
INTRODUCTION
Frostbite is considered as traumatic injury and may lead to death [1–5]. Severe cold leads to hypothermia and damages in skin and other tissues or organs, which may result in limb amputations, organ damages, and death. Here, we propose that strategies can be developed to utilize the basic concept of frostbite, along with UV irradiation, to treat skin and other types of cancer.
Both extrinsic and intrinsic pathways are involved in the programmed cell death [6–8]. The extrinsic pathway is death receptor dependent. For example, stimulation of tumor necrosis factor (TNF) receptor by TNF-α results in activation of the downstream death pathway [9, 10]. In contrast, when cells possess damaged DNA or are subjected to apoptotic stress, the intrinsic pathway is activated. Cytochrome c release from the mitochondria into the cytosol occurs. Cytochrome c binds and causes the aggregation of the adaptor protein Apaf-1. Apaf-1 binds procaspase-9 molecules, which leads to assembly of pentameric Apaf-1/caspase-9. Further activation of downstream caspases 3 and 7 induces nuclear damage [11].
In this study we simulated frostbite in vitro. Cells were subjected to UV irradiation and then brief exposure to freezing conditions. The UV energy was at UVC range (wavelength at 265 nm). Gas formation was initiated in the nucleus. It appears that the UV energy was absorbed by the cells that allow relocation of many cytosolic proteins to the nucleus, which subsequently leads to generation of nitric oxide (NO)-containing gas in the nucleus. In most cases, the gas pushed out to form a short tunnel through the nuclear and the plasma membranes to form a single large bubble. In contrast, when cells were exposed to UV irradiation and then cultured at 37°C, the rate of bubble formation in these cells was significantly retarded, suggesting that functional proteins to block bubbling are at work at 37°C. We determined that tumor suppressor WW domain-containing oxidoreductase, designated human WWOX or FOR or mouse WOX1, participates in the UV irradiation-mediated apoptosis [12–15]. Here, we examined how WWOX/WOX1 acted in response to UV and cold shock, and determined whether components in the signal pathway, including NOS2, p53 and TRAF2 [7, 8], are involved in the bubbling death.
RESULTS
UV irradiation and cold shock induce “bubbling death” effectively at 4 and 22°C
When cells were subjected to UV irradiation and/or cold shock and then incubated at 4, 22 or 37°C for indicated durations, formation of a large bubble from the nucleus per cell was counted. UVC was used for all experiments. For example, when COS7 fibroblasts were overexpressed with EGFP (enhanced green fluorescent protein), followed by exposure to UV irradiation and then cold shock at 4°C for 5 min. Time-lapse microscopy was carried out at 22°C or room temperature for 2 hr. Gas was generated in the nucleus and pushed the nuclear and cytoplasmic membranes to pop out a big bubble (Figure 1A; Supplementary Figure S1 for enlarged pictures with better resolution). Nuclear EGFP relocated to the gas bubble with time of incubation. Each cell generated and released one bubble only (Supplementary Figures S1–S3). Neighboring cells, without expressing EGFP, also formed bubbles in a synchronized manner similar to that of the EGFP-expressing cells (Figure 1A). Indeed, upon exposure to UV, all cells underwent bubbling death in a synchronized manner (Supplementary Figure S3). Cold shock for 5 min at 4°C enhances the generation of bubbles. Herein, we designated the event as “bubbling death”.
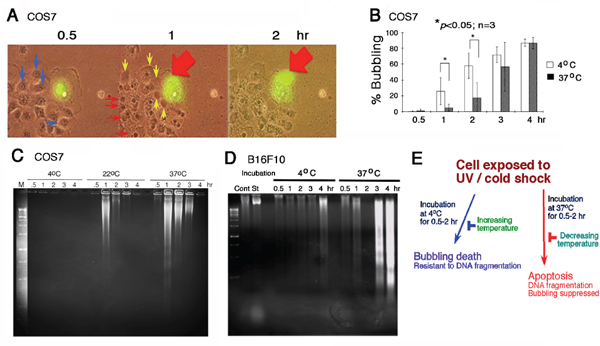
Figure 1: UV/cold shock induces nuclear membrane bubbling. (A) COS7 cells, with or without expressing EGFP, were exposed to UV irradiation at 480 mJoule/cm2 and then incubated at 4°C for 5 minutes, prior to imaging by time-lapse microscopy at room temperature. Bubble formation (big red and small yellow arrows) from the nucleus is shown. Nuclear EGFP leaked into the gas bubble. See the enlarged pictures are at Supplementary Figure S1 for better resolution. (B) Under similar conditions, COS7 cells were exposed to UV irradiation (480 mJoule/cm2) and then cold shock (4°C for 5 min). The cells were then cultured at 4°C and 37°C, respectively, for indicated times. The extent of bubbling was counted. (C, D) Similarly, COS7 and B16F10 cells were exposed to UV (480 mJoule/cm2)/cold shock (4°C for 5 min) and then incubated at indicated temperatures for 0.5–4 hr. Internucleosomal DNA fragmentation was determined by agarose gel electrophoresis. (E) UV irradiation/cold shock-induced apoptosis is switched to bubbling cell death at low temperature, and vice versa.
UV alone induces bubbling of COS7 cells in a synchronized manner (Supplementary Figure S3), and cold shock could dramatically enhance the bubbling. For example, it took almost 1.5–2 hr for inducing 50% bubbling up to ~10 μm in diameter of the bubbles in UV only-treated cells, whereas it dropped down to 10–30 min when cells were exposed to UV/cold shock.
In similar experiments, COS7 cells were transiently overexpressed with ECFP or the WW domain of WWOX/WOX1 tagged with EGFP (EGFP-WWOXww), and subjected to UV irradiation/cold shock (4°C, 5 min). By time-lapse microscopy at room temperature, release of the ectopic ECFP or EGFP-WW domain from the nuclei to the bubble is shown (Supplementary Videos 1, 2). When cells were treated with UV/cold shock and then incubated at 4°C, 22°C, or 37°C for various durations, no major differences were observed regarding the morphological changes leading to bubbling death. When overexpressed, the ectopic WW domain protein localizes in the nucleus [15]. Also, generation of exosome-like particles of less than 1 μm in diameter [16], either intracellular or extracellular, is shown with time of bubbling (see the background in Supplementary Videos 1, 2).
UV/cold shock-treated cells undergo bubbling death at low temperatures without DNA fragmentation
When COS7 cells were subjected to UV irradiation/cold shock and then cultured at 37°C, bubble formation was significantly retarded (Figure 1B). Similar results were observed by testing tumor necrosis factor (TNF)-sensitive L929 fibroblasts [15] (Supplementary Figure S4). That is, L929 cells were exposed to UV alone and then cold shock, followed by incubation at 4 and 37°C, respectively. In addition, when melanoma B16F10 cells were exposed to UV and then cold shock (4°C for 5 min), or cold shock first and then UV exposure. These cells were then cultured at 4, 22, or 37°C. Changes in the order of exposure to UV or cold shock did not alter the results of bubbling death at 4 or 22°C (data not shown). However, the bubbling event was totally blocked at 37°C (> 99%). We examined more than 10 cancer cell lines, including skin, breast and other cancer cells. In most cases, the bubbling death was retarded at 37°C (data not shown).
In stark contrast, we found that UV/cold shock-induced DNA fragmentation is blocked at low temperature but functions normally at 37°C. COS7 and B16F10 cells were exposed to UV/cold shock and then incubated at 4, 22, and 37°C for indicated times, followed by processing DNA fragmentation by agarose gel electrophoresis (Figure 1C, 1D). DNA fragmentation was blocked in COS7 cells at 4°C, but occurred at 22 and 37°C (Figure 1C). UV/cold shock-treated B16F10 cells were resistant to DNA fragmentation at 4°C, (Figure 1D) but exhibited bubble formation (data not shown). In contrast, DNA fragmentation was restored in UV/cold shock-treated B16F10 cells upon incubation at 37°C (Figure 1D), whereas bubbling was blocked totally at 37°C (Figure 1D). Taken together, UV/cold shock induces cells to undergo bubbling but resist DNA fragmentation at 4°C (Figure 1E). Upon increasing the temperatures to 37°C, bubbling death is retarded and DNA fragmentation occurs. The observations imply that switching from apoptosis to bubbling death is controlled by decreasing temperatures, and vice versa (Figure 1E).
A nuclear gas bubble is generated per cell
It appears that under stimulation with UV/cold shock, the nuclear gas pushed forward as a jet stream in a single direction, so as to form a short stalk (~1–2 μm in length) to connect the bubble and the cell (see arrow; Figure 2A). Nucleoli were then released into the bubbles (see arrows; Figure 2B; Supplementary Video 3). Post bubbling for 30 min to 1 hr, cells were essentially dead as they failed to exclude trypan blue stain (> 95% cells) and their nuclei became positive for the propidium iodide stain (Figure 2C). A schematic graph shows a putative gas jet stream is formed and pops out to form a bubble. The wall of the bubble is probably enclosed by the outer plasma membrane and the inner nuclear membrane (Figure 2D).
Finally, we attempted to verify the gas one-hit theory that a gas jet stream is formed in the nucleus and pops off to form one bubble per cell. COS7 cells were subjected to UV/cold shock, and then fixed for electron microscopy analysis [17]. In controls, COS7 cells received no UV/cold shock and their nuclei and nuclear pores were intact (see red arrows; Figure 3A–3B). When cells were subjected to UV/cold shock, a representative damaged nucleus underwent condensation and possessed an enlarged burst of the nuclear membrane (see the red arrow; Figure 3C–3D), suggesting that a gas jet stream hits the nuclear wall or pushes apart of a single nuclear pore that results in nuclear burst.
Bubbling from nucleus is not identical to membrane blebbing
Bubbling from the nucleus cannot be considered the same as membrane blebbing. The membrane blebs are protrusions of the cell membrane, which exhibits many irregular bulges from the cell surface due to localized decoupling of the cytoskeleton from the plasma membrane [18, 19]. This event is reversible for the generation of small membrane blebs, whereas bubbling is irreversible for the formation of a final large bubble. We have previously demonstrated that U0126, an MEK1 inhibitor, prevents phorbol myristate acetate (PMA)-induced apoptosis of Jurkat T cells by causing membrane blebbing [20]. U0126 induces cells to undergo membrane blebbing to resist death [20].
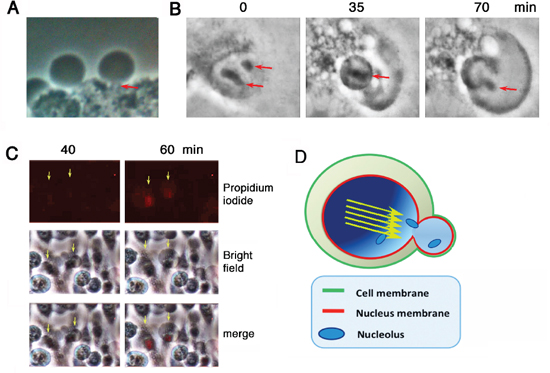
Figure 2: Bubbling from a gas jet stream. COS7 cells were subjected to UV/cold shock and imaging by time-lapse microscopy at room temperature. (A) A representative COS7 cell exhibits a short gas tunnel or stalk (~1 μm in length; red arrow) connecting the cell and the bubble. (B) Release of nucleoli from a burst nucleus into the gas bubble is shown (red arrows). (C) Bubbling occurred prior to pickup of propidium iodide by the condensed nuclei. (D) A schematic model shows that the gas drives through the nucleus as a jet stream (yellow arrows) that punches out a single hole for generating a single bubble. The wall of the bubble is composed of the inner nuclear and outer cytoplasmic membranes.
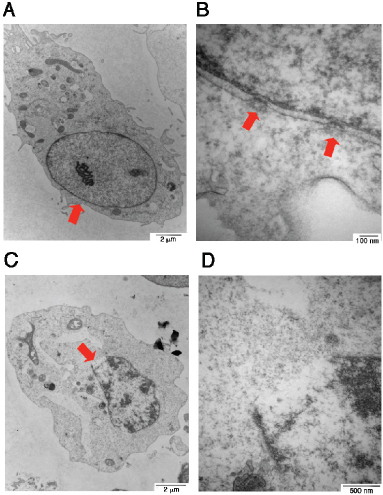
Figure 3: Electron microscopy of UV/cold shock-induced gas formation to cause damage to the nucleus. COS7 cells were exposed to UV irradiation (480 mJoule/cm2) and then incubated at 4°C for 3 hours. Cells were fixed and prepared for electron microscopy analysis. (A–B) COS7 cells were without treatment. Intact nuclear pores are shown (see arrows). (C–D) A single gas hit to the nuclear wall is shown, which results in burst of the nucleus with a single opening (red arrow).
Bubbling death dose not exhibit flip-over of membrane phosphatidylserine (PS)
In contrast to apoptosis, there was no initial flip-over of phosphatidylserine (PS) toward the surface of cell membrane. For example, wild type Wwox MEF cells were exposed to UV/cold shock, and then incubated at 4°C for 30 min to 3 hr. No detectable positive signals were obtained using Annexin V stain (Supplementary Figures S5A). Similar results were observed using COS7 cells (data not shown). In positive controls, COS7 cells were transiently overexpressed with a Zfra expression construct by electroporation and then cultured for 24 hr. Flip-over of PS onto cell surface was shown as determined by Annexin V staining using flow cytometry (Supplementary Figure S5B). Zfra, known as zinc finger-like protein that regulates apoptosis, is a 31-amino-acid naturally occurring small protein [21–24]. We have shown previously that staurosporine- and Zfra-induced apoptosis are involved in flip-over of PS toward the surface of the cell membrane [21–24].
To further verify the aforementioned results, time-lapse microscopy was carried out using melanoma B16F10 cells. Aliquots of green fluorescent Annexin V and red fluorescent propidium iodide were added to the culture of B16F10 cells. The cells were subjected to UV/cold shock and time-lapse microscopy at room temperature. Again, damage to the nucleus that led to bubble formation occurred in less than one hour, as revealed by accumulation of the red fluorescent propidium iodide in the nucleus, whereas no green fluorescence showing the flip-over of PS to the cell surface was observed (Figure 4A). To induce mitochondrial apoptosis, B16F10 cells were treated with betulinic acid [20, 21]. Time-lapse microscopy showed the presence of green fluorescence prior to the cell death (Figure 4B).
Bubbling death dose not cause damage to Golgi complex
COS7 cells were transiently overexpressed with ECFP specific for Golgi-targeting, followed by exposure to UV/cold shock. The ECFP protein was largely retained in the Golgi complex during bubbling death, as determined by time-lapse microscopy (Supplementary Videos 4A, 4B), suggesting that no damage to the Golgi complex. Again, exosome-like particles were released during bubbling death (Supplementary Videos 4A, 4B).
In comparison, COS7 cells were treated with staurosporine to induce apoptosis. A typical apoptotic cell death, including cell shrinkage and nuclear condensation, is shown, as determined by time-lapse microscopy (Supplementary Video 5).
Starving cells prefer autophagic death but not bubbling death upon UV/cold shock exposure
When COS7 cells were pre-starved for 24–48 hr and then exposed to UV/cold shock, these cells exhibited increasing numbers of cytosolic autophagic vacuoles with time, along with whole cell shrinkage and nuclear burst (Supplementary Video 6). In most cases, the nuclei appeared to burst from inside out without generation of bubbles.
Also, COS7 cells were transiently overexpressed with LC3 tagged with EGFP, cultured for 24 hr, and then exposed to UV/cold shock. Time-lapse microscopy at room temperature revealed that during bubbling death, EGFP-LC3 was released to the generated bubble and no generated LC3 aggregates in the cytoplasm were shown (Supplementary Video 7), suggesting that no autophagy has occurred to induce LC3 degradation and aggregation.
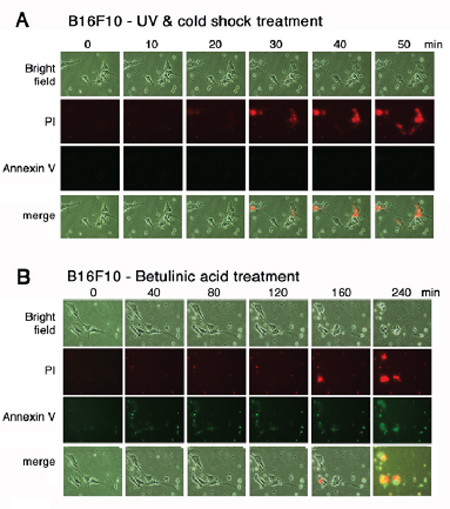
Figure 4: UV irradiation and cold shock did not cause flip-over of PS onto cell surface. (A) B16F10 cells in culture were added aliquots of PI and Annexin V stains, exposed to UV irradiation (480 mJoule/cm2) and cold shock (4°C for 5 min), and subjected to time-lapse imaging at room temperature (2 min per frame). Little or no Annexin V-positive cells were observed. (B) Similarly, B16F10 cells were treated with betulinic acid (100 μM) and subjected to time-lapse imaging at room temperature (2 min per frame).
UV/cold shock induces the generation of nitric oxide (NO)-containing gas bubbles
To determine the underlying mechanisms, Nω-nitro-L-arginine methyl ester hydrochloride (Nω-LAME), an analog of arginine, was shown to suppress the formation of gas bubbles (Figure 5A), suggesting that NO is in the generated gas. When COS7 cells were pretreated with Nω-LAME for 1 hr, and then exposed to UV and chilled at 4°C for indicated times, Nω-LAME blocked bubbling in a time- and a dose-dependent manner (Figure 5A). UV/cold shock rapidly induced the expression of nitric oxide synthase 2 (NOS2) in 30 min, as determined by Western blotting (Figure 5B). The cellular levels of α-tubulin were relatively unchanged (Figure 5B). Similarly, UV/cold shock significantly induced the generation of NOS2 in 5–10 min, as determined by immunofluorescence staining (Figure 5C and 5D). UV irradiation or cold shock alone was less effective (Figure 5C and 5D).
WWOX/WOX1 and p53 are essential for UV/cold shock-induced nuclear damage and bubbling death
We have previously demonstrated the essential role of tumor suppressors p53 and WWOX/WOX1 in causing UV irradiation-mediated apoptosis [12–14] and other events [21]. We established Wwox knockout mice and mouse embryonic fibroblasts (MEF) [25, 26]. When wild type Wwox+/+ MEF cells were subjected to UV/cold shock, these cells readily underwent bubbling death (Figure 6A and 6B; Supplementary Video 8). However, the knockout Wwox−/− MEF cells resisted bubbling death by UV/cold shock (Figure 6A and 6B; Supplementary Video 9), suggesting the essential role of WWOX/WOX1 in the bubbling death.
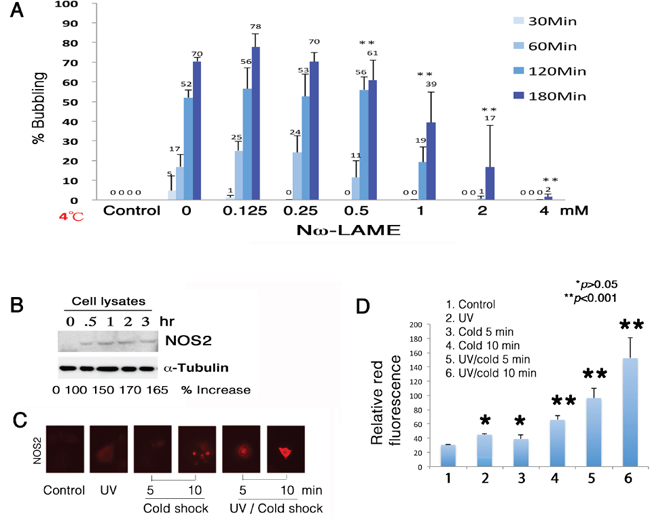
Figure 5: Suppression of bubbling by NOS2 inhibitor Nω-LAME. (A) COS7 cells were pretreated with Nω-LAME (0.125–4 mM) for 30 min, followed by exposure to UV irradiation (480 mJ/cm2) and cold shock (4°C) for 30 to 180 min. (B) Induction of NOS2 expression by UV/cold shock was shown in 30 min. The levels of intracellular NFκB (p65) and α-tubulin remained largely unchanged. A representative data is shown from 2 experiments. (C–D) By immunofluorescence microscopy, UV/cold shock together significantly induced the expression of NOS2 in COS7 cells (n = 10; mean ± standard deviation; Student's t-test).
Without WWOX/WOX1, p53 stability is dramatically reduced [13]. When wild type Wwox+/+ MEF cells were exposed to UV irradiation and cold shock at 4°C, these cells had an increased level of pS46–p53 by 15–50% (Figure 6C, 6D). However, pS46–p53 was not detectable in the knockout Wwox−/− MEF cells treated with or without UV/cold shock (Figure 6C and 6D). Ser46 phosphorylation is essential for p53-mediated apoptosis [13]. In comparison, UV/cold shock-treated COS7 cells had increased levels of activated WWOX/WOX1 and p53 with phosphorylation at Tyr33 and Ser46, respectively (Figure 6E). Phosphorylation at Tyr287 in WWOX/WOX1 also occurred (Figure 6E). The observations indicate that protein phosphorylation can occur at 4°C. The Tyr287 phosphorylation is known to increase ubiquitination and degradation of WWOX/WOX1 [27]. Importantly, ectopic dominant-negative p53 (S46G–p53) effectively blocked the bubbling death in COS7 cells (Figure 6F), supporting an essential role of p53 in the bubbling death. Previously, we have demonstrated that p53 and WWOX/WOX1 act synergistically in inducing apoptosis under UV irradiation or environmental stress [12–14, 18].
TRAF2 significantly retards bubbling death
UVB activates TNF receptor-associated factor-2 (TRAF2), p53 and WWOX/WOX1 in the skin [12–14, 20, 28]. In COS7 cells, UV irradiation rapidly induced the expression of endogenous TRAF2 in the cytoplasm, and that subsequent cold shock caused upregulation and colocalization of TRAF2 and NOS2 in the nucleus in 10 min, as determined by immunofluorescence microscopy (Figure 7A). The nuclear accumulation of TRAF2 and NOS2 was greater than 95% (~100 cells counted; n = 3).
When Wwox wild type MEF cells were exposed to UV irradiation followed by cold shock for 30 min to 3 hr, both antiapoptotic TRAF2 and NF-κB/p65 were down-regulated, whereas the levels of proapoptotic TRADD were not reduced (Figure 7B). Under similar conditions, the level of TRAF2 was increased by 2–3 fold in Wwox knockout MEF cells (Figure 7B). The knockout cells were highly resistant to bubbling death by UV/cold shock (Figure 6A, 6B). By immunohistochemistry, normal human skin expressed a relative low level of WWOX/WOX1, but a high level of TRAF2 (Figure 7C). When human skin was subjected to frostbite, the WWOX/WOX1 level was dramatically increased (> 80% increase), and the TRAF2 level reduced by greater than 90% (Figure 7C).
To investigate the role of TRAF2 in regulating UV/cold shock, COS7 cells were transiently transfected with expression constructs of ECFP, truncated ECFP-TRAF2(113–312), and 2 identical full-length ECFP-TRAF2, respectively. The extent of protein expression was about 50–60%, as determined by fluorescence microscopy. When full-length TRAF2-expressing COS7 cells were subjected to UV/cold shock, bubbling death was significantly retarded, as compared to the ECFP control cells (Figure 7D). The N-terminal ring domain (amino acid #1–113) and C-terminal coiled coil and MATH domains (amino acid #313–501) were removed from the full-length TRAF2. The resulting ECFP-TRAF2(113–312) had a reduced suppression of bubbling death (Figure 7D). In these experiments, only cells expressing the indicated protein were counted.
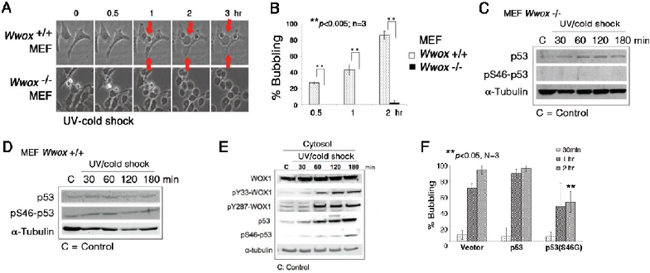
Figure 6: Wwox knockout MEF cells resist bubbling death. (A) MEF Wwox+/+ and Wwox−/− cells were exposed to UV irradiation (480 mJoule/cm2) and then cold shock at 4°C for 5 min, followed by chasing bubble formation at room temperature by time-lapse microscopy. (B) Similar experiments were carried out by treating MEF Wwox+/+ and Wwox−/− cells with UV/cold shock, and incubated at room temperature for 0.5, 1 and 2 hr, respectively (~100 cells counted; n = 3; mean ± standard deviation; Student's t-test). (C–D) Again, Wwox+/+ and Wwox−/− MEF cells were exposed to UV/cold shock, followed by incubation at 4°C for indicated durations. Expression of p53 and pS46–p53 is shown. α-tubulin is a loading control. A representative data is shown from 2 experiments. (E) In similar experiments, COS7 cells were subjected to UV/cold shock, and shown to have increased levels of cytosolic pY33-WOX1(WWOX), pY287-WOX1(WWOX), p53, and pS46–p53. (F) COS7 cells were transiently overexpressed with an empty vector, wild type p53, or dominant negative p53 (S46G–p53). These cells were cultured overnight and then exposed to UV/cold shock, followed by incubation at 4°C for indicated durations. The extent of bubbling death was counted.
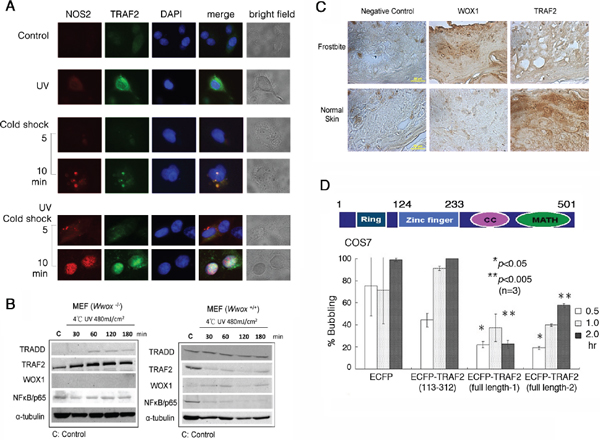
Figure 7: UV/cold shock induces NOS2 and TRAF2 expression. (A) COS7 cell were exposed to UV irradiation (480 mJoule/cm2) and subsequent incubation at 4°C for 5 and 10 min. Cold shock alone weakly induced the expression of both endogenous NOS2 and TRAF2. Pre-exposure of cells to UV and then cold shock enhanced the generation of both proteins. UV alone rapidly induced the expression of TRAF2. (B) Down-regulation of antiapoptotic TRAF2 and NF-κB/p65 occurred when Wwox wild type MEF cells were exposed to UV (480 mJoule/cm2) and subsequent cold shock for 30 min to 3 hr. However, TRAF2 was upregulated in UV/cold shock-treated Wwox knockout MEF cells. (C) By immunohistochemistry, the expression of WWOX/WOX1 and TRAF2 in normal human skin and frostbitten skin is shown. Scale bars: 20 μm. (D) A schematic graph for the TRAF2 domains is shown. Ring = Ring-type zinc finger domain; CC = Coiled coil domain; MATH = MATH/TRAF domain. COS7 cells were transiently overexpressed with ECFP, truncated ECFP-TRAF2(113–312), and 2 identical full-length ECFP-TRAF2, respectively. UV (480 mJoule/cm2)/cold shock (4°C for 5 min)-induced bubbling death in cells expressing the indicated protein was counted (~100 cells counted; n = 3).
DISCUSSION
Summary
We have discovered for the first time that UV irradiation/cold shock induces a novel type of programmed cell death, and this may provide a feasible approach for skin cancer treatment. Our key discovery is that UVC energy, coupled with low temperatures, mediates upregulation and accumulation of NOS2 in the nucleus, so as to generate a NO-containing gas per cell. The bubbling process does not require ATP (data not shown). The UV/cold shock-induced bubbling death functions effectively at low temperatures (4–22°C). Most strikingly, these cells resist DNA fragmentation at low temperatures, unless the temperatures go up to 37°C. At 37°C, bubbling death is dramatically suppressed in the UV/cold shock-pretreated cells. The observations imply that switching from apoptosis to bubbling death can be achieved by reducing temperatures, and vice versa.
We believe that NO, ROS, and other oxidizing gases may cause protein modifications. We found that cytosolic proteins undergo aggregation during bubbling death (data not shown). Also, there is a strong redox activity during the early stage of UV/cold shock exposure, prior to bubble formation. We have tried to isolate bubbles and determined the nature of proteins in the bubbles. This has been really difficult, as the bubbles are readily broken during sample preparation. Thus, the functional relevance of the nuclear proteins in the bubbles is unknown.
Role of NO, NOS2, p53, WWOX and TRAF2
NO, NOS2, p53 and WWOX/WOX1 are crucial in the bubbling death, whereas the antiapoptotic TRAF2 blocks the cell death event. Suppression of NOS2 blocks the bubbling death. WWOX and p53 function in a synergistic manner in causing apoptosis under many types of stress conditions, including UV irradiation [12–15]. Both activated proteins physically interact using motifs surrounding the p-Tyr33 and p-Ser46 areas, and translocate to the mitochondria or nucleus to work synergistically in mediating apoptosis [12–15]. Whether WWOX and p53 bind and counteract the protective function of TRAF2 is unknown. In the absence of p53 and WWOX, the bubble formation is suppressed. The observations suggest that the nuclear accumulation of p53 and WWOX is needed to activate NOS2 to induce formation of NO.
In the frostbitten skin tissues, we showed that there is a greater than 90% downregulation of TRAF2, along with a dramatic upregulation of WWOX in humans. Unlike typical apoptosis, we did not observe PS flip-over onto the outer cell surface, mitochondrial apoptosis, and DNA fragmentation in the UV/cold shock-induced cell death at low temperatures. Unlike a recent report using ionizing irradiation [29], UV/cold shock-induced nuclear burst is not involved in activation of DNA damage-sensing proteins such as ATR, ATM, Chk1, and Chk2, as determined by Western blotting using specific antibodies (data not shown). Moreover, induction of DNA damage by an inducer, neocarzinostatin (10–40 μM) [29, 30], failed to cause bubbling death.
Mechanistic insight
UV irradiation/cold shock rapidly causes upregulation and relocation of many cytosolic proteins to the nucleus. UV energy increases the expression of antiapoptotic TRAF2 and NF-κB/p65 and proapoptotic WWOX and p53, along with cytosolic NOS2 and membrane Hyal-2 and C1qBP (data not shown). Subsequent exposure of UV-treated cells to cold shock enhances accumulation of these proteins in the nucleus. NF-κB/p65 is a downstream effector of the TNF signaling, which blocks TNF-mediated cell death and thereby provides cancer survival [31, 32]. Hyal-2 acts as an alternative receptor for TGF-β1 and a binding protein for WWOX [17]. Serum complement C1q participates in cancer suppression via interaction with C1qBP and downstream WWOX [33]. WWOX plays a role in the hypoxic response [34]. Whether HIF1α is involved in the UV/cold shock response is unknown.
NOS2 and TRAF2
NOS2 is one of the nitric oxide synthase family proteins [35–37], and is responsible for the generation of NO-containing gas in the nucleus. Functional nitric oxide synthase has been shown in the nucleus [37]. In response to UV/cold shock, NOS2 becomes colocalized with TRAF2. Whether NOS2 and TRAF2 interact with each other and co-translocate to the nucleus remains to be established. Also, whether NOS2 binds p53 and WWOX is unknown. NOS2-regulated NO formation needs further conformation by using other specific methods. Similarly, how the jet stream pushes out in a directional manner through the nuclear and cell membranes is unknown and remains to be established.
Cold shock and protein relocation
UV alone is able to upregulate WWOX/WOX1, p53, NF-κ/p65, and TRAF2, plus C1qBP and Hyal-2 (data not shown). In contrast, cold shock alone is sufficient to induce the expression of NOS2, indicating the synthesis of mRNA and proteins at low temperature in mammalian cells. In combination, UV irradiation with subsequent cold shock promotes the accumulation of the aforementioned proteins in the nucleus. How the cold shock drives proteins to relocate to the nucleus is very intriguing. Yet, UV/cold shock induces NO-containing gas generation and bubble formation in an ATP-independent manner, which leads to cell death in vitro and may provide explanation to our understanding of the clinical feature of frostbite in humans.
Translocation of nucleolar proteins and exosome release
As determined by time-lapse microscopy, a majority of the nuclear and nucleolar proteins go to the bubble, and a minor portion may leak to the cytoplasm. We have tried to isolate bubbles and determined the nature of proteins in the bubbles. This has been really difficult, as the bubbles go readily broken during sample preparation. The functional relevance of the nuclear proteins in the bubbles is unknown. Furthermore, where there is a connection between the nuclear events of bubbling to the cytosolic membrane event of exosome release has yet to be established. Generation of exosomes could occur as the side effect (or counteractive forces) generated by the gas jet stream.
Inhibition of bubbling death by TRAF2
TNF-induced apoptosis can be turned off by activating NF-κB through TRAF2 [28]. TRAF2 has an N-terminal RING-type zinc finger domain, followed by a zinc finger domain, a coiled coil domain, and a MATH/TRAF domain at the C-terminus. Deletion of the N-terminal RING-type zinc finger domain of TRAF2 resulted in partial loss of its activity in blocking UV/cold shock-induced bubbling. Further analysis showed that the central zinc finger domain confers resistance to the effect of UV/cold shock. It has been reported that TRADD directly interacts with TRAF2, and then activates NF-κB for inducing the antiapoptotic event [8, 38–40]. In UV-activated cell apoptosis, TRAF2 promotes cell survival via activating NF-κB and inhibiting p53 from binding to the mitochondria and blocking the release of cytochrome C [7, 28, 41].
Conclusion
UV/cold shock induces bubbling death of cells at low temperatures (4–22°C). The event is p53- and WWOX-dependent. There is little or no chromosomal DNA fragmentation at low temperatures, indicating that apoptotic death is blocked. Upon raising the temperature up to 37°C, bubbling death is dramatically retarded in many types of cells, whereas DNA fragmentation occurs. That is, switching from bubbling death to apoptosis can be achieved by raising temperatures. Overall, we have discovered a novel bubbling death operating at low temperatures, and that apoptosis is blocked.
MATERIALS AND METHODS
Cell lines, transient gene expression in cells, and stable gene transfectants
Monkey kidney COS7 fibroblasts, murine L929 fibroblasts, murine melanoma B16F10, human squamous cell carcinoma SCC9 and SCC15, human colon HCT116, MEF Wwox−/− and Wwox+/+ cells, and 10 other types of cancer cell lines were cultured and used for experiments [12, 13, 15, 25]. COS7 and L929 were mainly used for experiments. Where indicated, cells were transfected by electroporation (BTX ECM 630 electroporator, Genetronics) with a specific mammalian expression construct for transient expression [12, 13, 15]. Or, the cells were treated with G418 for establishing stable gene transfecctants [12, 13, 15].
cDNA expression constructs
A variety of WWOX/WOX1 and p53 cDNA expression constructs, in pEGFP-C1, pEYFP-C1, pECFP-C1, and pDsRed monomer, were made as described [12, 13, 15, 19]. The following primers were used to construct TRAF2 cDNAs: 1) TRAF2 (full-length)-pEGFP-C1, forward: CAAGCTTGCATGG CTGCAGCCAGTGTGACTTC CCCTGGCTCCC; reverse: GCGCGAA GCTTCTAGAGTCCTGTTAGGT CCACAATAGCTTTGATG. 2) TRAF2(124–233)-pEGFP-C1, forward: ATTGAATTCTATGTGCCACGA AGGAC; reverse: AATGAATTCTTACT GCAGGTTCT CAGTCTCC.
Chemicals and antibodies
Polyclonal antibodies against WWOX, p53, pS46–p53, TRADD, FADD, TRAF2, ERK, and pERK, and monoclonal antibodies against NOS2, C1qBP, NF-κB/p65 and p53 were purchased from Santa Cruz Biotechnology. Lamin A/C polyclonal antibody and an antibody kit against proteins in sensing DNA damage (e.g. ATR, ATM, Chk1, Chk2, and others) were from Cell Signaling. Homemade polyclonal antibodies against WWOX/WOX1 and pTyr33-WOX1 were generated in rabbits as described [12, 13, 15]. Phospho-Ser14 WWOX/WOX1 polyclonal antibodies were made in rabbits against a synthetic peptide CAGLDDTD-pS-EDELPPG (amino acid sequence #7–21; Genemed Synthesis) [12, 13, 15]. The cysteine residue was added to the N-terminus to facilitate conjugation of the peptide to Keyhole Limpet Hemocyanin (KLH). Similarly, polyclonal antibodies against the phospho-Tyr287 WOX1 antibody were also made (amino acid sequence #286 to 299) using a synthetic peptide D-pY-WAMLAYNRSKLC. Two corresponding peptides WOX1(7–21) and WOX1(285–299) without phosphorylation were also made, respectively, and used for antibody production. An approved protocol for animal use was obtained from the Institutional Animal Care and Use Committee of the National Cheng Kung University Medical College. Nω-LAME, DAPI, and staurosporine were from Calbiochem.
Immunofluorescence microscopy, electron microscopy, and time-lapse microscopy
COS7 cells were irradiated with UV 480 mJ/cm2 and then subjected to cold shock for 30, 60, 120, 180 minutes at 4°C, respectively. Where indicated, the cells were incubated at room temperature or 37°C. In controls, cells were not treated or exposed to UV or cold shock only. Immunostaining was carried out using specific antibodies against TRAF2 and WWOX/WOX1, or other combinations. Electron microscopy was carried out as described [17]. Time-lapse microscopy was also carried out for UV/cold shock-treated COS7 and other cells. Confocal microscopy was then carried out using Nikon C1-Si Laser Scanning Confocal Microscope [17].
Immunohistochemistry (IHC), co-immunoprecipitation and Western blotting
IHC, co-immunoprecipitation and Western blotting were carried out, as described [12–15, 17]. Antibody against WWOX/WOX1 was used for co-immunoprecipitation [12–15, 17, 22]. For IHC, skin tissue sections from patient were immersed in blocking solution (2% BSA) for 1 hr, followed by adding an aliquot of antibody against WWOX/WOX1 or TRAF2 for 2 hr incubation at room temperature. Following washing with PBS for 3 times, the tissue sections were incubated with aliquots of horseradish peroxidase (HRP)-conjugated secondary antibodies, and the resulting color was then developed using an immunohistochemical staining kit (Dako). In controls, no primary antibodies were used. The images were analyzed by an NIH Image J program.
ACKNOWLEDGMENTS
This research was supported, in part, by the Department of Defense USA (W81XWH-08–1-0682), the Research the National Science Council, Taiwan, ROC (NSC98–2628-B-006–041-MY3, 99–2320-B-006–012-MY3, 102–2320-B-006–018-, 102–2320-B-006–030-, and 102–3011-P-006–005-), the National Health Research Institute, Taiwan, ROC (NHRI-EX101–10102BI, and NHRI-EX102–10102BI), the National Cheng Kung University Landmark Projects (C0167) and the Department of Health, Taiwan, ROC (DOH101-TD-PB-111-TM010) (to NS Chang).
Abbreviations
EGFP - enhanced green fluorescent protein; PS, phosphatidylserine; TRAF2 - TNF receptor-associated factor 2; TNF - tumor necrosis factor; WWOX (WOX1, FOR), WW domain-containing oxidoreductase; NOS2 - nitric oxide synthase 2; NO - nitric oxide; Nω-LAME - Nω-nitro-L-arginine methyl ester hydrochloride.
CONFLICT OF INTEREST
The authors declare no conflict of interest
Authors’ contributions
SJC, PWL, HPL and SSH: carried out experiments and worked on graphic arts; LJH: established Wwox knockout mice; FJL and HMS: provided human skin tissue sections; SJC, SSH, LJH, FJH and HMS: proofread and discussed the manuscript; NSC: wrote the manuscript, conceived ideas, designed experiments and performed fluorescence microscopy and time-lapse microscopy.
REFERENCES
1. Ikaheimo TM, Hassi J. Frostbites in circumpolar areas. Glob Health Action. 2011; 4:10.3402/gha.v4i0.8456. (http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3191821/).
2. Alibardi L. Adaptation to the land: the skin of reptiles in comparison to that of amphibians and endotherm amniotes. J Exp Zool B Mol Dev Evol. 2003; 298:12–41.
3. Breitkreutz D, Mirancea N, Nischt R. Basement membranes in skin: unique matrix structures with diverse functions? Histochem Cell Biol. 2009; 132:1–10.
4. Iozzo RV. Basement membrane proteoglycans: from cellar to ceiling. Nat Rev Mol Cell Biol. 2005; 6:646–656.
5. Auerbach LJ, Galvez MG, De Clerck BK, Glotzbach J, Wehner MR, Chang EI, Gurtner GC, Auerbach PS. A novel mouse model for frostbite injury. Wilderness Environ Med. 2013; 24:94–104.
6. Chandrasekar B, Vemula K, Surabhi RM, Li-Weber M, Owen-Schaub LB, Jensen LE, Mummidi S. Activation of intrinsic and extrinsic proapoptotic signaling pathways in interleukin-18-mediated human cardiac endothelial cell death. J Biol Chem. 2004; 279:20221–20233.
7. Cabal-Hierro L, Lazo PS. Signal transduction by tumor necrosis factor receptors. Cell Signal. 2012; 24:1297–1305.
8. Wajant H, Scheurich P. TNFR1-induced activation of the classical NF-kappaB pathway. FEBS J. 2011; 278:862–876.
9. Grell M, Zimmermann G, Gottfried E, Chen CM, Grunwald U, Huang DC, Wu Lee YH, Dürkop H, Engelmann H, Scheurich P, Wajant H, Strasser A. Induction of cell death by tumour necrosis factor (TNF) receptor 2, CD40 and CD30: a role for TNF-R1 activation by endogenous membrane-anchored TNF. EMBO J. 1999; 18:3034–3043.
10. Terry Powers JL, Mace KE, Parfrey H, Lee SJ, Zhang G, Riches DW. TNF receptor-1 (TNF-R1) ubiquitous scaffolding and signaling protein interacts with TNF-R1 and TRAF2 via an N-terminal docking interface. Biochemistry. 2010; 49:7821–7829.
11. Hedrych-Ozimina A, Behrendt K, Hao Z, Pofahl R, Ussath D, Knaup R, Krieg T, Haase I. Enhanced contact allergen- and UVB-induced keratinocyte apoptosis in the absence of CD95/Fas/Apo-1. Cell Death Differ. 2010; 18:155–163.
12. Chang NS, Doherty J, Ensign A. JNK1 physically interacts with WW domain-containing oxidoreductase (WOX1) and inhibits WOX1-mediated apoptosis. J Biol Chem. 2003; 278:9195–9202.
13. Chang NS, Doherty J, Ensign A, Schultz L, Hsu LJ, Hong Q. WOX1 is essential for tumor necrosis factor-, UV light-, staurosporine-, and p53-mediated cell death, and its tyrosine 33-phosphorylated form binds and stabilizes serine 46-phosphorylated p53. J Biol Chem. 2005; 280:43100–43108.
14. Lai FJ, Cheng CL, Chen ST, Wu CH, Hsu LJ, Lee JY, Chao SC, Sheen MC, Shen CL, Chang NS, Sheu HM. WOX1 is essential for UVB irradiation-induced apoptosis and down-regulated via translational blockade in UVB-induced cutaneous squamous cell carcinoma in vivo. Clin Cancer Res. 2005; 11:5769–5777.
15. Chang NS, Pratt N, Heath J, Schultz L, Sleve D, Carey GB, Zevotek N. Hyaluronidase induction of a WW domain-containing oxidoreductase that enhances tumor necrosis factor cytotoxicity. J Biol Chem. 2001; 276:3361–3370.
16. Tsai CC, Chen-Han Yang CH, Chang NS. Role of exosomes in intercellular signaling, maintenance of normal physiology, and their therapeutic potentials (commentary). Clin Exp Pharmacol. 2014; 4:154.
17. Hsu LJ, Schultz L, Hong Q, Van Moer K, Heath J, Li MY, Lai FJ, Lin SR, Lee MH, Lo CP, Lin YS, Chen ST, Chang NS. Transforming growth factor beta1 signaling via interaction with cell surface Hyal-2 and recruitment of WWOX/WOX1. J Biol Chem. 2009; 284:16049–16059.
18. Fackler OT, Grosse R. Cell motility through plasma membrane blebbing. J Cell Biol. 2008; 181:879–884.
19. Charras GT. A short history of blebbing. J Microsc. 2008; 231:466–478.
20. Lin HP, Chang JY, Lin SR, Lee MH, Huang SS, Hsu LJ, Chang NS. Identification of an in vivo MEK/WOX1 complex as a master switch for apoptosis in T cell leukemia. Genes Cancer. 2011; 2:550–562.
21. Hsu LJ, Hong Q, Schultz L, Kuo E, Lin SR, Lee MH, Lin YS, Chang NS. Zfra is an inhibitor of Bcl-2 expression and cytochrome c release from the mitochondria. Cell Signal. 2008; 20:1303–1312.
22. Hong Q, Hsu LJ, Schultz L, Pratt N, Mattison J, Chang NS. Zfra affects TNF-mediated cell death by interacting with death domain protein TRADD and negatively regulates the activation of NF-kappaB, JNK1, p53 and WOX1 during stress response. BMC Mol Biol. 2007; 8:50.
23. Dudekula S, Lee MH, Hsu LJ, Chen SJ, Chang NS. Zfra is a small wizard in the mitochondrial apoptosis. Aging (Albany NY). 2010; 2:1023–1029.
24. Lee MH, Su WP, Wang WJ, Lin SR, Lu CY, Chen YA, Chang JY, Huang SS, Chou PY, Ye SR, Chen SJ, He H, et al. Zfra activates memory Hyal-2+ CD3- CD19- spleen cells to block cancer growth, stemness, and metastasis in vivo. Oncotarget. 2014; in press.
25. Tsai CW, Lai FJ, Sheu HM, Lin YS, Chang TH, Jan MS, Chen SM, Hsu PC, Huang TT, Huang TC, Sheen MC, Chen ST, Chang WC, Chang NS, Hsu LJ. WWOX suppresses autophagy for inducing apoptosis in methotrexate-treated human squamous cell carcinoma. Cell Death Dis. 2013; 4:e792.
26. Chang JY, Lee MH, Lin SR, Yang LY, Chen SJ, Sun HS, Sze CI, Hong Q, Lin YS, Chou YT, Gong CX, Hsu LJ, Chang NS. Trafficking protein particle complex 6A delta (TRAPPC6AΔ) is a plaque-forming protein in the brain extracellular matrix. Oncotarget. 2014; in press.
27. Mahajan NP, Whang YE, Mohler JL, Earp HS. Activated tyrosine kinase Ack1 promotes prostate tumorigenesis: role of Ack1 in polyubiquitination of tumor suppressor Wwox. Cancer Res. 2005; 65:10514–10523.
28. Tobin D, van Hogerlinden M, Toftgard R. UVB-induced association of tumor necrosis factor (TNF) receptor 1/TNF receptor-associated factor-2 mediates activation of Rel proteins. Proc Natl Acad Sci U S A. 1998; 95:565–569.
29. Abu-Odeh M, Salah Z, Herbel C, Hofmann TG, Aqeilan RI. WWOX, the common fragile site FRA16D gene product, regulates ATM activation and the DNA damage response. Proc Natl Acad Sci U S A. 2014; 111:E4716–4725.
30. Mallon R, Feldberg L, Kim S, Collins K, Wojciechowicz D, Kohler C, Kovacs D, Discafani C, Zhang N, Wu B, Floyd B, Powell D, Berger D. Identification of 4-anilino-3-quinolinecarbonitrile inhibitors of mitogen-activated protein/extracellular signal-regulated kinase 1 kinase. Mol Cancer Ther. 2004; 3:755–762.
31. Mauro C, Zazzeroni F, Papa S, Bubici C, Franzoso G. The NF-kappaB transcription factor pathway as a therapeutic target in cancer: methods for detection of NF-kappaB activity. Methods Mol Biol. 2009; 512:169–207.
32. Chen SJ, Huang SS, Chang NS. Role of WWOX and NF-κB in lung cancer progression. Transl Respir Med. 2014; 1:15.
33. Hong Q, Sze CI, Lin SR, Lee MH, He RY, Schultz L, Chang JY, Chen SJ, Boackle RJ, Hsu LJ, Chang NS. Complement C1q activates tumor suppressor WWOX to induce apoptosis in prostate cancer cells. PLoS One. 2009; 4:e5755.
34. Abu-Remaileh M, Aqeilan RI. Tumor suppressor WWOX regulates glucose metabolism via HIF1α modulation. Cell Death Differ. 2014; 21:1805–1814.
35. Geller DA, Billiar TR. Molecular biology of nitric oxide synthases. Cancer Metastasis Rev. 1998; 17:7–23.
36. Weinberg JB. Nitric oxide synthase 2 and cyclooxygenase 2 interactions in inflammation. Immunol Res. 2000; 22:319–341.
37. Aquilano K, Baldelli S, Ciriolo MR. Nuclear recruitment of neuronal nitric-oxide synthase by alpha-syntrophin is crucial for the induction of mitochondrial biogenesis. J Biol Chem. 2014; 289:365–378.
38. Baker SJ, Reddy EP. Transducers of life and death: TNF receptor superfamily and associated proteins. Oncogene. 1996; 12:1–9.
39. Heyninck K, Beyaert R. Crosstalk between NF-kappaB-activating and apoptosis-inducing proteins of the TNF-receptor complex. Mol Cell Biol Res Commun. 2001; 4:259–265.
40. Sheikh MS, Huang Y. Death receptor activation complexes: it takes two to activate TNF receptor 1. Cell Cycle. 2003; 2:550–552.
41. Wan YS, Wang ZQ, Shao Y, Voorhees JJ, Fisher GJ. Ultraviolet irradiation activates PI 3-kinase/AKT survival pathway via EGF receptors in human skin in vivo. Int J Oncol. 2001; 18:461–466.