM).
Cyclin D1KE induces aneuploidy
Spectral karyotyping (SKY) was conducted comparing cyclin D1-/-KE Rescue vs. cyclin D1-/-Control cells. Aneuploidy refers to the loss or gain of whole or partial chromosomes resulting in a complement that differs from an exact multiple of the haploid number. In order to assess the role of cyclin D1KE in aneuploidy we performed SKY analysis at 72 hours and 120 hours after rescue of cyclin D1-/- MEFs. Representative metaphase spreads are shown from analysis of all metaphases (Figures 2A–2C and Supplementary Figure S3A–S3C). At 72 hours cyclin D1-/-KE Rescue induced aneuploidy in 42% of cells, compared to 7% in cyclin D1-/-Control cells. At 120 hours, 100% of cyclin D1-/-KE Rescue cells demonstrated aneuploidy compared to 70% in cyclin D1-/-Control MEFs (Figure 2D, and 2F). Therefore, induction of aneuploidy by cyclin D1 is kinase-independent. SKY analysis assigns chromosomal rearrangements classified as deletions, duplications and translocations. There was no significant difference in chromosomal rearrangements between cyclin D1-/-Control and cyclin D1-/-KE Rescue MEFs.
To further assess the role of cyclin D1 kinase activity in aneuploidy induction we transduced MEFs with cyclin D1KE in the presence and absence of a CDK4/6 antagonist, PD0332991, and assessed the induction of aneuploidy. Karyotyping was conducted comparing cyclin D1-/-KE Rescue vs cyclin D1-/-Control cells. Western blot analysis confirmed CDK4/6 antagonist PD0332991 diminished phosphorylation of pRB at S780 in cyclin D1-/-Control and cyclin D1-/-KE Rescue MEFs; cyclin D1-/-D1 Rescue 3T3 cells were used as a positive control for induction of phosphorylation of pRB at S780 (Supplementary Figure S4A). Representative metaphase spreads and numerical quantitation are shown from analysis of all metaphases from PD0332991 and vehicle treated MEFs (Supplementary Figure S4B). At 72 hours in presence of vehicle cyclin D1-/-KE Rescue induced aneuploidy in 67% (p = 0.027) of MEF cells, compared to 33% in cyclin D1-/-Control cells (Supplementary Figure S4C and S4D). At 72 hours in presence of PD0332991, cyclin D1-/-KE Rescue induced aneuploidy in 87% of cells (p < 0.001), compared to 20% in cyclin D1-/-Control cells (Supplementary Figure S4C and S4E).
In addition to using a CDK4/6 antagonist we also investigated the induction of aneuploidy by cyclin D1 in cdk4/6-/- 3T3 cells. Cdk4/6-/- 3T3 cells were transduced with cyclin D1WT and cyclin D1KE and we assessed the induction of aneuploidy. Western blot analysis of the cell lysates confirmed the cells were cdk4-/- and expressed exogenous cyclin D1WT and cyclin D1KE (Supplementary Figure S5A). At 72 hours in cdk4/6-/- 3T3 cells cyclin D1WT and cyclin D1KE induced aneuploidy in 67% of cells (p = 0.045) and 83% of cells respectively (p = 0.002), compared to 44% in cdk4/6-/-Control cells (Supplementary Figure S5B, S5C, and S5D). Therefore induction of aneuploidy by cyclin D1WT and cyclin D1KE is CDK independent.
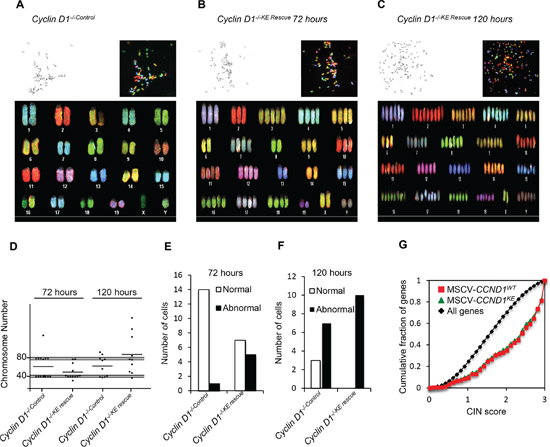
Figure 2: Cyclin D1 kinase-independent induction of aneuploidy. Representative metaphases from spectral karyotyping (SKY) on MEFs of cyclin D1-/-Control at 72 hours (P6) (A), cyclin D1-/-KE Rescue at 72 hours (P6) (B) and cyclin D1-/-KE Rescue at 120 hours (C). Each panel contains the following images: inverted 4’,6-diamidino-2-phenylindole (DAPI) image of the metaphase (top left corner), raw spectral image of the metaphase (top right) and classified metaphase of the same metaphase (lower panel). (D) Scatter plots of chromosomal number across metaphase spreads from cyclin D1-/-Control and cyclin D1-/-KE Rescue cells showing the total number of chromosomes at 72 hours and 120 hours from cells with the noted genotype. The grey shaded bar represents expected deviation from normal at 2N and 4N (+/– 2 chromosomes). Applying the chi-square test of association by comparing cyclin D1-/- versus the cyclin D1-/-D1 Rescue MEFs, and cyclin D1-/-KE Rescue cells yields p < 0.001. (E and F) Bar graphs showing the number of normal and abnormal karyotypes comparing cyclin D1-/-Control and cyclin D1-/-KE Rescue at 72 hours and 120 hours post transduction. (G) An expression profile for cyclin D1-/-D1 Rescue (red line) and cyclin D1-/-KE Rescue (green line) induced genes [16] enriched for high CIN score (p < 0.0001).
Analysis of microarray data of cyclin D1-/-KE Rescue and cyclin D1-/-D1 Rescue vs. cyclin D1-/-Control MEFs demonstrated increased expression of genes associated with a high CIN score [30]. The CIN score was derived by a computational approach to define a gene expression signature that correlates with functional aneuploidy in tumors. The signature predicted poor outcome in 12 cancer data sets from six cancer types. The higher CIN score genes regulate the DNA damage checkpoint, spindle checkpoint and spindle assembly. The induction of high CIN score genes by cyclin D1 was independent of its kinase function (Figure 2G).
Acute induction of Cyclin D1KE leads to expression of high CIN score genes in vivo
To directly determine the role of cyclin D1-mediated kinase activity in promoting mammary tumorigenesis, transgenic mouse models were deployed using either the tetracycline-inducible cyclin D1 transgenic mice (rtTA/CCND1), the Ponasterone inducible mammary epithelial cell targeted cyclin D1-antisense or the MMTV-cyclin D1 transgenic mouse model [12, 22] (Supplementary Figures S6A and S6C). Mammary-targeted expression of cyclin D1 was achieved by crossing transgenic mice carrying a mammary gland targeted recombinant Tetracycline transcription factor (rtTA-Tet ON system) to transgenic mice bearing an rtTA-responsive promoter driving either cyclin D1WT or cyclin D1KE (PTet-CCND1WT and PTet-CCND1KE). The resulting offspring double positive for the transgenes were designated rtTA/CCND1WT and rtTA/CCND1KE (Supplementary Figure S6B). Pregnant females (14 days post coitus) were treated with tetracycline for 7 days, followed by sacrifice of the animals and removal of the thoracic mammary glands for further studies. Western blot analysis verified the induction of the cyclin D1 transgene (Figure 3A). Microarray analysis for gene expression profiles of the mammary glands identified gene clusters regulated by cyclin D1WT and cyclin D1KE (Figure 3B, Supplementary Figure S7A and Dataset S1). There was significant overlap between the gene expression profile regulated by cyclin D1WT and cyclin D1KE (p < 1 × 10-10). Pathway analysis of the genes in common between rtTA/CCND1WT and rtTA/CCND1KE revealed many functional terms previously identified as being cyclin D1 regulated including cell cycle and mitosis (Supplementary Figure S7B). Notably, the rtTA/CCND1WT gene profile was enriched for high CIN score genes to a similar level as the rtTA/CCND1KE gene profile (Figure 3C). Therefore, acute expression of cyclin D1KE was sufficient to induce CIN gene expression profiles within 7 days.
Sustained mammary gland expression of cyclin D1KE induces tumors independent of kinase
Next we employed mammary gland targeted cyclin D1 for a sustained expression study (Supplementary Figure S6C). MMTV-cyclin D1KE and MMTV-cyclin D1WT transgenic mice were monitored twice weekly for the development of mammary tumors. All mice in the tumor kinetics study were nulliparous, thus eliminating any potentially confounding effects of parity on tumor development in the FVB strain. Mice that developed palpable tumors were sacrificed within a week of tumor detection. MMTV-cyclin D1KE tumor incidence (43.8%; n = 32 mice) was similar to MMTV-cyclin D1WT (33.3%; n = 48 mice) (p = 0.358) with a 4-fold (p = 0.0002) and 3-fold (p = 0.0002) greater incidence, respectively, compared to the wild type mice (n = 92 mice) (Figure 3D). A Kaplan–Meier survival (Mammary gland tumor free survival) plot and analyses with a logrank test for curve comparisons were performed among all three lines and between paired lines. The event plotted was the date of sacrifice of the mice that developed tumors. Mice were censored on the date at which they were no longer followed. This included, 1) those that died unrelated to tumor prior to 760 days (censored on the date of death) and 2) those alive without tumor at the end of the study (censored on day 760). Kaplan–Meier survival plots demonstrated kinetics that was similar for both MMTV-cyclin D1KE and MMTV-cyclin D1WT animals (logrank p = 0.237) but significantly different from wild type mice (p < 0.0001 and p = 0.0037, respectively) (Figure 3E). Next, we performed histological analysis of the tumors from MMTV-cyclin D1WT and MMTV-cyclin D1KE mice. The spectrum of histological subtypes of the mammary cancers was similar between MMTV-cyclin D1WT and MMTV-cyclin D1KE mice (Supplementary Table S1). Indeed, it’s the same spectrum that is seen in the ‘background’ of mammary cancers developing spontaneously in wild type mice.
Protein abundance from MMTV-cyclin D1KE transgene in the mammary tumors was similar to MMTV-cyclin D1WT in the mammary gland (Supplementary Figure S8A). The phosphorylation status of a CDK4/6 target site in pRB was substantially reduced in mammary gland tumors of MMTV-cyclin D1KE compared to MMTV-cyclin D1WT tumors (Supplementary Figure S8B). Gene expression profiles of the mammary tumors for MMTV-cyclin D1KE and MMTV-cyclin D1WT mice showed highly significant overlap (p < 1 × 10-10) (Supplementary Figure S9A–S9C and Dataset S2). Furthermore, enrichment for CIN gene expression was observed with both MMTV-cyclin D1WT and MMTV-cyclin D1KE (Figure 3F). There were no significant differences in the CIN score between the MMTV-cyclin D1WT and MMTV-cyclin D1KE tumors. Conversely, mammary epithelial cells from transgenic mice with targeted cyclin D1 anti-sense induced by ponasterone [12] showed a reciprocal change in CIN gene expression (Figure 3F), highlighting a role for endogenous cyclin D1 in maintaining basal CIN gene expression. Reintroduction of either cyclin D1WT or cyclin D1KE into cyclin D1-/- MEFs, transient expression in the mammary gland in transgenic mice, or sustained expression under control of the MMTV promoter, was sufficient for the induction of CIN gene expression; therefore these functions of cyclin D1 are kinase-independent.
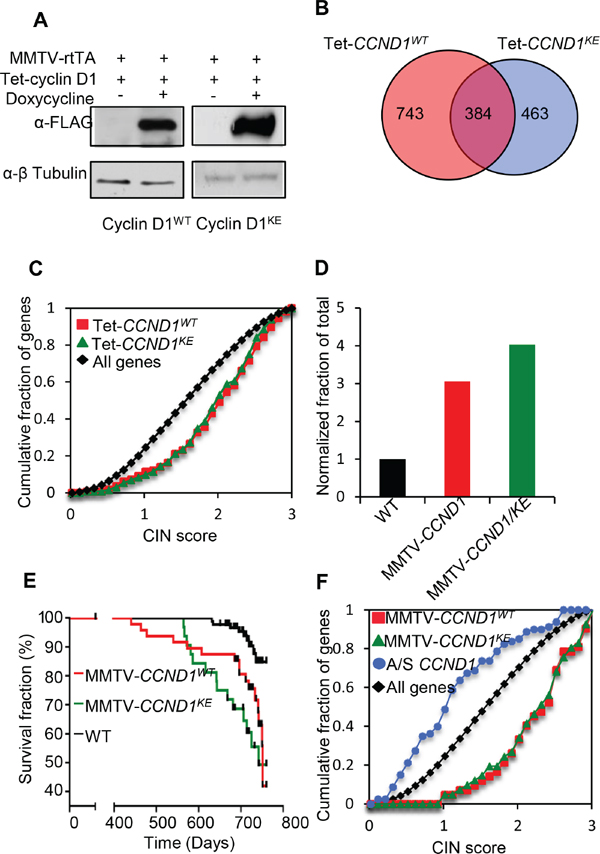
Figure 3: Cyclin D1 induces CIN genes in vivo and mouse mammary tumorigenesis independent of its kinase activity. (A) Western blot using anti-FLAG of mammary gland protein lysates from Tet-CCND1WT and Tet-CCND1KE mice treated with doxycycline compared to control (Left panel). (B) Venn diagram representing genes differentially regulated by Tet-CCND1WT (n = 3) and Tet-CCND1KE (n = 3) (Right panel). 1-way ANOVA was used to evaluate the significance of differential expression between biological conditions. Data represents p < 0.05 and fold change in gene expression >1.5. (C) The most highly differentially regulated genes (Fold >2, p < 0.05) for Tet-CCND1WT (red line) and Tet-CCND1KE (green line) induced genes [16] are enriched for high CIN score (p < 0.0001). (D) Tumor incidence was markedly increased in MMTV-CCND1WT mice and MMTV-CCND1KE mice compared to WT mice. (E) Kaplan–Meier survival curves from mammary tumors of MMTV-CCND1WT (red line) and MMTV-CCND1KE (green line). (F) The most highly differentially regulated genes (Fold >2, B >3) for MMTV-CCND1WT (red line) and MMTV-CCND1KE (green line) induced genes are enriched for high CIN score (p < 0.0001). mRNA from the mammary glands of ponasterone A inducible cyclin D1 antisense mice [12] were subjected to microarray analysis demonstrated reduced CIN gene expression for cyclin D1 induced genes (p < 0.0001).
Recruitment of cyclin D1 to local chromatin is kinase-independent
Cyclin D1 regulates transcription factor (TF) occupancy in chromatin and a cyclin D1-DNA bound form occupies promoter-regulatory regions in the context of local chromatin [21, 23]. In order to determine whether DNA association in chromatin was kinase-dependent we conducted genome wide analysis comparing the cyclin D1WT and cyclin D1KE mutant using ChIP-Seq analysis. The distribution of binding sites by ChIP-Seq in relation to the transcriptional start sites demonstrated binding of active regions within the promoter-region and beyond 10 kb, consistent with a model in which cyclin D1 localizes to both very distal elements and promoter proximal regulatory elements (Figure 4A and 4B) (Supplementary Table S2 and Dataset S3). The tag density profiles for cyclin D1WT and cyclin D1KE demonstrated a similar distribution of genomic association when comparing location at the promoter, within a gene or downstream of the transcriptional start site (Figure 4B). In addition, as in cyclin D1WT, the tag density profiles for cyclin D1KE were enriched at the transcriptional start sites (Figure 4C). Chip-Seq analysis demonstrated significant overlap between cyclin D1WT and cyclin D1KE gene occupancy (1068 genes in common, p = 4.48 × 10-11). Comparison to a previously published gene set from cyclin D1 associated genes by ChIP-ChIP also showed a significant overlap (1505 intervals in common, p = 0.0018, 1144 genes in common, p = 1.61 × 10-12 [21]).
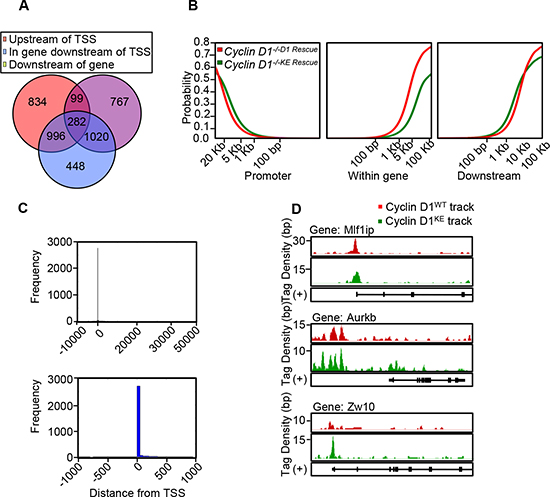
Figure 4: Chip-Seq demonstrates similar characteristics of genomic occupancy for cyclin D1KE and cyclin D1WT. (A) Venn diagram showing distribution of the 4446 intervals with respect to neighboring genes. The interval is depicted in relation to transcriptional start site (TSS). Upstream of TSS defined as –10 kb to 0 kb. Downstream defined as 0 kb to +10 kb after transcriptional stop site. (B) The cumulative fraction of intervals from cyclin D1WT and cyclin D1KE mutant that are within the upstream, intergenic or downstream regions of a gene. (C) Histogram of cyclin D1 bound regions relative to transcriptional start point at –10 kb to +50 kb (Upper panel) and –1 kb to +1 kb (Lower panel). (D) Integrated genome browser visualization of tag density profiles for ChIP-Seq cyclin D1WT and ChIP-Seq cyclin D1KE. Selected genes are, MLF1 interacting protein (Mlf1ip-a kinetochore platform protein), aurora kinase B (AurkB-member of chromosomal passenger complex) and zeste white 10 homolog (Zw10-mitotic check point protein).
Select CIN associated genes showed similar ChIP-Seq tag density profiles for cyclin D1WT and cyclin D1KE (Figure 4D). ChIP analysis of selected target genes governing CIN demonstrated similar relative occupancy for cyclin D1WT and cyclin D1KE (Figure 5A). We then analyzed a broader array of genes governing CIN by QT-PCR, demonstrating similar upregulation of the transcript level by cyclin D1WT and cyclin D1KE (Figure 5B). The enrichment for transcription factor (TF) binding sites identified TF motifs and their statistical significance for the cyclin D1WT and cyclin D1KE (Supplementary Figure 6A and Supplementary Table S3). For the examples shown the prevalence of the TF binding site was similar and significant for both cyclin D1WT and cyclin D1KE. Representative TF motifs most significantly enriched in the cyclin D1WT intervals are shown for the cyclin D1KE intervals (Figure 6B). In addition to associating with the TF motifs, we verified that cyclin D1KE regulated the reporter activity of selected TF responsive elements in a similar manner to cyclin D1WT (Figure 6C).
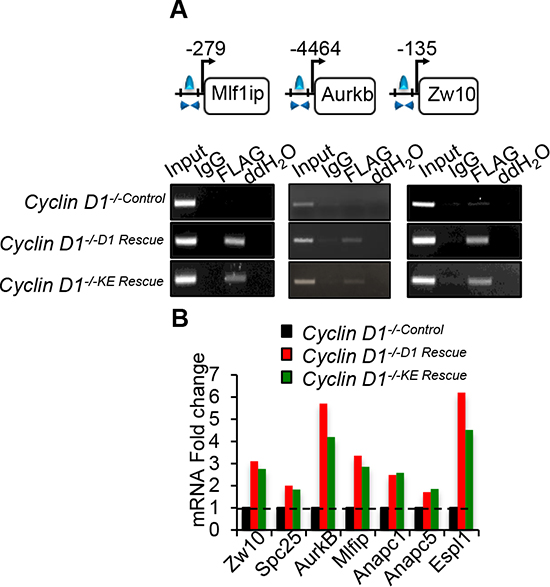
Figure 5: Cyclin D1WT and cyclin D1KE associate with and promotes expression of genes involved in mitosis. (A) Chromatin immunoprecipitation (ChIP) assay performed to assess the association of cyclin D1WT and cyclin D1KE mutant on the promoter regions of selected genes. (B) Quantitative PCR on target mRNAs selected based on cyclin D1KE associated genes. Normalized expression ratio of cyclin D1-/- cells with MSCV-FLAG/CCND1 compared to MSCV-control.
DISCUSSION
The current studies demonstrate that transient cyclin D1 overexpression induces CIN gene expression both in fibroblasts and in the mammary gland in vivo. Previous studies had carefully characterized a mutant of cyclin D1 (cyclin D1KE) demonstrating that it has substantially decreased cyclin-dependent kinase activity using pRb as a substrate [28, 29]. In the current studies, the cyclin D1KE mutant was used to either rescue cyclin D1-/- MEF or was expressed in the mammary gland of transgenic mice. Either reintroduction of cyclin D1KE into cyclin D1-/- MEF, transient expression in the mammary gland in transgenic mice, or sustained expression under control of the MMTV promoter, was sufficient for the induction of CIN gene expression. The induction of CIN gene expression by cyclin D1KE was indistinguishable from the induction of CIN gene expression by cyclin D1WT.
D-type cyclins have been shown to physically bind and to either activate or repress activity of transcription factors [32–34]. In reporter gene assays this function was independent of the CDK activating function [34]. In vivo using cyclin D1-/- mice, the abundance of cyclin D1 was shown to be limiting in the recruitment of transcription factors in the context of local chromatin using ChIP assays [19]. ChIP identified cyclin D1 at transcription factor binding sites of endogenous gene promoters, associated with the recruitment of SUV39, HP1α, HDAC 1, 2, and p300 [20, 31]. Cyclin D1 determined the local acetylation and both di- and tri- methylation of histones [31]. Using serial ChIP analysis of non-coding miRNA regulatory regions, cyclin D1 was identified at the regulatory region of miR17/20 [35]. The current studies are consistent with a role for a DNA bound form of cyclin D1 governing gene expression independent of its kinase function. Furthermore these studies show through quantitative ChIP-Seq studies similar binding patterns for cyclin D1 independent of its kinase function to similar regions of the genome, and similar levels of binding to individual genes governing CIN.
In the current studies sustained expression of either cyclin D1WT or cyclin D1KE induced mammary tumors in transgenic mice with similar kinetics. Consistent with this experimental evidence for cdk independence of cyclin D1’s role as a driver oncogene, human breast cancers overexpressing cyclin D1 do not show high levels of the canonical E2F target gene cyclin E [4, 5] and exhibit relatively normal proliferation rates compared to tumors with genetic deletion of pRb [4–6, 36]. Furthermore, cyclin D1 levels in tumors do not correlate with the marker of proliferating cells, Ki67 [36, 37]. The current studies demonstrate that forced expression of either cyclin D1WT or cyclin D1KE give very similar expression patterns of downstream gene expression, and raise the intriguing possibility that cyclin D1 primarily contributes to oncogenesis through regulating a transcriptional program implicated in CIN.
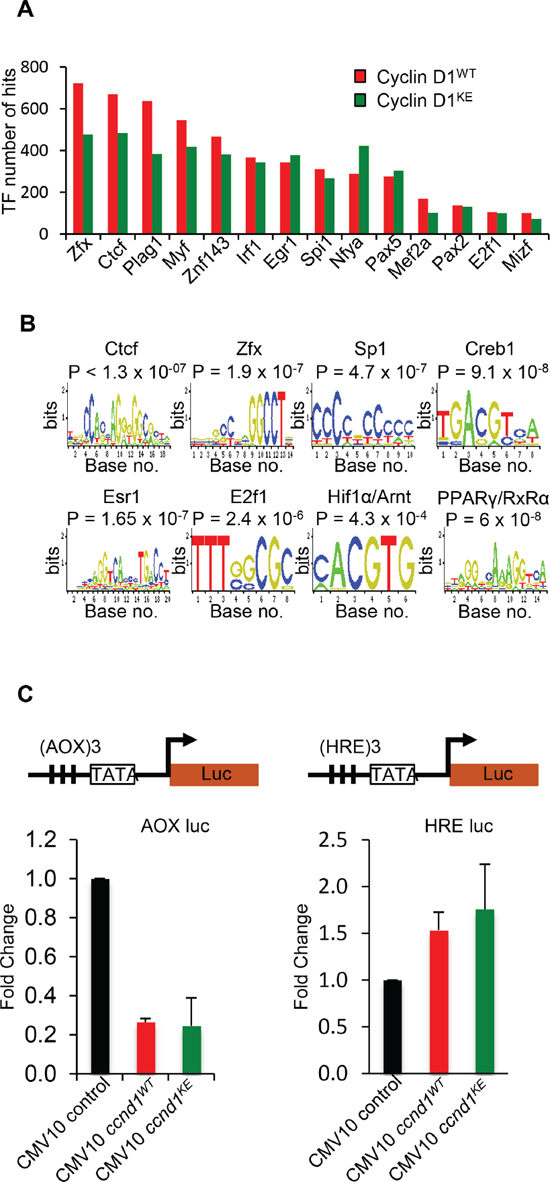
Figure 6: Identification of transcription factor motifs found in cyclin D1WT and cyclin D1KE interval sequences. (A) Selection of transcription factor motif hits common between Cyclin D1WT and cyclin D1KE interval sequences (B) Representative TF motifs found in the interval regions associated with cyclin D1WT and cyclin D1KE (C) Luciferase reporter gene assays were conducted using the Peroxisome Proliferator-Activated Receptor γ (AOX-LUC) (left panel) and Hypoxia Responsive Element (HRE-LUC) (right panel) luciferase reporter constructs. The number of responsive elements for each construct is depicted in the reporter schematic. HEK293T cells were co-transfected with cyclin D1 (50 ng). Data are of n = 2 separate experiments, mean ± SEM.
In contrast, in certain model systems cyclin D1 serves as a mediator of mammary tumorigenesis induced by other oncogenes such as ErbB2, the role of cyclin D1 is CDK-dependent. For example, CDK4-/- mice and cyclin D1KE knock-in mice are resistant to ErbB2-induced mammary tumorigenesis [14, 29, 38]. Together, these studies may illustrate two distinct scenarios reflecting two distinct clinical-pathological settings. Cyclin D1 is overexpressed in the majority of human breast tumors, many of these representing downstream effects through induction of cyclin D1 by oncogenic signals (Ras, MAPK [39]), Src [40], ErbB2 [41], STATs [42], Notch [43], NFκB [44]. Such tumors rely on kinase activity of cyclin D1, and tumor growth could be abrogated not only by inactivation of cyclin D1 but also by CDK4/6 inhibition. CDK4/6 inhibitors (Palbociclib, LEE011, LY2835219), currently in various stages of phase clinical trial, are showing promise as potential therapies in a range of human malignancies [45].
In contrast, cyclin D1 is often overexpressed as a function of genomic rearrangement or amplification. In this setting cyclin D1 is a primary driver oncogene and is experimentally recapitulated by targeted cyclin D1 transgene overexpression. Thus, the present evidence for a CDK-independent role of cyclin D1 in driving mammary tumorigenesis may be especially relevant to human breast cancer, particularly the large subset with clonally selected cyclin D1 gene amplification and potentially the multiple other types of human tumors similarly driven by cyclin D1 amplification or rearrangement [1]. Accordingly, for these tumors, direct therapeutic targeting of cyclin D1 would be predicted to have more efficacy than CDK inhibitors.
MATERIALS AND METHODS
Cell culture and cell lines
The MSCV-IRES-GFP retroviral vector and cyclin D1 wild-type constructs were previously described [46]. Cyclin D1+/+ and cyclin D1−/− primary MEF cultures were prepared as described previously. Cells were maintained in DMEM supplemented with 10% fetal bovine serum, 100 μg/ml each of penicillin and streptomycin. Cdk4/6-/- 3T3 cells were a gift from Dr. M. Barbacid.
Generation of transgenic mice
Two 8 amino acid FLAG tagged constructs were prepared using either human cyclin D1 cDNA (pPL-8) [2] or an otherwise identical cyclin D1 cDNA bearing the “KE” mutation - an AAG to GAG that changes K (lysine) to E (glutamic acid) at amino acid 112, blocking cyclin D1 associated kinase activity. These constructs were inserted into the previously described MMTV-Sv40-BSSk vector [3] (see Supplementary Figure 1A and 5C) and its SalI—SpeI linearized fragment which included MMTV-LTR, the FLAG tagged construct, plus SV40 intron and polyadenylation signals, was microinjected into fertilized FVB/N mouse oocytes and implanted into pseudopregnant FVB fosters using standard methods. Pups were examined for successful insertion of the respective transgenes using tail genomic DNA and PCR primers for the SV40 cassette with confirmation by Southern blotting as described [3]. From these founders and progeny, two independent lines, called MFD1 and MFD1-KE, with robust and comparable levels of transgene expression in mammary tissue as determined by Northern blotting with a human cyclin D1 cDNA probe [1, 3], were selected for expansion and long-term analyses of tumor kinetics. The previously described MMTV-cyclin D1 (no FLAG tag) line MP1 [3] and FVB wild type (WT) mice were used as controls as indicated below.
The cDNA of human cyclin D1 including 3xFLAG sequence was amplified by PCR using p3xFLAG CMV 10-cyclin D1 as template. The restriction sites (Xho I/Not I) were introduced to the primers. The PCR fragment was cloned into pF43 vector. To prepare the DNA fragment for making transgenic mice, the pF43–3xFLAG-cyclin D1 vector was digested with Xho I/Not I/Pvu I. A 2.4-kb DNA fragment was recovered from agarose gel and purified for injection. Transgenic founder lines were backcrossed with wild type FVB mouse for three generations to obtain the stably inherited transgene line, followed by cross mating with MMTV-rtTA line (from Dr. Lewis Chodosh’s lab) to obtain cyclin D1+/+ rtTA+/+ mice (Supplementary Figure 6A and 6B). 6–8 weeks-old female double transgenic mouse was used for further experiments. 8-week-old tetracycline-inducible cyclin D1/rtTA bi-transgenic pregnant female mice (12 days postcoitus) were administered doxycycline in the drinking water to a final concentration of 2 mg/ml. Following 7 days of doxycycline treatment, the mice were sacrificed and mammary glands extracted for tissue fixation and RNA/ protein isolation.
Retrovirus production and infection
Retroviral production and infection of cyclin D1−/− MEFs cells were described in detail previously [46].
ChIP-Seq analysis and transcription factor enrichment
Detailed methods of chromatin preparation, labeling and construction of libraries have been documented previously [22]. For ChIP-Seq analysis, the 35-nt sequence reads (“tags”) identified by Illumina’s Genome Analyzer 2 are mapped to the genome using the ELAND algorithm. Only tags that map uniquely, have no more than 2 mismatches, and that pass quality control filtering are used in the subsequent analysis. Since the 5’-ends of the sequence tags represent the end of ChIP/IP-fragments, the tags are extended in silico (Genpathway software) at their 3’-ends to a length of 110 bp, which is the average fragment length in the size selected library. To identify the density of fragments (extended tags) along the genome, the genome is divided into 32-nt bins and the number of fragments in each bin is determined. The ChIP-Seq peak intervals were determined using the MACs version 1.4 algorithm. We used the default values and provided the FLAG experiment with IgG as a background dataset. We used a p-value of 1.0E-5 as the cutoff for peak detection, which identified 4296 intervals. Supplementary Dataset 1 provides a further summary of the number of intervals found and their position relative to mouse genes. We used the DAVID Functional Annotation Tool to annotate functional enrichment. Transcription factor binding sites were computed as previously described [22].
In order to find the transcription factor binding sites we downloaded the latest version of the mouse genome, mm9, which was released in July 2007 from the USCS Main Genome Browser [47]. Using the Galaxy Toolbox [48] we extracted the sequence 10 kb upstream and downstream of each gene and submitted them to the Jasper server [49] with the default parameters to find all vertabrate transcription factor binding sites. We then assessed the overlap between these transcription factor binding sites and the cyclin D1KE peak intervals. We used a permutation test initially proposed by Tuteja et al, [50]. In brief, this test involves creating psuedo-random in silico ChIP-Seq experiments that accurately reflects a null model of random binding. We shuffled the locations of the windows obtained from the cyclin D1KE ChIP-Seq experiment and then counted the observed number of transcription factor binding sites. We calculate the p-value as the fraction of times in which the random count is larger then the observed count. For this experiment we performed 1.0E + 9 random permutations.
To determining overlap between cyclin D1WT and cyclin D1KE cyclin D1 binding-sites we used both a gene-based method and an interval based method. For the interval based method we used the same permutation method as described for the transcription factor enrichment to determine the overlap between the cyclin D1WT and the cyclin D1KE mutant binding sites. We used the intervals published previously for the cyclin D1WT intervals [22]. Significance of overlap between cyclin D1KE set and ChIP-ChIP data set [21] calculated using the same approach. For the gene-based method we used a hypergeometric test to determine the probability that cyclin D1WT and cyclin D1KE intervals are located in the promoter region or 2 kb upstream of the same genes.
In order to further examine the similarity of the enriched transcription factors we examined the number of transcription factor binding sites within the promoter region of each gene and in the cyclin D1WT or cyclin D1KE intervals. We then fit the difference in the counts between the corresponding transcription factors to a distribution using a Gaussian kernel density estimator [51]. Due to the discrete nature of the distribution we truncated to the maximum difference, 855 counts in this case, and re-normalized the distribution. We then calculated the p-value for each transcription factor as 1-cdf (delta).
Chromatin immuno-precipitation assay (ChIP)
ChIP material was prepared in accordance with the Magna ChIP (Millipore) manufacturer’s guidelines. Briefly, 3 × 10 cm plates of actively growing late passage MEFs cyclin D1-/- MSCV-IRES-cyclin D1 were fixed for 10 min with paraformaldehyde 37% (final concentration 1%). Unreacted formaldehyde was quenched with 1 ml of 10 × glycine. The 3 plates were washed twice with ice cold PBS and the pellets harvested in 1 ml of PBS with protease inhibitor cocktail and pooled together in a 15 ml tube in order to obtain 1.5 × 106 cells. DNA fragmentation of the pellets was achieved by sonication, 35 cycles of 20 seconds each at maximum speed using OMNI-Ruptor 4000 (OMNI International, Inc, Kennesaw, GA). Immuno-precipitation (IP) was performed with 10 μg of M2 FLAG antibody (Sigma-Aldrich, St. Louis, MO) and equivalent amount of mouse IgG as negative control. Washes and elution of the IP-DNA were performed according to the protocol. PCR primers were designed based on the peak interval sequence associated with cyclin D1 and the PCR products were visualized by agarose gel electrophoresis.
ChIP-DNA quantitation was conducted in an Agilent 2100 bio analyzer (Agilent Technologies, Palo Alto, CA), using Power SYBR Green (AB biosciences, Allston, MA) according to the manufacturer’s guidelines. Equal quantities of ChIP-DNA were used for the real-time PCR quantitation. Ct values were used to calculate the relative fold enrichment (2-ΔCt, ΔCt = Ctinput–CtIgG). A one way ANOVA followed by Student’s t-test comparison was performed to compare the relative fold enrichment (n = 3).
Karyotype analysis
For SKY analysis, fluorescence color images of chromosomes stained by Rhodamine, Texas Red, Cy5, FITC and Cy5.5 were captured under a Nikon microscope equipped with a spectral cube and Interferometer module. SKY View software (version 1.62), was used to analyze chromosomal number and structural alterations of chromosomes, including simple balanced translocations, unbalanced (or nonreciprocal) translocations, deletions and duplications. At least 10 metaphases were analyzed per sample. Statistical significance calculated using chi-square test of association (Pearson).
Real-time PCR
RNA quantitation was conducted in an Agilent 2100 bio analyzer (Agilent Technologies, Palo Alto, CA), using Power SYBR Green (AB biosciences, Allston, MA) according to the manufacturer’s guidelines. Equal quantities of RNA were used for the reverse transcription reactions. Primers for all the genes were designed using GenScript’s bioinformatics tools (GenScript, Piscataway, NJ).
Microarray analysis
Genes with differential expression p-value ≤ 0.01 and absolute fold change ≥ 1.25). Mouse MG_U74Av2 microarrays were used for MSCV-rescued MEFs, Mouse 430A_2 microarrays were used for MMTV-Cyclin D1 model (GEO accession number—GSE43216). Arrays were processed as previously described [22]. CIN score enrichment was conducted as described, the comparison to CIN curves from Tet-CCND1WT and MMTV-CCND1WT has been published previously.
Western blotting and luciferase assays
The following antibodies were used for Western blotting: Guanine Nucleotide Dissociation Inhibitor (GDI) [40], Cyclin D1 (NeoMarkers, MS-210-P), FLAG M2 antibody (Sigma Aldrich, #F1804), β-Tubulin (Sigma Aldrich, T4026), phosphorylated RB (S780) (Cell Signaling), cdk4 (H-22) (Santa Cruz Biotechnology Inc.). Luciferase assays were conducted as described previously described [52]. Assays were conducted using 50 ng of plasmid DNA and 100 ng reporter plasmid.
Immunofluorescence and confocal analysis
Immunofluorescence was performed as described previously [22]. Cyclin D1-/-Control, cyclin D1-/-D1 Rescue and cyclin D1-/-KE Rescue subcellular localization was determined using the M2 anti-FLAG antibody (Sigma-Aldrich, #F1804). The whole cell fluorescence intensity per pixel2 was normalized to WT signal intensity. A one-way ANOVA followed by Student’s t-test comparison was performed to compare percentage of fluorescence intensity for cyclin D1 abundance between cyclin D1-/-D1 Rescue and cyclin D1-/-KE Rescue cells (n = 20).
Study approval
Animal studies were approved by the appropriate institutional animal care and oversight committees of the University of Connecticut and Thomas Jefferson University.
Statistical analyses
MACs algorithm was employed to determine number of ChIP-Seq peaks (FDR = 4.35%). Analysis of transcription factor enrichment within the interval sequences produced by the ChIP-Seq data was computed using a permutation test. Enrichment for high CIN scoring genes [30] between two sets compared using Wilcoxon matched paired test. Kaplan–Meier plots were compared by log-rank test. For comparison between two independent groups, the Student’s t-test was used (p < 0.05). Significance of karyotype analysis conducted using Chi-squared test of association.
ACKNOWLEDGMENTS
This work was supported in part by awards from the Susan Komen Breast Cancer Foundation [BCTR0504227 to C.W.] and to A.A.], National Institutes of Health [R01CA70896, R01CA75503 and R01CA86072 to R.G.P.] [R01CA55909 to A.A] [RO1 CA12934 to E.S.K] and the Breast Cancer Research Foundation (R.G.P.). Work conducted at the Sidney Kimmel Cancer Center was supported by the NIH Cancer Center Core grant [P30CA56036 to R.G.P.]. This project was also supported in part from the Dr Ralph and Marian C. Falk medical research trust (to R.G.P.). This project is funded in part by the Pennsylvania Department of Health grant [C.W., R.G.P.] and the Murray–Heilig Fund in Molecular Medicine [A.A]. Supported in part by an American-Italian Cancer Foundation Post-Doctoral Research Fellowship [G.D.]. The Department of Health specifically disclaims responsibility for any analyses, interpretations or conclusions.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
REFERENCES
1. Arnold A, Papanikolaou A. Cyclin d1 in breast cancer pathogenesis. Journal of Clinical Oncology. 2005; 23:4215–4224.
2. Motokura T, Bloom T, Kim HG, Juppner H, Ruderman JV, Kronenberg HM, Arnold A. A novel cyclin encoded by a bcl1-linked candidate oncogene. Nature. 1991; 350:512–515.
3. Wang TC, Cardiff RD, Zukerberg L, Lees E, Arnold A, Schmidt EV. Mammary hyperplasia and carcinoma in MMTV-cyclin D1 transgenic mice. Nature. 1994; 369:669–671.
4. Zukerberg LR, Yang WI, Gadd M, Thor AD, Koerner FC, Schmidt EV, Arnold A. Cyclin D1 (PRAD1) protein expression in breast cancer: approximately one-third of infiltrating mammary carcinomas show overexpression of the cyclin D1 oncogene. Modern Pathology. 1995; 8:560–567.
5. Nielsen NH, Loden M, Cajander J, Emdin SO, Landberg G. G1-S transition defects occur in most breast cancers and predict outcome. Breast Cancer Research and Treatment. 1999; 56:105–112.
6. Jares P, Rey MJ, Fernandez PL, Campo E, Nadal A, Munoz M, Mallofre C, Muntane J, Nayach I, Estape J, et al. Cyclin D1 and retinoblastoma gene expression in human breast carcinoma: correlation with tumour proliferation and oestrogen receptor status. Journal of Pathology. 1997; 182:160–166.
7. Solomon DA, Wang Y, Fox SR, Lambeck TC, Giesting S, Lan Z, Senderowicz AM, Knudsen ES. Cyclin D1 splice variants. Differential effects on localization, RB phosphorylation, and cellular transformation. Journal Biological Chemistry. 2003; 278:30339–30347.
8. Lu F, Gladden AB, Diehl JA. An alternatively spliced cyclin D1 isoform, cyclin D1b, is a nuclear oncogene. Cancer Research. 2003; 63:7056–7061.
9. Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes and Development. 2004; 18:2699–2711.
10. Kato J, Matsushime H, Hiebert SW, Ewen ME, Sherr CJ. Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes and Development. 1993; 7:331–342.
11. Ewen ME, Sluss HK, Sherr CJ, Matsushime H, Kato J, Livingston DM. Functional interactions of the retinoblastoma protein with mammalian D-type cyclins. Cell. 1993; 73:487–497.
12. Sakamaki T, Casimiro MC, Ju X, Quong AA, Katiyar S, Liu M, Jiao X, Li A, Zhang X, Lu Y, et al. Cyclin D1 determines mitochondrial function in vivo. Mol Cell Biol. 2006; 26:5449–5469.
13. Wang C, Li Z, Lu Y, Du R, Katiyar S, Yang J, Fu M, Leader JE, Quong A, Novikoff PM, et al. Cyclin D1 repression of nuclear respiratory factor 1 integrates nuclear DNA synthesis and mitochondrial function. Proc Natl Acad Sci U S A. 2006; 103:11567–11572.
14. Jeselsohn R, Brown NE, Arendt L, Klebba I, Hu MG, Kuperwasser C, Hinds PW. Cyclin D1 kinase activity is required for the self-renewal of mammary stem and progenitor cells that are targets of MMTV-ErbB2 tumorigenesis. Cancer Cell. 2010; 17:65–76.
15. Albanese C, D’Amico M, Reutens AT, Fu M, Watanabe G, Lee RJ, Kitsis RN, Henglein B, Avantaggiati M, Somasundaram K, et al. Activation of the cyclin D1 gene by the E1A-associated protein p300 through AP-1 inhibits cellular apoptosis. Journal of Biological Chemistry. 1999; 274:34186–34195.
16. Li Z, Wang C, Jiao X, Katiyar S, Casimiro MC, Prendergast GC, Powell MJ, Pestell RG. Alternate Cyclin D1 mRNA Splicing Modulates p27KIP1 Binding and Cell Migration. The Journal of Biological Chemistry. 2008; 283:7007–7015.
17. Neumeister P, Pixley FJ, Xiong Y, Xie H, Wu K, Ashton A, Cammer M, Chan A, Symons M, Stanley ER, et al. Cyclin D1 governs adhesion and motility of macrophages. Molecular Biology of the Cell. 2003; 14:2005–2015.
18. Jirawatnotai S, Hu Y, Michowski W, Elias JE, Becks L, Bienvenu F, Zagozdzon A, Goswami T, Wang YE, Clark AB, et al. A function for cyclin D1 in DNA repair uncovered by protein interactome analyses in human cancers. Nature. 2011; 474:230–234.
19. Hulit J, Wang C, Li Z, Albanese C, Rao M, Di Vizio D, Shah S, Byers SW, Mahmood R, Augenlicht LH, et al. Cyclin D1 genetic heterozygosity regulates colonic epithelial cell differentiation and tumor number in ApcMin mice. Molecular and Cellular Biology. 2004; 24:7598–7611.
20. Fu M, Wang C, Rao M, Wu X, Bouras T, Zhang X, Li Z, Jiao X, Yang J, Li A, et al. Cyclin D1 represses p300 transactivation through a cyclin-dependent kinase-independent mechanism. The Journal of Biological Chemistry. 2005; 280:29728–29742.
21. Bienvenu F, Jirawatnotai S, Elias JE, Meyer CA, Mizeracka K, Marson A, Frampton GM, Cole MF, Odom DT, Odajima J, et al. Transcriptional role of cyclin D1 in development revealed by a genetic-proteomic screen. Nature. 2010; 463:374–378.
22. Casimiro MC, Crosariol M, Loro E, Ertel A, Yu Z, Dampier W, Saria EA, Papanikolaou A, Stanek TJ, Li Z, et al. ChIP sequencing of cyclin D1 reveals a transcriptional role in chromosomal instability in mice. The Journal of Clinical Investigation. 2012; 122:833–843.
23. Thompson SL, Bakhoum SF, Compton DA. Mechanisms of chromosomal instability. Current Biology. 2010; 20:R285–295.
24. Gordon DJ, Resio B, Pellman D. Causes, consequences of aneuploidy in cancer. Nature Reviews Genetics. 2012; 13:189–203.
25. Gollin SM. Mechanisms leading to chromosomal instability. Seminars in Cancer Biology. 2005; 15:33–42.
26. Draviam VM, Xie S, Sorger PK. Chromosome segregation and genomic stability. Current Opinions in Genetics Development. 2004; 14:120–125.
27. Hinds PW, Dowdy SF, Eaton EN, Arnold A, Weinberg RA. Function of a human cyclin gene as an oncogene. Proceedings of the National Academy of Sciences. USA. 1994; 91:709–713.
28. Baker GL, Landis MW, Hinds PW. Multiple functions of D-type cyclins can antagonize pRb-mediated suppression of proliferation. Cell Cycle. 2005; 4:330–338.
29. Landis MW, Pawlyk BS, Li T, Sicinski P, Hinds PW. Cyclin D1-dependent kinase activity in murine development and mammary tumorigenesis. Cancer Cell. 2006; 9:13–22.
30. Carter SL, Eklund AC, Kohane IS, Harris LN, Szallasi Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nature Genetics. 2006; 38:1043–1048.
31. Fu M, Rao M, Bouras T, Wang C, Wu K, Zhang X, Li Z, Yao TP, Pestell RG. Cyclin D1 inhibits peroxisome proliferator-activated receptor gamma-mediated adipogenesis through histone deacetylase recruitment. The Journal of Biological Chemistry. 2005; 280:16934–16941.
32. Pestell RG, Albanese C, Reutens AT, Segall JE, Lee RJ, Arnold A. The cyclins and cyclin-dependent kinase inhibitors in hormonal regulation of proliferation and differentiation. Endocrine Reviews. 1999; 20:501–534.
33. Zwijsen RM, Buckle RS, Hijmans EM, Loomans CJ, Bernards R. Ligand-independent recruitment of steroid receptor coactivators to estrogen receptor by cyclin D1. Genes Development. 1998; 12:3488–3498.
34. Fu M, Wang C, Li Z, Sakamaki T, Pestell RG. Minireview: Cyclin D1: Normal and Abnormal Functions. Endocrinology. 2004; 145:5439–5447.
35. Yu Z, Willmarth NE, Zhou J, Katiyar S, Wang M, Liu Y, McCue PA, Quong AA, Lisanti MP, Pestell RG. microRNA 17/20 inhibits cellular invasion and tumor metastasis in breast cancer by heterotypic signaling. Proceedings of the National Academy of Sciences USA. 2010; 107:8231–8236.
36. Oyama T, Kashiwabara K, Yoshimoto K, Arnold A, Koerner F. Frequent overexpression of the cyclin D1 oncogene in invasive lobular carcinoma of the breast. Cancer Research. 1998; 58:2876–2880.
37. Shoker BS, Jarvis C, Davies MP, Iqbal M, Sibson DR, Sloane JP. Immunodetectable cyclin D(1)is associated with oestrogen receptor but not Ki67 in normal, cancerous and precancerous breast lesions. British Journal of Cancer. 2001; 84:1064–1069.
38. Yu Q, Sicinska E, Geng Y, Ahnstrom M, Zagozdzon A, Kong Y, Gardner H, Kiyokawa H, Harris LN, Stal O, et al. Requirement for CDK4 kinase function in breast cancer. Cancer Cell. 2006; 9:23–32.
39. Albanese C, Johnson J, Watanabe G, Eklund N, Vu D, Arnold A, Pestell RG. Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. The Journal of Biological Chemistry. 1995; 270:23589–23597.
40. Lee RJ, Albanese C, Stenger RJ, Watanabe G, Inghirami G, Haines GK 3rd, Webster M, Muller WJ, Brugge JS, Davis RJ, et al. pp60(v-src) induction of cyclin D1 requires collaborative interactions between the extracellular signal-regulated kinase, p38, and Jun kinase pathways. A role for cAMP response element-binding protein and activating transcription factor-2 in pp60(v-src) signaling in breast cancer cells. The Journal of Biological Chemistry. 1999; 274:7341–7350.
41. Lee RJ, Albanese C, Fu M, D’Amico M, Lin B, Watanabe G, Haines GK 3rd, Siegel PM, Hung MC, Yarden Y, et al. Cyclin D1 is required for transformation by activated Neu and is induced through an E2F-dependent signaling pathway. Molecular and Cellular Biology. 2000; 20:672–683.
42. Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE Jr. Stat3 as an oncogene. Cell. 1999; 98:295–303.
43. Stahl M, Ge C, Shi S, Pestell RG, Stanley P. Notch1-induced transformation of RKE-1 cells requires up-regulation of cyclin D1. Cancer Research. 2006; 66:7562–7570.
44. Joyce D, Bouzahzah B, Fu M, Albanese C, D’Amico M, Steer J, Klein JU, Lee RJ, Segall JE, Westwick JK, et al. Integration of Rac-dependent regulation of cyclin D1 transcription through a nuclear factor-kappaB-dependent pathway. The Journal of Biological Chemistry. 1999; 274:25245–25249.
45. Casimiro MC, Velasco-Velazquez M, Aguirre-Alvarado C, Pestell RG. Overview of cyclins D1 function in cancer and the CDK inhibitor landscape: past and present. Expert Opinion on Investigational Drugs. 2014; 23:295–304.
46. Li Z, Wang C, Jiao X, Lu Y, Fu M, Quong AA, Dye C, Yang J, Dai M, Ju X, et al. Cyclin D1 regulates cellular migration through the inhibition of thrombospondin 1 and ROCK signaling. Molecular and Cell Biology. 2006; 26:4240–4256.
47. Kuhn RM, Haussler D, Kent WJ. The UCSC genome browser and associated tools. Briefings in Bioinformatics. 2013; 14:144–161.
48. Taylor J, Schenck I, Blankenberg D, Nekrutenko A. Using galaxy to perform large-scale interactive data analyses. Current Protocols in Bioinformatics. 2007; Chapter 10:Unit 10 15.
49. Portales-Casamar E, Thongjuea S, Kwon AT, Arenillas D, Zhao X, Valen E, Yusuf D, Lenhard B, Wasserman WW, Sandelin A. JASPAR 2010: the greatly expanded open-access database of transcription factor binding profiles. Nucleic Acids Research. 2010; 38:D105–110.
50. Tuteja G, White P, Schug J, Kaestner KH. Extracting transcription factor targets from ChIP-Seq data. Nucleic Acids Research. 2009; 37:e113.
51. Silverman BW. (1986). Density Estimation for Statistics and Data Analysis. (London: Chapman and Hall).
52. Wu K, Jiao X, Li Z, Katiyar S, Casimiro MC, Yang W, Zhang Q, Willmarth NE, Chepelev I, Crosariol M, et al. Cell fate determination factor Dachshund reprograms breast cancer stem cell function. The Journal of Biological Chemistry. 2011; 286:2132–2142.