
INTRODUCTION
Pseudomyxoma peritonei (PMP) is a rare epithelial neoplasm, of mainly appendiceal origin [1], which if untreated, is characterized by disseminated peritoneal mucinous tumor deposition and progressive accumulation of mucinous ascites [2]. Distant and nodal metastases are the exception. PMP has an estimated incidence of 1–2 per million in Western populations [3]. In nationally representative series [4, 5], 30–70% of patients at presentation are suitable to undergo loco-regional surgical treatment involving the combination of macroscopic tumour excision, described as cytoreductive surgery (CRS), and hyperthermic intraperitoneal chemotherapy (HIPEC)—often referred to as the “Sugarbaker procedure”, following his work in developing the techniques involved [6].
Due to the rarity of this tumor, biological understanding is limited. There have been a small number of reports on PMP that focused on: (i) selected pathways determining protein expression, for example, mucins [7–9]; epithelial-mesenchymal transition proteins [7, 8]; and CDX2 expression [10]; and (ii) on selected genetic mutations, for example, quantifying the proportion of samples with K-RAS [11, 12], GNAS [13], and p53 mutations [14]—but no global characterisation approaches.
For chemotherapy options, PMP is generally considered to be resistant. During HIPEC administration, the commonest used agent is mitomycin C [15], though other agents including oxaliplatin and cisplatin, with and without concurrent systemic 5-fluorocuracil (a fluoropyrimidine), are also administered [6]. By the intra-peritoneal route, these agents are delivered in concentrations considerably higher than those used systemically. The rationale for their selection is based on empirical extrapolation from treatments of colorectal cancer. Despite the radicality of CRS and HIPEC, there is a recognized propensity for disease recurrence and progression. For the latter, and in patients deemed unsuitable for initial radical surgery, the natural history is characterized by high levels of morbidity (e.g. abdominal distension, discomfort, fistulation), and demise due to disease progression. We have previously reported a phase II trial in this setting, using systemically combined mitomycin and oral fluoropyrimidine-based chemotherapy, capecitabine (MCap), but with short-term stabilization of disease of a few months in only a third of patients [16]. Against this background, there is a clear need to improve the effectiveness of current chemotherapy regimens and/or develop new anti-PMP agents.
In this study, we address the two aforementioned needs in translational research for PMP. First, we performed exon array analysis from laser micro-dissected PMP tissue and comparative normal colonic epithelia; identified and confirmed differential expression of the candidate genes and their protein products in tissue. In parallel, we established two primary PMP cell lines. From our previously experiments [7], we learnt that primary PMP cell lines are slow-growing cells, with limited viability, and unfit for high-throughput experiments. Thus, here, we immortalized these cell lines with an SV40 T-antigen lentiviral vector, and cross-checked for differentially expressed genes, from the array analyses, using qPCR.
RESULTS
It is technically challenging to work with PMP epithelial tissue as it exists in small clusters in an ‘ocean’ of mucin. We developed laser capture micro-dissection methods to maximize epithelial yield from specimens that were confirmed histologically as PMP (Figure 1A).
Gene microarray analysis
We performed exon-array analysis comparing three disseminated (all omentum) plus one appendiceal PMP samples versus three samples of normal colonic mucosa. Initial PCA plots of the expressed genes demonstrated that the normal versus omental samples clustered to distinct populations at both the probeset and gene level (Figure 2A). These differences were not due to adipose tissue contamination of the omentum samples, as when the appendiceal PMP sample was added, it clustered with the omentum samples suggesting true differences between normal and diseased states. Overall, there was a high level of homogeneity (see Figure 1 Legend).
The differences in expression of the identified genes with greater than two fold changes were visualized using a heat-map (Figure 2B). For disease PMP tissue versus normal, 450 genes were identified as differentially expressed. The differential expressions were similar whether or not the appendiceal sample was included. After adjustment for multiple testing, 27 genes up-regulated in PMP were statistically significant with p values less than 0.05; thirty four genes were significantly down-regulated. These are listed, with descriptions of their main biological functions, in Supplementary Table S1. From these lists, we selected to explore in greater detail eight genes, based on (i) statistical significance; (ii) known biological function; and (iii) availability of probes and antibodies for validation (Figure 2C). The selected up-regulated genes were: SLC16A4, a proton-linked monocarboxylate transporter; DSC3 (desmocollin 3), a component of intercellular desmosome junctions; ALDOB, a fructose-1,6-bisphosphate aldolase; EPHX4, a hydrolase; and ARHGAP24, a Rho GTPase-activating protein involved in cell polarity, cell morphology and cytoskeletal organization. The commonly used PMP marker, MUC2, was increased by 1.8 log fold increase in the PMP samples compared with normal colonic mucosa expression, just outside our a priori 2-fold cut-off. The down-regulated genes were: MS4A12, a component of a multimeric receptor complex; TMIGD1, a transmembrane and immunoglobulin domain-containing protein-1; and Caspase-5, a mediator of apoptosis.
Gene and protein product expression in PMP tissue
To validate the above observed differences in gene expression, and in the absence of a suitable antibody, we performed in-situ hybridization (ISH) for the top up-regulated gene, SLC16A4, on PMP (n = 3) versus normal colonic mucosa (n = 3) samples (Figure 3). This confirmed our findings from the exon array analysis, that expression of SLC16A4 is elevated in PMP.
For immunohistochemical (IHC) staining, there was no difference in expression intensities between PMP and normal colonic epithelia for the clinically used tissue markers, CDX2, MUC2 and KRT20. However, the distribution of MUC2 is altered in PMP: MUC2 expression limited to goblet cells in normal intestinal tissue, but universally expressed in PMP cells, consistent with the notion that PMP is a goblet cell neoplastic process. Additionally, whereas KRT20 is limited to the colonic table in normal colonic epithelia, its expression is ubiquitous in PMP (Supplementary Figure S1).
Image analysis of the staining for DSC3 (desmocollin 3), Aldolase B, EPHX4 and ARHGAP24 generally agreed with the exon array demonstrating significant increases in PMP tissue (3 diseased appendix; 6 disseminated disease) compared with either normal colonic (n = 7) or normal appendiceal (n = 2) epithelia (Figure 4A). IHC staining for the top down-regulated gene, MS4A12, revealed apical membrane staining of the colonocytes in normal colon samples, as previously described [17], and an absence of staining in the PMP samples consistent with the exon array results (Figure 4B). Comparative expressions for other markers (TMIGD1 and Caspase-5), which were down-regulated in the gene analysis, were equivocal on IHC due to the low levels of expression in the normal tissue and background staining of the mucin component of the PMP samples, and no formal statistics were performed.
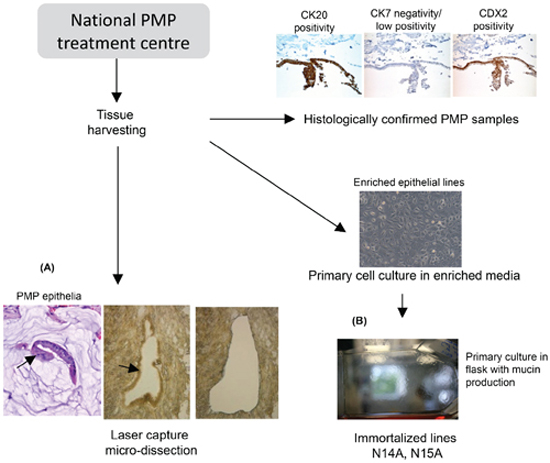
Figure 1: Overview flow diagram of the tissue harvest and cell line studies. It is technically challenging to work with PMP epithelial tissue as it exists in small clusters in an ‘ocean’ of mucin (arrow in H&E section). (A) We developed laser capture micro-dissection methods to maximise epithelial yield from specimens that were confirmed histologically as PMP. In parallel, we generated primary PMP cell cultures from confirmed PMP tissue. (B) shows that the immortalized cells exhibit the characteristic “cobblestone” epithelial morphology cells, and produce a layer of mucin on the flask—equivalent to the clinical phenotype.
Establishment of two immortalized PMP cell lines
In parallel, we developed cell lines from samples from patients with histological confirmed PMP. Lines were established from multiple patients and multiple disease sites per patient resulting in approximately 50 primary cell lines. Of these, two sustainable cell lines underwent further characterization, N14A and N15A, both derived from primary appendiceal disease tissue, each from separate patients. Immortalized cells exhibit the characteristic “cobblestone” epithelial morphology cells, produce a layer of mucin on the flask (Figure 1B) (but do not stain for MUC2 due to the highly glycosylated nature of the mucin), and have a doubling time of approximately 48 h after clonal expansion. The short tandem repeats (STR) ‘finger printing’ profile of the cell lines was matched to genomic DNA isolated from FFPE samples from the same patients, and confirm identical sources. Additionally, the STR profiles did not match known cell line profiles from the ATCC (data available from authors on request).
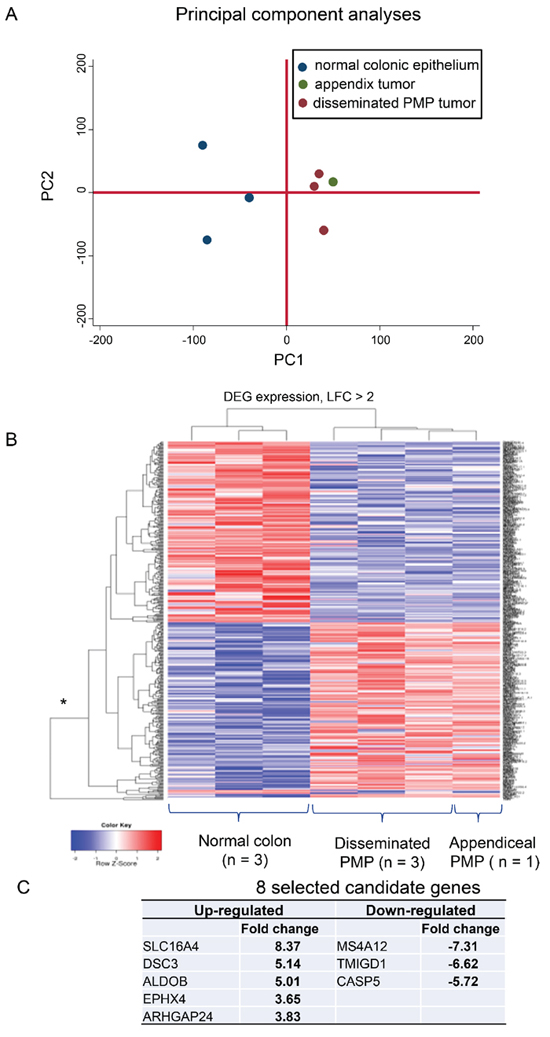
Figure 2: Exon array analysis identified 27 up-regulated and 34 down-regulated genes in PMP epithelial tissue (p < 0.05, adjusted for multiple testing) compared with normal colonic mucosa. There was a high levels of homogeneity demonstrated by (A) the distinctive clustering between normal and neoplastic tissue in the principal component analysis and (B) the shallow heights of the DEG expression bars. (C) Eight candidate genes were selected and explored in greater detail. DEG: differentially expressed genes; LFC: log fold change.
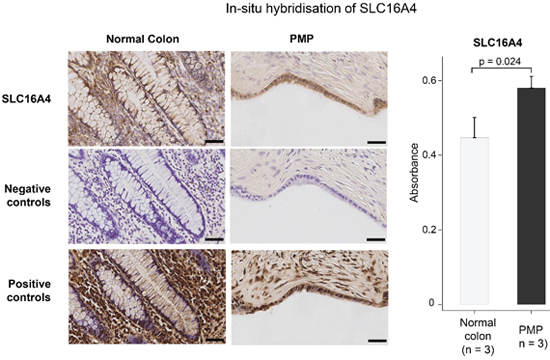
Figure 3: In-situ hybridization staining for gene expression of up-regulated gene, SLC16A4, in normal colon and disseminated PMP, with positive and negative control probes. Scale bar represents 100 μm on low power images and 50 μm on high power images. Data in the graphs represent the mean ± SD for SLC16A4 expression by PMP versus normal colonic mucosa. Absorbance determined using the Definiens software. For the PMP tissue, a correction was made to account for the dilutional effect of mucin. p value derived from Student t-test.
Cross-validation of differential gene expression in cell lines
The PMP cell lines were screened by qPCR for levels of our candidate eight genes. In the absence of a widely available human normal intestinal lines, there is no natural comparator group. We thus compared with the same gene expressions (up-regulated transcripts: SLC16A4, DSC3, ALDOB, EPHX4, ARHGAP24; and down-regulated transcripts (MS4A12, TMIGD1, CASP5) in 5 colon cancer lines: HCT 116, Caco2, HT-29, Lovo, and C32. In general, gene expression of the candidates were similar between the PMP cell lines, but within the colon cancer cell panel, there was considerable variation of the expression of these candidate genes, and in turn, several differed to the PMP lines—as illustrated in Figure 5. These experiments also confirmed that gene expression of ARHGAP24 is highly elevated among the PMP lines compared with the 5 colon cancer cell lines.
Gene profiles differ in PMP compared with colorectal neoplasia
The transcription profiles of PMP were compared with those of colorectal neoplasia (colorectal adenocarcinoma, mucinous adenocarcinoma and adenoma) by comparing changes in transcript level in this study with those reported in equivalent studies in the Oncomine database. The fold change observed for each transcript was then plotted for each comparison on the log scale. We compared the top nine up-regulated transcripts (SLC16A4, DSC3, ALDOB, EPHX4, SKAP1, ARHGAP24, NTE5, SNHG8, ADAM9) and top eleven down-regulated transcripts (MS4A12, TMIGD1, CASP5, BEST4, GPR98, DHRS11, PAG1, HHLA2, PRR5L, SEMA6D, ZG16) from our PMP gene list, which were also present within the Oncomine database. Several transcripts were up-regulated in PMP and in colorectal tumors, but the extent of the changes were distinct. Thus, up-regulated expression was greater in PMP for SLC16A4, DSC3, ALDOB compared with colorectal tumors. However, three transcripts (SKAP1, ARHGAP24, and NT5E) were up-regulated in PMP but generally down-regulated in colorectal tumors (Figure 6A). For down-regulated transcripts, there was commonality with colorectal tumors, although the extent of reduced expression was generally greater in colorectal tumors compared with PMP (Figure 6B).
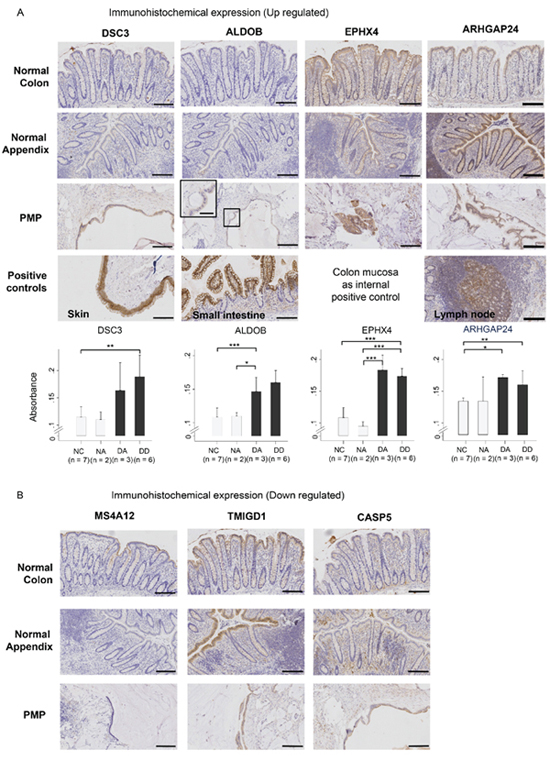
Figure 4: Immuno-histochemical staining for protein product expression of (A) up-regulated genes (DSC3, ALDOB, EPHX4, ARHGAP24) and (B) down-regulated genes (MS4A12, TMIGD1, CASP5) in normal colon (NC) and normal appendiceal (NA) mucosa, diseased appendix (DA), and disseminated disease (DD), with positive and negative controls. Data in the graphs represent the mean ± SD, and differences tested by Student’s t-test between comparative pairs from either diseased groups (DA, DD) versus normal epithelia groups (NC, NA) (*p < 0.01; **p < 0.001; ***p < 0.0001). Absorbance determined using the Definiens software. Scale bar represents 100 μm on low power images and 50 μm on high power images.
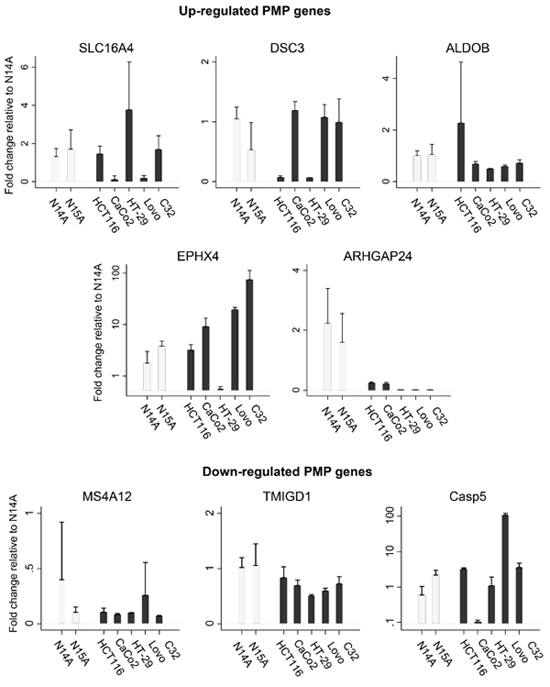
Figure 5: In order to validate the comparative analyses observed in the Oncomine database, we determined qPCR gene expression for candidate up-regulated and down-regulated PMP genes in N14A and N15A relative to 5 colon cancer cell lines. In the absence of a widely available human normal intestinal lines, there is no natural comparator group. Nonetheless, the gene expression is relatively consistent for the two PMP cell lines, but differ considerable compared with colon cell lines for each candidate gene. For plots of up-regulated PMP genes, data have been standardised against ALDOB N14A; for plots of down-regulated PMP genes, data have been standardised against TMIGD1 N14A. Note different y-axis scales.
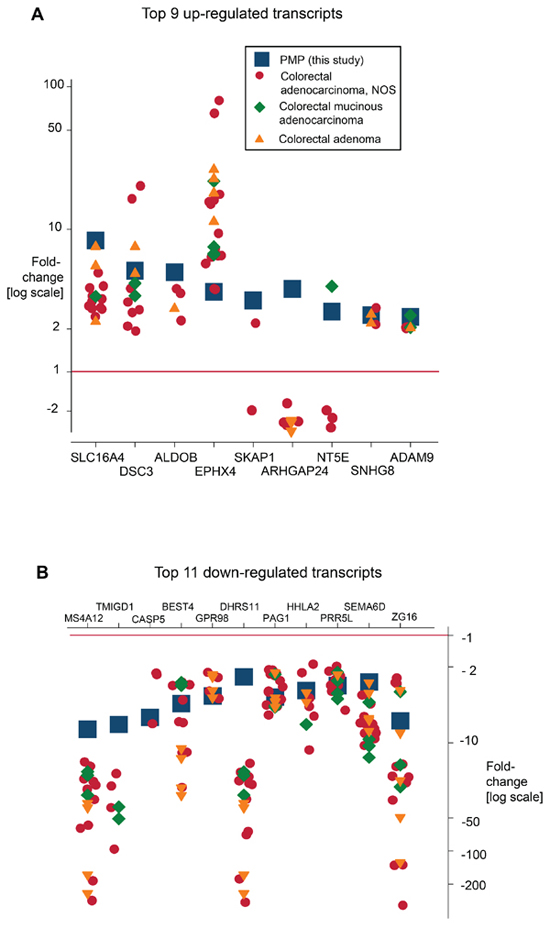
Figure 6: The transcription profile of PMP was compared with those of colorectal tumours (colorectal adenocarcinoma, mucinous adenocarcinoma and adenoma) by comparing changes in transcript level in the present study with those reported in similar studies in the Oncomine database. Changes were reported using similar comparisons (normal vs tumor, mRNA/cDNA input) and similar array platforms were recorded. The fold change observed for each transcript was then plotted for each comparison on the log scale. For PMP, (A) nine up-regulated transcripts (SLC16A4, DSC3, ALDOB, EPHX4, SKAP1, ARHGAP24, NTE5, SNHG8, ADAM9), and (B) eleven down-regulated transcripts (MS4A12, TMIGD1, CASP5, BEST4, GPR98, DHRS11, PAG1, HHLA2, PRR5L, SEMA6D, ZG16) were compared with the same gene expressions in colorectal tumors. Clear patterns emerged (summarized in main manuscript).
Preliminary cytotoxicity testing
To test the utility of the cell lines, N14A and N15A, as pre-clinical models of PMP, the sensitivity of these cells to Mitomycin C (MMC) and Oxaliplatin, agents currently used in the treatment of PMP, were determined (Figure 7). The dose response curves (using doses spanning the typical clinical maximum dose range) and IC50 values indicate that the two PMP cells were more sensitive to MMC than Oxaliplatin, and that the sensitivities were similar for MMC and slightly less for Oxaliplatin, compared with the same agents against the colon cell line, HT-29. As a proof of principle that these cells might be used as a pre-clinical tool for novel agents, we tested the sensitivity of the PMP cell to phloretin, an apple metabolite previously reported as an inhibitor of lactate and pyruvate transport [18] and used to inhibit monocarboxylate transporters (similar to SLC16A4) [19]. We found IC50 values equivalent to those for Oxaliplatin, and again, the sensitivities were very similar to those against the colon cell line, HT-29.
DISCUSSION
Main findings
We have undertaken a global gene exon array analysis in histologically confirmed PMP tissue, and identified differentially genes as candidates that may underpin the unique natural history of this malignancy, and shown that these dysregulated genes and their protein products are representative in a series of human PMP samples. For the first time, we additionally developed two immortalized PMP cell lines, N14A and N15A, and established a platform for future pre-clinical anti-tumor drug testing and oncogene discovery in PMP. We cross-checked these novel cell lines against the differentially expressed genes (from the array analyses) using qPCR and found consistencies. Finally, we demonstrated that the gene profiles in PMP are distinct from those of colorectal tumor libraries and commonly used colon cell lines.
Context of literature
The basic pathobiology of PMP is understudied, reflecting its rarity and its historic pathological confusion with other neoplastic processes, namely ovarian cancer. Previous studies have reported on altered expression of selected protein pathways [7–10] and selected genetic mutations [11–14]. One recent study [20] reported a genomic profiling analysis in 19 PMP specimens using next-generation sequencing, but this was limited to a 48 cancer-related gene amplicon. By contrast, this current study performed a global expression analysis and identified a number of potential candidate genes. First, SLC16A4, a monocarboxylate transporter (of lactate, pyruvate and other monocarboxylates), was the most significantly overexpressed gene in our analysis. SLC16A4 is expressed in the intestine [21, 22] and is a potential therapeutic target in conditions characterized by hypoxia [23]. Whilst we speculate that hypoxia is not a dominant characteristic of PMP, glucose and fatty acid metabolism of PMP cells might be dysregulated, and may in part explain the production of the large amount of mucus observed in this disease. Highly elevated SLC16A4 expression appears to be PMP specific, with equivalent elevated levels limited to only some colorectal tumors and cancer cell lines. The second overexpressed candidate gene was desmocollin-3 (DSC3), a member of the cadherin family expressed in epithelial cells [24]. Switching from desmocollin-2 to desmocillin-3 has been noted in colorectal cancer development [25] and the methylation of the DSC3 promoter is a prognostic marker of colorectal cancer [26]. PMP is unique in its pattern of growth and dissemination as it exists as strings of cells within pools of mucus loosely attached to surrounding tissue. Overexpression of DSC3 might maintain the cell-to-cell contacts leading to the string-like morphology. Third was Aldolase B (ALDOB), an enzyme involved in fructose metabolism, which is known to be overexpressed in ulcerative colitis [27]. Loss of ALDOB is associated with early recurrence and poor prognosis in hepatocellular carcinoma [28]. The up-regulated expression of ALDOB in PMP may reflect the potential high metabolic requirements to sustain the production of large amounts of mucin. Fourth was epoxide hydrolase 4 (EPHX4), an enzyme involved in carcinogen inactivation with reduced levels of epoxide hydrolase activity and linked to increased colorectal cancer risk [29]. Elevated levels of EPHX4 in PMP may reflect its relatively differentiated state.
Of the down-regulated gene, MS4A12 is a colon selective calcium channel with expression normally restricted to the apical membrane of colonocytes. Loss of MS4A12 in Lovo cells attenuates EGFR mediated effects, resulting in reduced invasion, chemotactic motility and proliferation [17], which matches with the known phenotype of PMP (low mitogenic potential and no distant metastasis formed) [7]. MS4A12 is elevated in colorectal cancer, but down-regulated in our PMP samples despite being a known CDX2 transcriptional target, often up-regulated in PMP [30]. This may reflect different pathways initiated by CDX2 in appendiceal tissue in comparison with colonic tissue. TMIGD1, (transmembrane and immunoglobulin domain containing protein-1) is a relatively unknown protein, though with known expression in the colon (Unigene database). Caspase-5 is involved in the inflammatory response and frameshifts are often found in endometrial [31] and lung [32] cancers. In addition, caspase-5 cleaves Max, part of the Myc network of transcription factors, and therefore, may play a role in tumorigenesis [33].
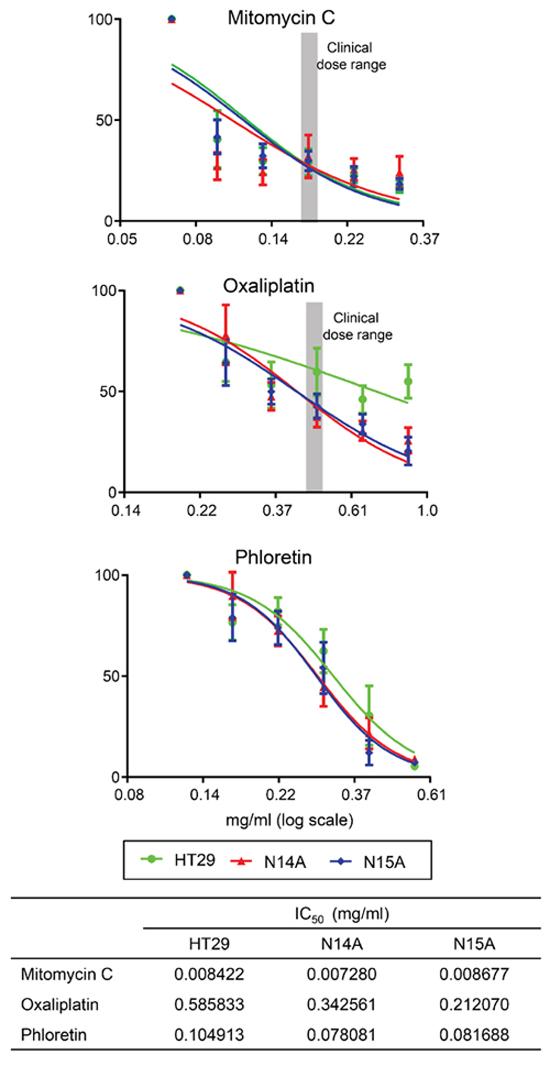
Figure 7: We tested the in vitro sensitivity of the immortalized cell lines (N14A, N15A) and a comparator colon cancer cell line, HT-29, against commonly used chemotherapy agents—mitomycin C and oxaliplatin—and Phloretin, an apple metabolite previously reported as an inhibitor of lactate and pyruvate transport, and used here as an example of a pathway inhibitor. To determine IC50 values, cells were seeded in complete medium and allowed to adhere for 24 h prior to drug treatment. Media were replaced following 24 h exposure to the drugs and the cells were incubated under normal culture conditions for 4 days before the MMT assay. For mitomycin and oxaliplatin, we selected a range of concentrations centred round clinically relevant values (grey-shaded area). The cytotoxic efficacy was evaluated after determining respective IC50 values, the concentrations that reduced growth by 50%, using GraphPad Prism software (bottom panel). All experiments were performed at 37°C. Data in the graphs represent the mean ± SD of at least three independent experiments performed in triplicate.
Limitations and strengths
There are potential limitations to this study. First, PMP is a heterogeneous clinico-pathological disease, such that our modest sample size may not have captured its diversity. Nonetheless, the homogeneity of the gene microarray analysis was noteworthy and allowed us to derive and generate hypotheses based on small sample numbers. Second, we did not measure mRNA expression, other than for SLC16A4. However, previous studies have noted strong correlations between protein and mRNA expression in colorectal tissue [34], and we propose a similar association in PMP tissue. Third, we utilized normal colonic epithelia in our micro-array analyses which may differ in its gene expression patterns to normal appendiceal epithelia. However, we confirmed similarities in the expression of the protein products of our candidate genes in both colonic and appendiceal tissue. Finally, a potential limitation was the use of the SV40 large T antigen to immortalize the cell lines, which may modify the genetic characteristics of these cells. Nonetheless, this technology is commonly used in other cell line systems, and for our PMP lines, the expressions of the key candidate genes mirrored those from the in vivo human PMP samples.
Strengths of the study are the following. First, working within the framework of national treatment center, we optimized protocols for the collection of fresh ‘disease-rich’ tissue and pathological confirmation. Second, as part of our previous published work [7], a learning set of PMP samples was used to explore histopathological features for IHC. This is critical, as the cellular components on PMP are sparse and many sections are required to establish optimal representation. Third, previous attempts to (spontaneously) develop a PMP cell line have resulted in limited success [7]; we have improved on this with the immortalization of the cells using a lentivirus to express the entire SV40 genome; successfully used in other intestinal in vitro systems, and here, achieving viability to over 20 passages. Fourth, to validate our exon array findings, we tested gene and protein product expression in a panel of representative PMP samples. Finally, we demonstrated that the genetic make-up of PMP is distinct from that for colorectal adenoma, colorectal adenocarcinoma, and a number of colon cancer cell line.
Clinical implications and future research
For patients with PMP, there are broadly two clinical scenarios in which chemotherapy may have a role: in patients with advanced unresectable disease, and as adjuvant to cytoreductive surgery, typically administered as HIPEC, at the time of surgery. For the former, clinical series demonstrate poor survival (median survival ranges from 12 to 18 months) after debulking surgery [4, 5, 15], reflecting poor response to systemic chemotherapy. We previously reported a phase II trial in this patient group with only modest benefit [16]. Thus, there is a clear clinical need to develop and test systemic agents with proven pre-clinical efficacy. But until now, we had no pre-clinical in vitro models for such testing.
For the clinical scenario of intra-operative chemotherapy perfusion, there is considerable discussion around maximizing agent delivery (within the abdominal cavity), while minimizing systemic absorption [35, 36]. Yet, there is a lack of data on the cytotoxic characteristics of the chemotherapy agents used. We can now address this using these pre-clinical in vitro models.
The establishment of two immortalized PMP lines provides a platform to undertake pre-clinical testing at several levels. First, one can optimize existing clinically-used protocols, and for example, expand the experiments with MMC and oxaliplatin reported in this study. Second, one may test for novel anti-PMP agents, either as selected mechanism-driven approaches, such as the phloretin experiment, or undertake high-throughput screening for multiple agents. Third, as other investigators develop new agents for mucinous subtypes of breast, colorectal and ovarian cancers, these can be rapidly tested in the PMP cell lines. Fourth, these cells will offer a sustainable source for animal xenograft studies, which until now have relied upon primary cells for each experiment [37]. Finally, this platform will allow the expansion of mechanistic in vitro studies identifying the key molecular pathology underpinning the initiation and development of PMP.
MATERIALS AND METHODS
Patients and controls
The primary cell cultures and PMP tissue samples were from histological confirmed cases undergoing cytoreductive surgery at the Manchester Peritoneal Tumour Service, The Christie NHS Foundation Trust, United Kingdom, a national center for the treatment of tumors of appendiceal origin [5]. Comparative tissues were: normal colonic (n = 8) and appendiceal (n = 2) mucosa versus neoplastic appendiceal (n = 3) and disseminated (n = 7) PMP. Demographic and clinic-pathological characteristics are listed in Supplementary Table S2. Samples were subdivided as: (i) frozen; (ii) formalin-fixed, paraffin embedded tissue blocks; and (iii) taken from the PMP material for primary cell culture. Normal colonic and appendiceal tissues were orientated to produce maximal numbers of longitudinal crypts. Histological identification of PMP areas within specimens was confirmed by staining for cytokeratin 20 (KRT20), MUC2, and CDX2 [10]. All tissue work was performed under local ethics approval (ref 11/NW/0638).
Gene microarrays
For the gene microarrays, we compared three disseminated plus one appendiceal PMP samples with three samples of normal colonic mucosa. Fresh frozen samples were sectioned and stained with cresyl violet (LCM Staining Kit, Ambion, Life Technologies, Paisley, UK). Epithelial cells were captured from approximately 6 sections per sample using a Leica LMD6000 system, and to avoid any bias in cell lineage full length crypts from normal samples were captured wherever possible. RNA was isolated from each sample immediately after laser capture using a Qiagen RNAeasy plus micro kit (Qiagen, Crawley, West Sussex, UK) as per the manufacturer’s instructions. The isolated RNA was quantified using an Agilent bioanalyzer and amplified using a NuGEN WT-Ovation™ RNA Amplification System (NuGEN, Leek, The Netherlands) as recommended by the manufacturer. The amplified RNA was hybridized to Affymetrix Genechip Human Exon 1.0 ST Arrays at the Cancer Research UK Affymetrix Genechip Microarray Service. Probesets were filtered to retain exonic, reliable, uniquely mapping probesets and retained probesets were also filtered according to DABG score (detection above background), to keep only those probesets reliably detected in at least two of the 7 samples.
Principal Components Analysis (PCA) was used to explore overall data structure. Expression data were then summarized to provide a single expression measure per gene, by taking the median of the log2 expression of the filtered probesets mapping to a unique gene symbol. PCA was repeated for the gene level expression data and these data were taken forward for further analysis. The R package limma was used to identify differentially expressed genes between the diseased versus control groups. Genes were classed as differentially expressed if the log fold change difference between the two groups was greater than 2 and were statistically significant if p values were less than 0.05 after adjustment for multiple testing.
Genes were categorized based on key words using Ingenuity Pathway Analysis (IPA®) software (Ingenuity Systems, CA, USA http://www.ingenuity.com/products/ipa) and literature searches of the PubMed database using the gene name. The entire differentially regulated gene set was utilized for the analysis (fold change cut-off 2.0), but for presentational purposes, the reported analysis is limited to the top 40 ranked pathways.
Immunohistochemistry and in situ hybridization
All IHC and ISH were performed on 5 μm thick FFPE sections on a BOND-MAX stainer (a standardized automated system) using a Bond Polymer Refine Detection Kit (Leica Microsystems, Milton Keynes, UK). Antigen retrieval was performed using ER1, a pH6.0 HIER solution (Leica Microsystems) for 20 minutes and tissues were stained with antibodies to MUC2 (1/2000, Novacastra, Leica Microsystems), CDX2 (1/2000), Aldolase B (1/500) and Caspase-5 (1/100, Abcam, Cambridge, UK), KRT20 (1/3000) EPHX4 (1/50) and TMIGD1 (1/100, Sigma Aldrich, Poole, UK), MS4A12 (1/1500, Abnova, via Caltag Medsystems, Buckingham, UK), SLC16A4 (1/1500 Novus Biologicals, Cambridge, UK), ARHGAP24 (1/1000, Proteintech, Manchester, UK) and DSC3 (1/100, Acris via 2BScientific, Upper Heyford, UK) using IHC BOND-MAX protocol F, and appropriate positive and negative controls. Slides were scanned using a Leica SCN400 slide scanner. Images were analyzed using Definiens Tissue Studio 2.0 software. Briefly, the software was trained using composer to identify the epithelial component of the tissue and then to quantify the staining intensity within this component. All staining intensities reported are a mean whole cell intensity of those cells within the epithelial component of the entire tissue section. Software algorithms are available upon request.
ISH probes were oligonucleotides from Invitrogen (Paisley, UK) against SLC16A4 (5′-Flourescein-TAAGGGCTCCTGTCCAGTCATACA-3′) and were designed by the Sigma–Aldrich probe design service. Positive and negative control probes were purchased from Leica Microsystems. Probes were detected using antibodies against Fluorescein (Leica Microsystems) or Biotin (Life Technologies) using an optimized ISH protocol on a BOND-MAX autostainer.
Cell culture and characterization
Cell lines were cultured in a humidified environment containing 5% CO2 and were regularly monitored for mycoplasma infection, and found to be negative. PMP cell lines were isolated as follows. Surgical samples were collected and transported on ice in PBS containing 10 units Penicillin/0.1 mg Streptomycin per ml (Sigma Aldrich) and 0.25 μl/ml Amphotericin B (Life Technologies). Tissue was dissected macroscopically to remove excess mucous and normal tissue and then minced using two scalpels. The resulting tissue was cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM) (Sigma Aldrich) supplemented with 10% FCS, 25 mM HEPES, 5 μg/ml Insulin, 10 mM L-Glutamine, 10 units Penicillin/0.1 mg Streptomycin (all Sigma Aldrich) and 0.25 μl/ml Amphotericin B (Life Technologies). After 1 to 5 days (depending upon the amount and type of tissue) tissue fragments and debris was removed and fresh media added. Cells were harvested 24 h later by chelation using PBS containing 3 mM EDTA and 0.6 mM DTT to selectively detach epithelial cells. A proportion of the cells were plated into 6 well plates and the remainder cryogenically stored in FCS containing 10% DMSO. The cells remaining in culture were then infected with Lenti-SV40 lentivirus (NBS Biologicals, Huntingdon, UK) at an MOI > 10 as per the manufacturers’ instructions. Media was replaced after 16 h and then cells passaged three days later. After two passages a sample of the cells was cryogenically stored and the remainder plated at low density to allow clonal expansion. Individual colonies which exhibited epithelial morphology were then selected and expanded further for characterization.
Cell lines were validated by STR analysis by the Molecular Biology Core facility at the Paterson Institute for Cancer Research, Manchester, United Kingdom, using the Powerplex 21 system (Promega, Southampton, UK) and compared with the ATCC database and genomic DNA isolated from FFPE patient samples using a QIAamp DNA FFPE Tissue kit (Qiagen). Cell lines were tested for the presence of the PMP markers by immunoblotting using antibodies to CDX2 (Abcam, Cambridge, UK) and KRT20 (Sigma Aldrich). For additional characterization using qPCR RNA was isolated using a Qiagen RNeasey plus micro kit and reverse transcribed using a High Capacity RNA-to-cDNA kit (Life Technologies). The resulting cDNA was then used in Taqman gene expression assays for Beta-Actin (Hs99999903_m1) and SLC16A4 (Hs01006127_m1), using Taqman Universal PCR Master Mix on an ABI 7900HT Q-PCR machine.
For comparative experiments, five colon cancer cell lines were used: HCT 116, Caco2, HT-29, Lovo (all from American Type Culture Collection, ATCC) and C32 (C32 (a gift from Professor Mohammod Ilyas, Nottingham, UK) and maintained as previously described [38].
Oncomine data
The transcription profiles for PMP were compared with those for colorectal tumors by comparing changes in transcript levels in this study with those reported in studies reporting similar methodologies and platforms (normal vs tumor, mRNA/cDNA input) on colorectal neoplasia (adenomas, adenocarcinoma, and mucinous adenocarcinoma) identified in the Oncomine database (https://www.oncomine.org/resource/login.html). The fold change (up- and down-regulated) observed for each transcript was then plotted for each comparison.
Cytotoxicity testing
Cells were plated in 96 well plates and allowed to reach confluency (approximately 1 day) to enter the plateau phase of growth, mimicking the non-proliferating state of PMP in vivo, and the media replaced. Cells were then exposed to a dose range of Mitomycin C, Oxaliplatin (R&D Systems, Abingdon, UK), or Phloretin (Sigma Aldrich) for 60 minutes at 37°C. Media was then replaced and the cells cultured under normal conditions for a further 4 days. Cellular metabolism was assessed using the MTT assay by addition of 1/10 volume (10 μl) of 5 mg/ml Thiazolyl Blue Tetrazolium Bromide (Sigma Aldrich) and incubation for 4 h. 75 μl of media was then removed and 50 μl DMSO added to solubilize the MTT crystals. Plates were incubated at 37°C for 10 minutes and then absorbance measured at 540 nm. Growth curves were fitted using a four parameter sigmoidal algorithm in GraphPad Prism 6 (GraphPad Software Inc. La Jolla, CA, USA) and IC50 values derived.
Statistical analysis
Comparisons of continuous variables were performed using Student’s t-test (Stata™ 11.1, College Station, TX, USA).
ACKNOWLEDGMENTS
Jan Taylor, Dr Crispin Miller (bio-informatics), Dr Bipasha Chakrabarty (pathology), clinical and data management team of the Manchester Peritoneal Service, and Paterson core facilities (MBCF, AIF, Histology). We thank Dr John Brognard for his critical appraisal of the manuscript.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
FUNDING
This study was supported by a grant to AGR, STOD and PLS from the PMP Research Foundation through the National Organization for Rare Disorders (NORD), with additional support from the Christie Charitable Peritoneal Tumour Research fund. The laboratory work was undertaken within a Cancer Research UK funded laboratory.
Contributions
DLR: conception and design, data collection and analysis, manuscript writing. STOD: provision of study materials, final manuscript approval. PLS: conception and design, manuscript writing. AGR: conception and design, provision of study materials, manuscript writing.
CONFLICTS OF INTEREST
For all authors, there are no conflicts of interest.
REFERENCES
1. McDonald JR, O’Dwyer ST, Rout S, Chakrabarty B, Sikand K, Fulford PE, Wilson MS, Renehan AG. Classification of and cytoreductive surgery for low-grade appendiceal mucinous neoplasms. Br J Surg. 2012; 99:987–992.
2. Renehan AG, O’ Dwyer ST, Stern PL. Pseudomyxoma peritonei. In: Schwab M, ed. Encyclopedia of Cancer. (London: Springer), 2008, pp. 2134–2138.
3. van den Heuvel MG, Lemmens VE, Verhoeven RH, de Hingh IH. The incidence of mucinous appendiceal malignancies: a population-based study. Int J Colorectal Dis. 2013; 28:1307–1310.
4. Dayal S, Taflampas P, Riss S, Chandrakumaran K, Cecil TD, Mohamed F, Moran BJ. Complete cytoreduction for pseudomyxoma peritonei is optimal but maximal tumor debulking may be beneficial in patients in whom complete tumor removal cannot be achieved. Dis Colon Rectum. 2013; 56:1366–1372.
5. Rout S, Renehan AG, Parkinson MF, Saunders MP, Fulford PE, Wilson MS, O’Dwyer ST. Treatments and outcomes of peritoneal surface tumors through a centralized national service (United kingdom). Dis Colon Rectum. 2009; 52:1705–1714.
6. Sugarbaker PH. New standard of care for appendiceal epithelial neoplasms and pseudomyxoma peritonei syndrome? Lancet Oncol. 2006; 7:69–76.
7. Bibi R, Pranesh N, Saunders MP, Wilson MS, O’Dwyer ST, Stern PL, Renehan AG. A specific cadherin phenotype may characterise the disseminating yet non-metastatic behaviour of pseudomyxoma peritonei. Br J Cancer. 2006; 95:1258–1264.
8. Flatmark K, Davidson B, Kristian A, Stavnes HT, Forsund M, Reed W. Exploring the peritoneal surface malignancy phenotype-a pilot immunohistochemical study of human pseudomyxoma peritonei and derived animal models. Hum Pathol. 2010; 41:1109–1119.
9. O’Connell JT, Tomlinson JS, Roberts AA, McGonigle KF, Barsky SH. Pseudomyxoma peritonei is a disease of MUC2-expressing goblet cells. Am J Pathol. 2002; 161:551–564.
10. Nonaka D, Kusamura S, Baratti D, Casali P, Younan R, Deraco M. CDX-2 expression in pseudomyxoma peritonei: a clinicopathological study of 42 cases. Histopathology. 2006; 49:381–387.
11. Liu X, Mody K, de Abreu FB, Pipas JM, Peterson JD, Gallagher TL, Suriawinata AA, Ripple GH, Hourdequin KC, Smith KD, Barth RJ Jr., Colacchio TA, Tsapakos MJ, Zaki BI, Gardner TB, Gordon SR, et al. Molecular profiling of appendiceal epithelial tumors using massively parallel sequencing to identify somatic mutations. Clin Chem. 2014; 60:1004–1011.
12. Pai RK, Hartman DJ, Gonzalo DH, Lai KK, Downs-Kelly E, Goldblum JR, Liu X, Patil DT, Bennett AE, Plesec TP, Kuan SF, Nikiforova MN, Shadrach B, Pai RK. Serrated lesions of the appendix frequently harbor KRAS mutations and not BRAF mutations indicating a distinctly different serrated neoplastic pathway in the appendix. Hum Pathol. 2014; 45:227–235.
13. Nishikawa G, Sekine S, Ogawa R, Matsubara A, Mori T, Taniguchi H, Kushima R, Hiraoka N, Tsuta K, Tsuda H, Kanai Y. Frequent GNAS mutations in low-grade appendiceal mucinous neoplasms. Br J Cancer. 2013; 108:951–958.
14. Shetty S, Thomas P, Ramanan B, Sharma P, Govindarajan V, Loggie B. Kras mutations and p53 overexpression in pseudomyxoma peritonei: association with phenotype and prognosis. J Surg Res. 2013; 180:97–103.
15. Chua TC, Moran BJ, Sugarbaker PH, Levine EA, Glehen O, Gilly FN, Baratti D, Deraco M, Elias D, Sardi A, Liauw W, Yan TD, Barrios P, Gomez Portilla A, de Hingh IH, Ceelen WP, et al. Early- and long-term outcome data of patients with pseudomyxoma peritonei from appendiceal origin treated by a strategy of cytoreductive surgery and hyperthermic intraperitoneal chemotherapy. J Clin Oncol. 2012; 30:2449–2456.
16. Farquharson AL, Pranesh N, Witham G, Swindell R, Taylor MB, Renehan AG, Rout S, Wilson MS, O’Dwyer ST, Saunders MP. A phase II study evaluating the use of concurrent mitomycin C and capecitabine in patients with advanced unresectable pseudomyxoma peritonei. Br J Cancer. 2008; 99:591–596.
17. Koslowski M, Sahin U, Dhaene K, Huber C, Tureci O. MS4A12 is a colon-selective store-operated calcium channel promoting malignant cell processes. Cancer Res. 2008; 68:3458–3466.
18. Wang X, Poole RC, Halestrap AP, Levi AJ. Characterization of the inhibition by stilbene disulphonates and phloretin of lactate and pyruvate transport into rat and guinea-pig cardiac myocytes suggests the presence of two kinetically distinct carriers in heart cells. Biochem J. 1993; 290:249–258.
19. Wang Q, Morris ME. The role of monocarboxylate transporter 2 and 4 in the transport of gamma-hydroxybutyric acid in mammalian cells. Drug Metab Dispos. 2007; 35:1393–1399.
20. Nummela P, Saarinen L, Thiel A, Jarvinen P, Lehtonen R, Lepisto A, Jarvinen H, Aaltonen LA, Hautaniemi S, Ristimaki A. Genomic profile of pseudomyxoma peritonei analyzed using next-generation sequencing and immunohistochemistry. Int J Cancer. 2015; 136:E282–289.
21. Gill RK, Saksena S, Alrefai WA, Sarwar Z, Goldstein JL, Carroll RE, Ramaswamy K, Dudeja PK. Expression and membrane localization of MCT isoforms along the length of the human intestine. Am J Physiol Cell Physiol. 2005; 289:C846–C852.
22. Hilgendorf C, Ahlin G, Seithel A, Artursson P, Ungell A-L, Karlsson J. Expression of Thirty-six drug transporter genes in human intestine, liver, kidney, and organotypic cell lines. Drug Metab Dispos. 2007; 35:1333–1340.
23. Draoui N, Feron O. Lactate shuttles at a glance: from physiological paradigms to anti-cancer treatments. Dis Model Mech. 2011; 4:727–732.
24. Syed S-e-H, Trinnaman B, Martin S, Major S, Hutchinson J, Magee AI. Molecular interactions between desmosomal cadherins. Biochem J. 2002; 362:317–327.
25. Khan K, Hardy R, Haq A, Ogunbiyi O, Morton D, Chidgey M. Desmocollin switching in colorectal cancer. Br J Cancer. 2006; 95:1367–1370.
26. Cui T, Chen Y, Yang L, Knosel T, Zoller K, Huber O, Petersen I. DSC3 expression is regulated by p53, and methylation of DSC3 DNA is a prognostic marker in human colorectal cancer. Br J Cancer. 2011; 104:1013–1019.
27. Eriksson A, Flach C-F, Lindgren A, Kvifors E, Lange S. Five mucosal transcripts of interest in ulcerative colitis identified by quantitative real-time PCR: a prospective study. BMC Gastroenterol. 2008; 8:34.
28. Peng SY, Lai PL, Pan HW, Hsiao LP, Hsu HC. Aberrant expression of the glycolytic enzymes aldolase B and type II hexokinase in hepatocellular carcinoma are predictive markers for advanced stage, early recurrence and poor prognosis. Oncol Rep. 2008; 19:1045–1053.
29. Sheweita SA, Tilmisany AK. Cancer and phase II drug-metabolizing enzymes. Curr Drug Metab. 2003; 4:45–58.
30. Koslowski M, Tureci O, Huber C, Sahin U. Selective activation of tumor growth-promoting Ca2+ channel MS4A12 in colon cancer by caudal type homeobox transcription factor CDX2. Mol Cancer. 2009; 8:77.
31. Schwartz S Jr., Yamamoto H, Navarro M, Maestro M, Reventos J, Perucho M. Frameshift mutations at mononucleotide repeats in caspase-5 and other target genes in endometrial and gastrointestinal cancer of the microsatellite mutator phenotype. Cancer Res. 1999; 59:2995–3002.
32. Hosomi Y, Gemma A, Hosoya Y, Nara M, Okano T, Takenaka K, Yoshimura A, Koizumi K, Shimizu K, Kudoh S. Somatic mutation of the Caspase-5 gene in human lung cancer. Int J Mol Med. 2003; 12:443–446.
33. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell. 2004; 117:561–574.
34. Dorudi S, Sheffield JP, Poulsom R, Northover JM, Hart IR. E-cadherin expression in colorectal cancer. an immunocytochemical and in situ hybridization study. Am J Pathol. 1993; 142:981–986.
35. Van der Speeten K, Govaerts K, Stuart OA, Sugarbaker PH. Pharmacokinetics of the perioperative use of cancer chemotherapy in peritoneal surface malignancy patients. Gastroenterol Res Pract. 2012; 2012:378064.
36. Van der Speeten K, Stuart OA, Sugarbaker PH. Pharmacology of perioperative intraperitoneal and intravenous chemotherapy in patients with peritoneal surface malignancy. Surg Oncol Clin N Am. 2012; 21:577–597.
37. Flatmark K, Reed W, Halvorsen T, Sorensen O, Wiig JN, Larsen SG, Fodstad O, Giercksky KE. Pseudomyxoma peritonei-two novel orthotopic mouse models portray the PMCA-I histopathologic subtype. BMC Cancer. 2007; 7:116.
38. Baricevic I, Roberts DL, Renehan AG. Chronic insulin exposure does not cause insulin resistance but is associated with chemo-resistance in colon cancer cells. Horm Metab Res. 2014; 46:85–93.