
Introduction
Statins are a class of small fungal metabolites that inhibit the HMG-CoA reductase, an enzyme that catalyses the reduction of 3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA) to mevalonate [1]. This is a rate-limiting step of the cholesterol biosynthesis pathway. For this reason statins have been used extensively for the treatment of hypercholesterolemia. It has been appreciated for some time that statins also can induce growth arrest in several diverse cancer cell lines [2, 3]. Statin induced apoptosis has also been reported to occur in a subset of tumour cell types [3]. Importantly, normal cells appear to be more resistant to statin induced growth arrest and cytotoxicity. Thus statins have been reported to inhibit proliferation of karyotipically abnormal embryonic stem cells but not normal embryonic stem cells [4]. In addition, lipophilic statins were found to induce cell death in MCF-7 cells more efficiently than in non-cancerous MCF-10 epithelial cells [5]. In vivo, lovastatin has also been shown to prevent carcinogenesis in a rat model for liver cancer [6]. Furthermore, recent evidence indicates that statins can impinge on cancer development at different stages of progression. Thus statins have the potential to interfere with the invasion and migration of prostate cancer cells via negative regulation of phospholipase 2 group VII (PLA2G7) [7]. Interestingly, statins have also been shown to have an immunomodulatory function in spontaneous mouse mammary tumours by promoting T cell infiltration [8]. This activity was shown to lead to reduced formation of new lesions in a mouse genetic model for tumour development. Prospective observational studies in humans revealed a significant reduction in cancer risk (14%-28%) associated with Statin use [9].
These observations coupled with a well-established clinical history in the treatment of hypercholesterolemia have led to the proposition that statins may be used in the treatment of cancer. However, conclusive data from randomised control trials for statin use and cancer risk are currently lacking. In general statin treatment seems to be beneficial for the treatment of certain types of cancer but not for others [10]. Statins also target a broad range of cellular processes which may confound their efficacy [11]. Therefore a better understanding of the molecular pathways responsible for their ability to inhibit cell proliferation seems important.
Statins can lead to the induction of the cyclin dependent kinase inhibitors (CKI) p27Kip1 and p21Cip1, leading to decreased cyclin dependent kinase activity and subsequent cell cycle arrest in G1 phase [12-14]. The induction of p27 by lovastatin is regulated post-transcriptionally [15]. During normal cell cycle progression, p27Kip1 is ubiquitinated in late G1 phase by the SCFSkp2 ubiquitin ligase and thereby targeted for proteasomal degradation [16-19]. SCFSkp2 is also an ubiquitin ligase for p21Cip1 [20, 21]. It has proposed that lovastatin can up regulate p27Kip1 and p21Cip1 by directly inhibiting the 26S proteasome [22]. The presence of a lactone moiety in the closed ring prodrug form of lovastatin that can directly inhibit the chymotrypsin like catalytic activity of the proteasome has been posited to account for this G1 arresting activity [22, 23]. However other reports demonstrate that neither the prodrug nor the active dihydroxy acid form of lovastatin can inhibit the proteasome in SSC25, HeLa or 293T cells [24]. Indeed, one study suggests that the closed ring form can mildly stimulate the chymotrypsin like activity of purified bovine 20S proteasomes [25] .
We previously observed that the level of the Skp2 protein, a E3 ubiquitin ligase subunit, regulating p27 stability during the G1/S transition is specifically reduced significantly following lovastatin-induced cell cycle arrest, but not a cell cycle arrest induced by nocodazole or thymidine [26]. Skp2 is an oncogene that has been shown to be an independent marker for poor prognosis in several types of human cancer [27-29]. We wondered if the reduced Skp2 levels were causing the elevated levels of p27 in lovastatin treated cells and therefore driving cessation of proliferation. This could reconcile the contradictory reports on the effects of lovastatin on proteasome activity; rather than a general blockade of proteasome activity, we speculated that by down regulating Skp2, lovastatin might prevent the proteasomal degradation of p27 and p21, whilst not affecting proteasomal turnover in general.
Here we show that Skp2 is robustly and specifically down regulated in response to lovastatin treatment and that this precedes the p27 and p21 accumulation. Downregulation of Skp2 is cell cycle phase independent and therefore not a consequence of the cell cycle arrest in G1 phase. Furthermore, we establish that destabilisation of the protein is responsible for the downregulation of Skp2. Interestingly, the mechanism for degrading Skp2 is distinct from the previously reported Cdh1-dependent degradation or auto-ubiquitination [30-32], thus representing a novel pathway of Skp2 degradation. Lovastatin triggers this pathway by preventing the geranyl-geranylation of an as yet unknown protein, since geranylgeranyl pyrophosphate rescues the lovastatin arrest and inhibition of geranylgeranyl transferase I also induces downregulation of Skp2. An N-terminal domain in Skp2 is required and sufficient to permit its downregulation by lovastatin and inhibitors of geranylgeranyl transferases.
Results
Lovastatin induces downregulation of the Skp2 protein, but does not alter Skp2 mRNA level
As reported previously, treatment of cells with lovastatin leads to increased levels of p21 and p27 and an accumulation of cells in G1 phase of the cell cycle [12-14, 26, 33] (Fig. 1A, C). This is associated with significantly reduced Skp2 protein level, whereas the INK4 inhibitors p15 and p16, cyclins A and E as well as CDK level remain mostly unaltered (Fig. 1A). Since the level of Skp2 protein is normally low during early and mid G1 phase, the reduced amount of Skp2 protein following lovastatin treatment might be a consequence of the cell cycle arrest rather than its cause. Skp2 transcription and protein levels are increased in S-phase [32, 34]. To rule out a cell cycle positional effect, we first arrested HeLa cells in S-phase and subsequently treated the S-phase arrested cells with lovastatin. Lovastatin caused a clear decline in the Skp2 protein level even in S-phase arrested cells, where Skp2 usually is relatively stable (Fig. 1A, + thymidine), demonstrating that the reduction of Skp2 occurs independently of the G1 arrest caused by lovastatin. p21 and p27 did not accumulate in S-phase arrested cells, probably because Skp2 levels remain generally elevated and these remaining levels of Skp2 may be sufficient to promote p21 and p27 degradation. In fact, even though Skp2 levels in lovastatin-treated S-phase cells were significantly reduced, they remain similar to those of proliferating (untreated) cells. Thus the decrease in Skp2 protein following lovastatin treatment does not merely reflect the cell cycle position, but suggests a cell cycle independent direct pathway regulating Skp2 abundance upon lovastatin addition. This could cause the stabilisation of p27 and p21 and the subsequent cell cycle arrest.
Consistent with this model, we observed that the lovastatin-induced decrease in Skp2 precedes the accumulation of p27 (Fig. 1B, C). To directly investigate the hypothesis that Skp2 decline is driving accumulation of p27 and subsequent inhibition of proliferation, we generated virally transduced cell lines stably overexpressing the Skp2 cDNA. As reported previously, Skp2 overexpression is able to induce degradation of p27 [18, 26]. p27 is known to be phosphorylated on threonine residue 187 in lovastatin-arrested cells [18, 26], generating the phosphodegron which is a prerequisite for its SCF-initiated degradation. Overexpression of Skp2 reduced p27 levels in stably transduced cells in the presence and absence of lovastatin. Importantly, Skp2 overexpression could attenuate the lovastatin-induced accumulation of the CDK inhibitors with p27 level similar to those of proliferating cells (Fig. 1D). This corresponded with a decreased number of cells in G1-phase and increased number of cells in S-phase as revealed by FACS analysis (Fig. 1D), consistent with the hypothesis that the decline of Skp2 promotes the accumulation of p21 and p27 in lovastatin arrested cells. As a main substrate of Skp2 is the CDK inhibitor p27 [19], we investigated if cells lacking p27 expression are less sensitive to lovastatin-induced cell cycle arrest. Immortalised p27-/- mouse embryonic fibroblasts were incubated with lovastatin for 24 hrs and compared to wt MEFs. Untreated p27-/- MEFs are characterised by reduced cells in G1 phase and increased cells in S-phase (Fig. 1E). Upon incubation in lovastatin, less p27-/- cells accumulated in G1 phase (73% vs. 95%) and more cells remained in S-phase (19% of p27-/- compared to 2% of wt MEFs). These data further support our hypothesis that the Skp2-p27 axis plays an important role in the lovastatin-induced cell cycle arrest.
Of note, statins are also able to induce a decline of overexpressed Skp2. This suggests that lovastatin regulates Skp2 levels independently of transcription from the endogenous Skp2 gene, since overexpression was achieved by viral transduction of a Skp2 cDNA expressed under the control of the phosphoglycerate kinase (PGK) promoter. To investigate if the reduction of Skp2 is transcriptionally regulated or due to altered Skp2 mRNA stability, we compared Skp2 mRNA levels in lovastatin-arrested cells and untreated cells. In contrast to the protein level, no significant reduction of Skp2 mRNA was observed in asynchronous cells as well as in S-phase arrested cells (Fig. 1F). This excludes transcriptional mechanisms and the regulation of Skp2 mRNA stability as a cause of the Skp2 elimination.
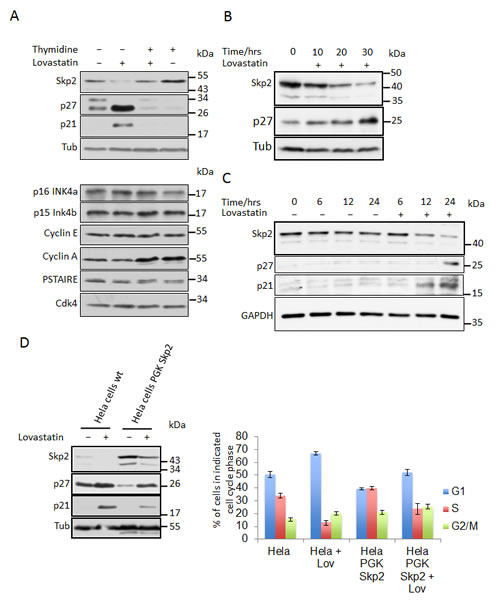
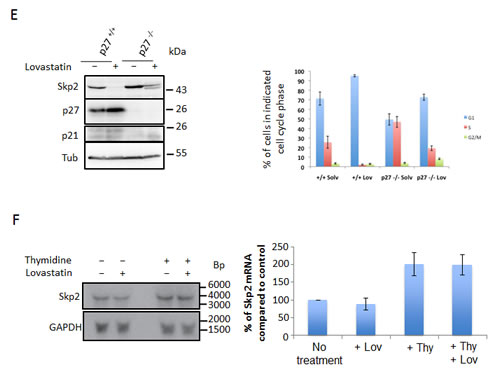
Figure 1: Lovastatin induces downregulation of the Skp2 protein. (A) HeLa cells growing asynchronously or arrested in S-Phase by treatment with 2mM thymidine for 24 hours, were then treated or left untreated with 40µM lovastatin for a further 24 hours and analysed by (A) immunoblotting. (B) LnCAP and HeLa cells (C) were incubated with 10µM and 40µM lovastatin respectively for the indicated times and analysed by immunoblotting. (D) HeLa cells stably expressing Skp2 from a PGK promoter were generated by lentiviral transfection. These cells were treated with 40µM lovastatin for 24 hours and then subjected to immunoblotting analysis. Percentages of cells in the different cell cycle phases were determined by flow cytometric analysis of three independent experiments using propidium iodine and BrdU double staining (Bar graph; data are represented as mean +/- SEM). (E) Control MEFs and p27 -/- MEFs were treated with 20µM lovastatin for 24 hours as indicated, harvested and analysed by western blotting analysis (left panel). Flow cytometry analysis of BrdU labeled cells to determine cell cycle phase distribution was performed. The percentage of cells in G1, S or G2/M phase is shown for at least three independent experiments (right panel). (F) Skp2 mRNA analysis of HeLa cells treated as described in Fig. 1A. Northern blots from three independent experiments were quantified using a PhosphorImager and the relative abundance of the Skp2 mRNA is shown ( bar graph; data are represented as mean +/- SEM).
Lovastatin induces degradation of Skp2 which is independent of APC/CCdh1
Northern blot analysis demonstrated that the decrease in Skp2 levels is based on a post-transcriptional mechanism. The Skp2 protein itself is known to undergo ubiquitin-dependent proteasomal degradation. In order to investigate if lovastatin might induce the degradation of the Skp2 protein, we treated cells with the cell permeable reversible proteasome inhibitor MG132 (carbobenzoxy-Leu-Leu-leucinal) and lovastatin and monitored the Skp2 decline. The peptide aldehyde MG132 prevents down-regulation of Skp2 following lovastatin treatment (Fig. 2A). This demonstrates that inhibition of proteolysis restores Skp2 expression, suggesting that lovastatin initiates the degradation of the protein. As expected, MG132 also induced stabilisation of p21 and p27, which are substrates of the ubiquitin-proteasome system (Fig. 2A).
The stability of Skp2 is cell cycle regulated. Another ubiquitin E3 ligase complex, APC/CCdh1 initiates its ubiquitin-mediated proteasomal degradation. We used mouse embryonic knockout fibroblasts for the APC subunit Cdh1 to determine if Skp2 degradation induced by lovastatin was dependent on the APC/CCdh1 ubiquitin ligase. As can be seen in (Fig. 2B), lovastatin treatment results in a decrease in Skp2 levels even in the absence of Cdh1. This was also observed in cells in which Cdh1 can be inducibly knocked down by doxycycline (Fig. 2C). Since we can also exclude autoubiquitination as a mechanism triggering the Skp2 elimination induced by statins (see below), a novel pathway is responsible for the statin-induced degradation of Skp2.
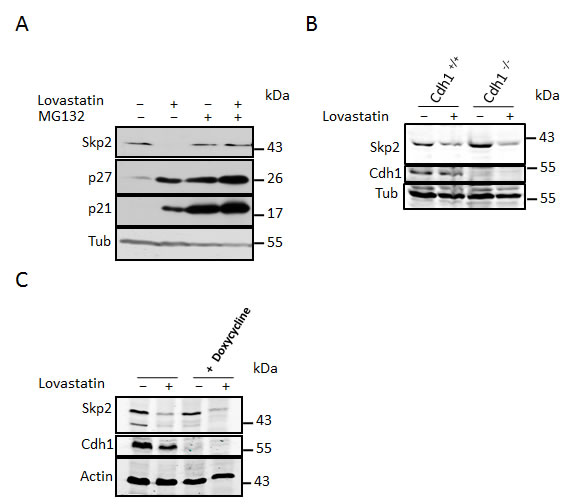
Figure 2: Lovastatin induces Skp2 protein degradation which is independent of its canonical E3 ubiquitin ligase APC/CCdh1. (A) HeLa cells were treated with 40µM Lovastatin in the presence or absence of 10µM MG132. After 24 hours, samples were harvested and analysed by western blotting. (B) Control MEFs and Cdh1 -/- MEFs were treated with 40µM lovastatin for 24 hours, harvested and subjected to western blotting analysis. (C) Utsh2 cells in which Cdh1 can be inducibly knocked down were treated with or without 1µg/ml doxycycline for 48 hours in order to induce Cdh1 knockdown after which they were replated in medium containing 100ng/ml doxycycline and incubated for a further 24 hours and then supplemented with 40µM lovastatin for 48 hours after which they were harvested and subjected to Western blotting analysis.
Skp2 downregulation is dependent on geranylgeranylation
Early intermediates of the cholesterol biosynthesis pathway such as mevalonate and isoprenoids can rescue statin mediated G1 arrest, whereas late intermediates such as squalene and cholesterol cannot [2]. Isoprenoid lipids are known to be required for the function of a number of proteins including oncogenic GTPases such as Ras and Rho proteins [35]. Furthermore, a number of studies have shown that inhibitors of isoprenylation can inhibit cell proliferation. We therefore tested the ability of geranylgeranyl pyrophosphate (GGPP) and farnesyl pyrophosphate (FPP) to prevent Skp2 down-regulation following lovastatin treatment. We found that GGPP could completely prevent the Skp2 decrease as well as the p27 and p21 increase, whereas FPP did not significantly alter the ability of lovastatin to down regulate Skp2 and increase p21 and p27 (Fig. 3A). As FPP is a precursor of GGPP, one might expect that FPP should also rescue the Lovastatin effect. However, in order to produce a 20 carbon geranylgeranylpyrophosphate from a 15 carbon FPP precursor, it requires the 5 carbon isopentenyl pyrophosphate. As lovastatin blocks the de novo synthesis of isopentyl pyrophosphate, which is synthesized from mevalonic acid, its depletion should inhibit the GGPP synthesis from FPP.
We speculated that inhibition of protein geranylgeranylation might cause the lovastatin induced Skp2 degradation. To investigate if blockade of protein geranylgeranylation might induce Skp2 degradation, we asked if inhibitors of geranylgeranylation could mimic the effects of lovastatin on Skp2. Inhibition of geranylgeranyltransferase I (GGTI) with the small molecule inhibitor GGTI-298 led to a dramatic decline in Skp2 levels and to an increase of p21 and p27 (Fig. 3B). In contrast, an inhibitor of farnesyltransferase, FTI-277, was not able to induce Skp2 down-regulation (Fig. 3B). These data indicate that lack of geranylgeranylation is responsible for the statin-induced Skp2 decline. Skp2 declined rapidly in response to GGTI-298 treatment with a significant decline occurring already at 6 hours of treatment (Fig. 3C). p27 accumulation commences after 12 hours and is clearly apparent after 24 hours. Thus the sequential order of Skp2 decline and p27 accumulation is conserved between GGTI-298 and lovastatin treatment. The Skp2 decline following GGTI-298 treatment is also prevented by MG132 treatment (Fig. 3D), supporting our model that lovastatin induces Skp2 degradation by inhibiting protein geranylgeranylation. As for lovastatin, knockout of p27 in mouse embryonic fibroblasts also attenuated G1 accumulation and inhibition of cell proliferation (Fig. 3E). Using the Cdh1-/- MEFs, we found that the GGTI-298 induced Skp2 decline (like that of lovastatin) still occurs in the absence of Cdh1 (Fig. 3F). Using Nocodazole treatment preceding GGTI-298 incubation we could also demonstrate that GGTI-298 can induce proteolytic destruction of Skp2 in G2/M arrested cells (Fig. 3G), confirming that this degradation pathway is independent from a G1 arrest and active in different phases of the cell cycle. Together, these data indicate that lovastatin and GGTI-298 promote the specific degradation of Skp2 and that this enables p27 to accumulate. The inhibition of geranylgeranyltransferases with GGTI-298 uses a highly conserved and robust mechanism to downregulate Skp2, as we observed this downregulation in all human and murine cell lines tested (Fig. 4 A, B).
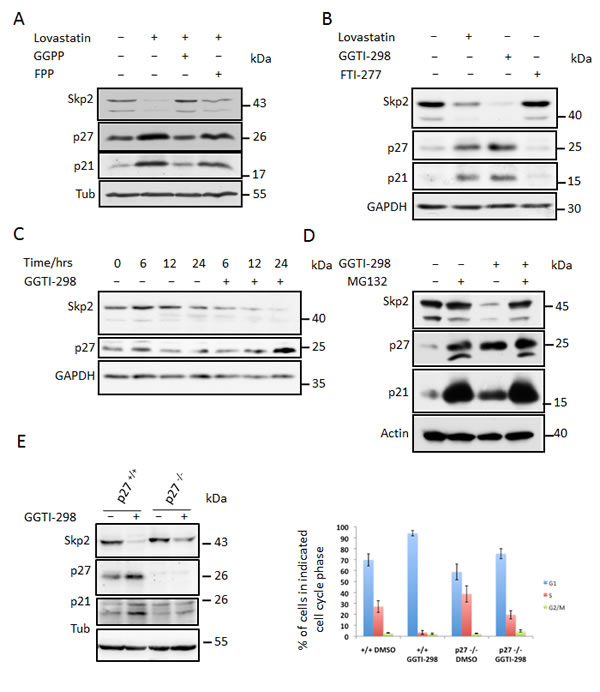
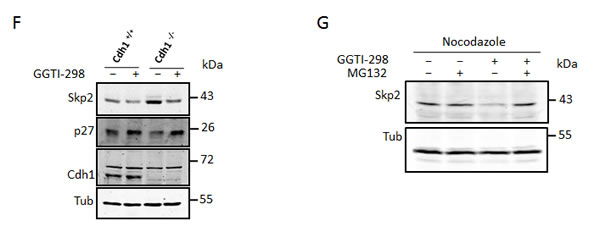
Figure 3: Skp2 decline is due to blockade of geranylgeranylation. (A) HeLa cells were treated with 40µM lovastatin in the presence of geranylgeranylpyrophosphate (GGPP) or farnesylpyrophosphate (FPP). After 24 hours incubation, cells were harvested and subjected to western blotting analysis. (B) HeLa cells were treated 20µM lovastatin, geranylgeranyltransferase inhibitor GGTI-298 or farnesyltransferase inhibitor FTI-277 for 36 hours and analysed by western blotting. (C) HeLa cells were treated with 10µM GGTI-298 for different times (as indicated) after which Skp2, p27 levels were determined by immunoblot analysis. (D) HeLa cells were treated with 10µM GGTI-298 in the presence and absence of 20mM MG132. After 24 hours, samples were harvested and analysed by western blotting. (E) Control MEFs and p27 -/- MEFs were treated with 10 µM GGTI-298 for 24 hours as indicated, harvested and analysed by western blotting analysis (left panel). Flow cytometry analysis of BrdU labeled cells to determine cell cycle phase distribution was performed. The percentage of cells in G1, S or G2/M phase is shown for at least three independent experiments (right panel). (F) Control MEFs and Cdh1-/- MEFs were treated with 10µM GGTI-298 for 24 hours then harvested and subjected to western blotting analysis. (G) Hela cells were treated with 50ng/ml Nocodazole for 18 hours after which they were supplemented with GGTI-298 at a final concentration of 10µM or solvent control. After a further 3 hours cells were supplemented with MG132 (20µM) or solvent and incubated for a further 9 hours. Cells were then harvested and processed for western blotting analysis.
The N-terminal domain of Skp2 is required for the down-regulation
We next aimed to identify a domain responsible for the statin-mediated degradation of Skp2. To identify sequence elements of Skp2 required for its downregulation by lovastatin or GGTI-298, we analysed several point mutants and deletion mutants for their ability to undergo GGTI-298 or lovastatin induced degradation. Structural [36] and functional [16, 18, 37] analysis of Skp2 has identified at least three separate domains: A N-terminal regulatory domain, the F-box domain required for interaction with the core SCF subunit Skp1 [38] and a C-terminal leucine-rich repeat region forming a concave surface that interacts with phosphorylated substrate proteins [36, 37, 39]. We generated a Skp2 mutant lacking the internal F-box domain [16], a deletion mutant of the N-terminal domain, (lacking amino acids 1-94: Skp2 ∆ NT), and a deletion mutant that lacks the C-terminal LRR domain (Skp2 ∆ CT) (Fig. 5A). These mutants were transfected into HeLa cells, which were then subsequently treated with GGTI-298 or lovastatin. The F-box of Skp2 is essential for its incorporation into the SCF complex [38]. A mutant lacking this domain was still regulated by GGTI-298 and lovastatin, indicating that incorporation of Skp2 into an SCF is not a prerequisite for its lovastatin or GGTI-298 induced degradation (Fig. 5B). Since the F-box domain is also required for the auto-ubiquitination of Skp2 by the bound SCF core complex [30], and since p27 inhibits Skp2 auto-ubiquitination in vivo [40], it is unlikely that auto-ubiquitination is contributing to the lovastatin or GGTI-298 induced elimination of Skp2.
The mutant lacking the C-terminal domain was down regulated to a similar extent as the wt protein following GGTI-298 or lovastatin addition (Fig. 5B). In contrast, the mutant lacking the first 94 amino acids is significantly stabilised (Fig. 5B), even though it contains the F-box domain.
These data suggested that an important determinant for lovastatin/GGTI-298 mediated Skp2 destabilisation resides in the N-terminus of the molecule. We therefore investigated if this N-terminal region might be sufficient to initiate degradation triggered by GGTI-298 or lovastatin. We fused this N-terminal domain fragment to enhanced YFP (eYFP) (Fig. 5A). The fusion protein, Skp2 ∆ NT and eYFP were expressed in HeLa cells. The levels of YFP alone or Skp2 lacking its N-terminus (Skp2 ∆ NT) are not appreciably reduced by either lovastatin or GGTI-298 treatment. However, the first 94 amino acids of Skp2 are sufficient to trigger degradation of the fusion protein upon inhibition of geranylgeranyl transferase I and, to a lesser extent by lovastatin (Fig. 5C). This is likely a consequence of the faster depletion of the geranylgeranylation by GGTI-298 compared to lovastatin (see above). Thus, the N-terminal region of Skp2 can function as a transferable degron in the context of GGTI-298 and lovastatin mediated Skp2 down-regulation. As this domain lacks the F-box domain and the C-terminus of Skp2, the statin-induced degradation of Skp2 does not require its incorporation into an SCF complex and therefore does not require auto-ubiquitination.
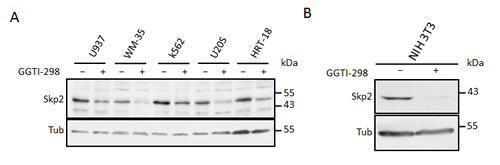
Figure 4: Inhibition of geranylgeranylation induces Skp2 degradation in many cell types. (A) A selection of tumour dervied human cell lines were incubated with 10µM GGTI-298 after which Skp2 levels were determined by immunoblotting. (B) NIH3T3 cells were incubated with 10µM GGTI-298 for 24 hours after which Skp2 levels were determined by immunoblotting.
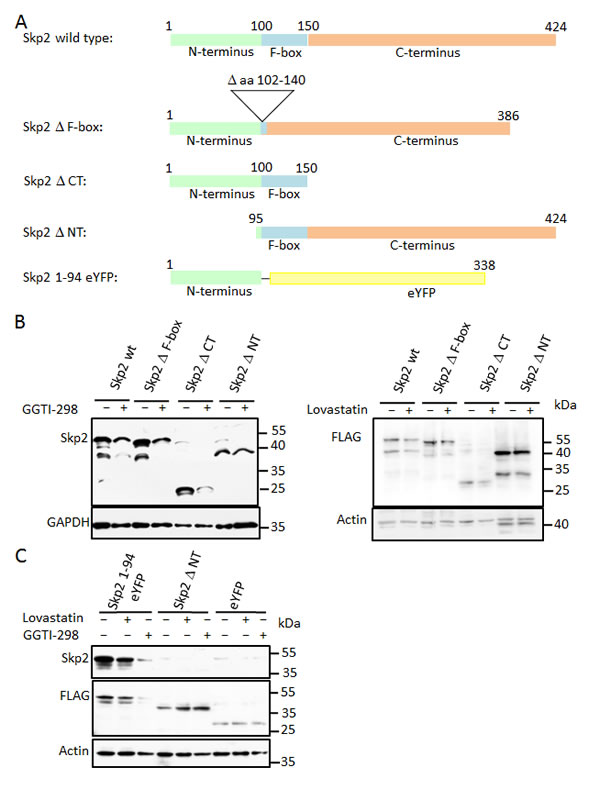
Figure 5: Skp2 degradation induced by GGTI-298/lovastatin depends on the N-terminal region which is sufficient to destabilise GFP in the presence of GGTI-298/lovastatin. (A) Schematic representation of Skp2 functional domain mutants. (B) HeLa cells were transfected with the indicated FLAG-tagged Skp2 constructs and 24 hours after transfection incubated in the presence of 11µM GGTI-298 (left panel) of 60µM lovastatin (right panel) for 24 hours, after which they were analysed by western blotting. (C) A FLAG-tagged fusion protein consisting of the N-terminus of Skp2 fused to eYFP was transfected into HeLa cells and after 24 hours incubation cells were then treated with 40µM lovastatin or 10µM GGTI-298 as indicated for an additional 24 hours. FLAG-tagged Skp2 ∆ NT and eYFP were transfected as controls. Cells were harvested and analysed by western blotting.
Discussion
Here we describe that the lovastatin-induced cell cycle arrest is caused by the degradation of Skp2 induced by the depletion of geranylgeranyl pyrophosphate. Skp2 is a subunit of the SCFSkp2 ubiquitin ligase and its statin mediated decline leads to an accumulation of the CDK inhibitors p27Kip1 and p21Cip1 and cell cycle arrest in G1 phase. The degradation of Skp2 triggered by statins is not a consequence of auto-ubiquitination or APC/CCdh1-mediated degradation. Instead it is mediated by a novel pathway initiating Skp2 degradation.
Some previous studies have suggested that the accumulation of p21 and p27 may result from direct inhibition of the proteasome by lovastatin. For example, it was observed that the inactive prodrug form of lovastatin could inhibit the proteasome [22]. Mevalonate, the product of the HMG-CoA reductase reaction, could activate proteasomal activity [41], explaining why mevalonic acid releases cells from the lovastatin arrest. But a later study found no evidence that the prodrug inhibits proteasomal activity [24]. Yet a further study found that the closed ring beta-lactone form of lovastatin induced the disappearance of p27. While this closed ring form was cytotoxic, no evidence of cytostatic activity could be found [25]. This study reported in contrast that mevalonate could inhibit the trypsin like activity of the proteasome. These experiments might have been complicated by the fact that serum is a potential source of carboxyesterases that can process the prodrug to its open ring form [42].
We observed no evidence that general and off target inhibition of the proteasome activity contributes to the lovastatin mediated G1 arrest. Our data rather suggest that the inhibition of p27 and p21 degradation is substrate specific and a consequence of the ability of lovastatin to deplete geranylgeranyl pyrophosphate. This depletion triggers a novel pathway that stabilises the CDK inhibitors p21 and p27 by degradation of the ubiquitin ligase subunit Skp2. Importantly, ubiquitin-mediated degradation of p27 and p21 could be partially restored by ectopic expression of Skp2 in the presence of lovastatin (Fig. 1D), clearly demonstrating that the proteasome remains active. Similarly, deletion of the central Skp2 substrate p27 attenuated the lovastatin-induced cell cycle arrest, confirming the important role of the Skp2/p27 axis in this arrest.
Interestingly, the geranylgeranylation-dependent degradation of Skp2 is functional in different cell lines (all of those that we tested) from human or mouse, reflecting the wide-ranging cell cycle arresting activity of lovastatin previously observed. In mouse models for tumorigenesis, it has been shown that geranylgeranyl-transferase-I ablation reduces tumour formation and improves survival [43], suggesting that geranylgeranylated proteins could also be important targets of lovastatin’s anti-tumour activity. Our observation that GGPP can rescue lovastatin induced Skp2 down-regulation and that inhibition of geranylgeranyl-transferase-I with GGTI-298 induces Skp2 downregulation suggests that a geranylgeranylated protein regulates Skp2 stability, whose function can be activated by inhibition of geranylgeranylation, leading to Skp2 degradation. It is tempting to speculate that this is not only a drug-specific pathway.
Geranylgeranylated Rho family GTPases, including RhoA and Rac1 have been shown to regulate G1 progression. Previous studies have suggested that geranylgeranylated Rho GTPases can destabilize p27 [44, 45] or that RhoA acts as positive regulator of cyclin E/CDK2 kinase activity [46]. Since Cyclin E /CDK2 phosphorylates p27 on T187 and this phosphorylation is required for its degradation, blocking of geranylgeranylation could inhibit p27 degradation [45, 47]. However, p27 is phosphorylated on threonine 187 in the presence of lovastatin [26] and overexpression of Skp2 can rescue p27 degradation in lovastatin-arrested cells. Other studies have suggested that the GTPase Rac1 can positively regulate Skp2 levels [48]. Inhibition of the geranylgeranylation of these small GTPases might therefore cause the decline of Skp2. However, we could not find any evidence that these GTPases or the Rho downstream kinase ROCK are involved in the statin-induced Skp2 degradation: the lovastatin/GGTI-298-induced Skp2 degradation was insensitive to the overexpression of wt or constitutively active GTPases including RhoA, Rac1A, and Rac1B; or by ROCK overexpression (JV, AM and LH, unpublished observations). In addition, lovastatin-induced Skp2 degradation was also not changed in the presence of cell permeable C3 toxin which inactivates RhoA, B and C [49].
We therefore speculate that an as yet unidentified geranylgeranylated protein is responsible for the Skp2 degradation initiated by lovastatin. However molecular details of this novel Skp2 degradation pathway remain to be elucidated in the future. Importantly, this novel pathway does not involve known mechanisms of Skp2 ubiquitin-mediated proteasomal degradation: The ubiquitin ligase APC/CCdh1 is necessary and sufficient for Skp2 degradation in early G1 phase [31, 32]. We can exclude involvement of APC/CCdh1 in statin-induced Skp2 degradation because lovastatin induced turnover of Skp2 also occurs in S-phase arrested cells, in which APC/CCdh1 is inactive [50], and because lovastatin and GGTI-298 can induce the degradation of Skp2 in cells lacking the Cdh1 protein.
In serum-deprived cells an auto-ubiquitination mechanism regulates Skp2 stability [30, 40]. This mechanism depends on Skp2 incorporation into an SCF complex. Since we observe efficient turnover of a Skp2 mutant lacking the F-box required for the incorporation into an SCF complex and which even functions as a dominant negative protein by stabilising p27 [16], we can exclude auto-ubiquitination as a mechanism for the lovastatin/GGTI-298 induced Skp2 degradation.
We therefore conclude that lovastatin activates a novel and as yet uncharacterised pathway of Skp2 degradation. It will be interesting to determine if this pathway is only activated by drugs inhibiting geranylgeranylation, or if alternative physiological mechanisms exist in normal cells that use this pathway to induce CDK inhibitors and cell cycle arrest.
The discovery of a novel degradation pathway for Skp2 induced by statins opens up the exiting possibility that this pathway may be targeted in cancer therapy. Whilst statins have shown potential in cancer prevention, their use as a therapy may be hampered by their pleiotropic effects and tendency to accumulate in the liver. Given that Skp2 overexpression is associated with increased malignancy and poor prognosis in several types of human cancer, the future elucidation of this conserved and robust antiproliferative pathway responsible for the elimination of the Skp2 may permit the identification of novel therapeutics that could be more specific and lack the side effects of statins.
Materials and methods
Reagents
Lovastatin (MERCK #L-154, Alfa Aesar # H52792), MG132 (Sigma-Aldrich #M7479), Thymidine (Sigma-Aldrich #T9250), GGPP (Sigma-Aldrich #G6025), FPP (Sigma-Aldrich #F6892), GGTI-298 (Sigma-Aldrich #G5169). FTI-277 (Sigma-Aldrich #F9803), BrdU (Sigma-Aldrich #B9285), propidium iodine (PI) (Sigma-Aldrich #81845). Nocodazole (Sigma-Aldrich # M1404). Lovastatin was obtained in the form of an inactive beta-lactone that was activated according to the following protocol: 208mg of lovastatin was dissolved in 5ml Ethanol and then supplemented with 7.5 ml 0.1M NaOH. The solution was incubated at 50oC for 2 hours after which it was allowed to cool to room temperature. Finally the pH was adjusted to 7.2 after with 1M HCl. Solvent control was prepared as above but with the omission of lovastatin.
Cell culture, transfections and lovastatin, GGTI-298, thymidine and Nocodazole treatments
The following cell lines were used in this study: Cdh1-/- MEFs and U2OS cells in which Cdh1 knockdown can be induced by tetracycline (Utsh2 cells) have been described previously [51]. Cells were cultured at 37 °C with 5% CO2 in DMEM (HeLa S3, HeLa, Cdh1-/- MEFs, Cdh1+/+ MEFs, NIH 3T3, p27-/- MEFs, p27+/+ MEFs, U20S and HRT-18) or RPMI (LNCAP, K562, WM35 and U937) medium containing 10% FCS. Transient transfection experiments were conducted using Effectene® (Qiagen) transfection reagent according to the manufacturers instructions. Lovastatin was added to cells to at final concentration of 40 µM unless otherwise indicated. Treatments were carried out for the times indicated in the figure legends. GGTI-298 was similarly added to cells at a final concentration of 20 or 10 µM as indicated and incubated for the times indicated in the figure legends. FTI-277 was used at a concentration of 20µM for 36 hours. Mg132 was added together with lovastatin for 18 hours at a final concentration of 10µM. Thymidine (Sigma) was added to cells at a final concentration of 2mM. Nocodazole (Sigma) was added to cells at a final concentration of 50ng/ml
Western blotting analysis
Cells were lysed in SDS buffer containing DTT, sonicated and incubated at 95 °C for 5 minutes. Proteins were loaded onto 13% SDS gels and resolved after which they were transferred onto PVDF membranes (Millipore) by western blotting. Bands were visualised by the ECL method (Fig. 1A, B, C, D, E; 2A, 3A-E, 4 and 5B, C) or using the Odyssey system (Licor) (Fig. 2B ,C and Fig.3F).
Generation of a Skp2 overexpressing HeLa cell line
The lentiviral Skp2 expression construct was cloned by recombining the human Skp2-cDNA (a generous gift of Wilhelm Krek) into the vector U509 (pHR-PGK-dest-bGH-polyA-SFFV-eGFP-rfa_verB generated by C. Ploner) using the Gateway® cloning technology (Life Technologies). Cells exhibiting stable Skp2 overexpression can be identified by their concomitant expression of GFP from the second promoter.
HeLa cells were transduced with lentiviral particles containing Skp2 according to the procedure previously described [52].
Generation of of pHR-PGK-bGH-pA-SFFV-eGFP (U509)
pHR-PGK-bGH-polyA-SFFV-eGFP was generated by multicloning strategy using pHR-SIN-CSGW (kindly provided by Mary Collins, London, UK) as starting vector. pHR-PGK-SFFV-eGFP was made by amplifying the PGK-promoter from pHR-frt-PGK-PURO-frt-SFFV-eGFP (kindly provided by S. Geley, Innsbruck, Austria) using the oligonucleotides 5-’tatagaattctaccgggtaggggaggc-3’ (sense) and 5’-TATAGAATTCCATATGGTTTAAACAC GCGTTACGTAGTCGAAAGGCCCGGAGATG-3’ (antisense). The EcoRI digested PCR product was subcloned into the EcoRI site of pHR-SIN-CSGW, thereby also inserting a multiple cloning site (SnaBI-MluI-PmeI-NdeI) downstream of the PGK-promoter. In a second step, the bovine growth hormone polyadenylation sequence (bGH-pA) was PCR amplified using the oligonucleotides 5’-tataacgcgtctagagctcgctgatcagcctc-3’ (sense) and 5’-TATACATATGGATCTCGAGCCCCAGCTGG-3’ (antisense) and pHR-frt-PGK-Puro-frt-SFFV-eGFP as template. The MluI/NdeI digested PCR product was inserted into the MluI/NdeI site of pHR-PGK-SFFV-eGFP generating pHR-PGK-bGH-polyA-SFFV-eGFP. Finally this vector was rendered GATEWAY-compatible by inserting a DEST – cassette (RfA, Invitrogen, Vienna, Austria) into the SnaBI site downstream of the PGK-promoter.
Plasmids and primers
The polymerase chain reaction was used to generate Skp2 with flanking AttB1 and AttB2. This PCR product was used in a gateway BP reaction with pDON207 GATEWAY® to generate a Skp2 entry clone. This was subsequently used in a LR reaction with delta T pDEST (no tag) GATEWAY® and deltaT-FLAG-DEST (NT flag tag) GATEWAY® [53] to generate vectors suitable for mammalian expression. Skp2 CT and NT domain mutants were generated as described above but using primer combinations in the initial step to amplify specific domains of Skp2. To generate Skp2 lacking the first 94 amino acids, a forward primer was used that commences at nucleotide 282 in combination with a reverse primer for the full length cDNA. In contrast to generate the CT delta mutant, the full length forward primer was used together with a reverse primer starting at nucleotide 465. To generate a variant of Skp2 lacking the F-box domain we used the approach described by Carrano et al. [16]. Thus a DNA fragment of pDON207 Skp2 corresponding to nucleotides 301-946 was removed by digesting with BSpE1 and Xba1. Then a PCR fragment containing nucleotides 420-946 and flanked by BSPe1 (5’) and Xba1 (3’) sites respectively was ligated into the cut vector generated a mutant lacking nucleotides 301-419, which correspond to the F-box region of the Skp2 protein.
To generate a Skp2 with a C-terminal eYFP fusion to PCR reactions were performed to amplify Skp2 with a 5’ attB1 site and a 3’ Spe1 restriction site and to amplify eYFP with a 5’ Spe1 site and a 3’ attB2. After Spe1 digestion of the resulting PCR products, a ligation was performed to generate Skp2 fused to eYFP with flanking attB1 and attB2 sites. This was used in a BP reaction and transferred into a Delta T – Flag pDEST for mammalian expression.
Northern blotting
Total RNA was isolated from HeLa cell pellets using the RNeasy Mini kit (Qiagen). The RNA was then subjected to electrophoretic separation using a 1% (wt/vol) agarose-formaldehyde (0.75% v/v) gel after which it was transferred to a positively charged Hybond membrane (GE healthcare). The membrane was UV irradiated in order to fix the RNA. 32P-labeled DNA probes for Skp2 and GAPDH were prepared using the Random primer DNA labeling system (Invitrogen) using restriction digest fragments of Skp2 and GAPDH cDNA. Hybridization was performed by incubating the membrane in ULTRAhyb® Ultrasensitive Hybridization Buffer (Ambion) containing the probe at 42 °C for 16 hours. After washing bands were visualized by autoradiography and quantification performed by phosphorimaging analysis.
Antibodies
Antibodies. The following antibodies were used in this study. Skp2 (Rabbit polyclonal H-435, Santa Cruz # sc-7164, 1:500), Skp2 (Mouse monoclonal A2, Santa Cruz #sc-74477 1:250), p27 (Mouse monoclonal, BD Transduction laboratoriesn#610242,1 in 250), p27 (rabbit polyclonal, C19 # sc-528, SantaCruz), p21 Mouse (Mouse monoclonal anti-human Cip1, BD Transduction laboratories, #610234), 1 in 250) p57 (C-20, Santa Cruz,sc-1040) p16 INK4a (Mouse monoclonal, G175-405, Pharmingen # 13251A, 1in 100), p15 Ink4b (Rabbit polyclonal, K-18, Santa Cruz, # sc-613) Cyclin E (mouse monoclonal HE12, Santa Cruz), Cyclin A (Polyclonal rabbit serum, produced in Hengst laboratory), PSTAIRE (Murine cell supernatant, Cdk4 (Rabbit Polyclonal, C-22, Santa Cruz, sc-260), Tubulin (mouse monoclonal, b-tubulin AA2 Sigma-Aldrich T8328), Cdh1/Fizzy related (homemade, rabbit 1 in 100), GAPDH mouse monoclonal (6C5, Calbiochem 1in 1000), Actin (I-19,Santa Cruz), GFP (Mouse Mixture of two monoclonal antibodies (7.1 and 13.1) Roche, # 11814460001, 1in 1000) Flag (mouse monoclonal, M2 Sigma Aldrich #F3165).
FACS
Cell cycle distribution was determined by using BrDU / propidium iodide double staining. Briefly, 20µM BrdU was added to cells approximately 1 hour before harvest. Cells were fixed in a 70% ethanol solution and after denaturation of DNA incubated with FITC labelled anti BrdU antibody and 0.5ug/ml propidium iodide. Cells were analysed using a BECKMANN COULTER EPICS XL MCL Flow Cytometer and cell cycle distribution determined using FlowJo cytometry data analysis software software.
Acknowledgements
We acknowledge Christian Ploner and Reinhard Kofler for generating and providing the plasmid U509 and Nisar P. Malek and James M. Roberts for the p27-/- MEFs. We would further like to acknowledge and Reinhard Sigl and Alexander Trockenbacher for providing reagents and all members of the Hengst lab for stimulating discussions. This work was funded by FWF grant P24031 to LH and the Graduate School for Molecular Cell Biology and Oncology (MCBO, FWF grant W1101).
Author contributions
JV designed the study, performed all of the experiments with the exception of Fig. 1B and supplementary Figure 1. He analyses the data, and wrote the manuscript. AM performed the experiment shown in Fig. 1B and supplementary Figure 1, analysed the data and provided intellectual input. MK generated reagents and provided intellectual input. SG provided CDH1 knockout and knockdown cell lines, CDH1 antibodies and intellectual input. CP generated the U509 vector subsequently used by MK to create a Skp2 overexpressing cell line. LH conceived and designed the study and wrote the manuscript.
Conflict of Interest
The authors declare that they have no conflict of interest.
References
1. Alberts AW, Chen J, Kuron G, Hunt V, Huff J, Hoffman C, Rothrock J, Lopez M, Joshua H, Harris E, Patchett A, Monaghan R, Currie S, Stapley E, Albers-Schonberg G, Hensens O, et al. Mevinolin: a highly potent competitive inhibitor of hydroxymethylglutaryl-coenzyme A reductase and a cholesterol-lowering agent. Proc Natl Acad Sci U S A. 1980; 77(7):3957-3961.
2. Keyomarsi K, Sandoval L, Band V and Pardee AB. Synchronization of tumor and normal cells from G1 to multiple cell cycles by lovastatin. Cancer Res. 1991; 51(13):3602-3609.
3. Dimitroulakos J, Ye LY, Benzaquen M, Moore MJ, Kamel-Reid S, Freedman MH, Yeger H and Penn LZ. Differential sensitivity of various pediatric cancers and squamous cell carcinomas to lovastatin-induced apoptosis: therapeutic implications. Clin Cancer Res. 2001; 7(1):158-167.
4. Gauthaman K, Manasi N and Bongso A. Statins inhibit the growth of variant human embryonic stem cells and cancer cells in vitro but not normal human embryonic stem cells. Br J Pharmacol. 2009; 157(6):962-973.
5. Kotamraju S, Williams CL and Kalyanaraman B. Statin-induced breast cancer cell death: role of inducible nitric oxide and arginase-dependent pathways. Cancer Res. 2007; 67(15):7386-7394.
6. Bjorkhem-Bergman L, Acimovic J, Torndal UB, Parini P and Eriksson LC. Lovastatin prevents carcinogenesis in a rat model for liver cancer. Effects of ubiquinone supplementation. Anticancer Res. 2010; 30(4):1105-1112.
7. Vainio P LL, Mirtti T, Hilvo M, Seppänen-Laakso T, Virtanen J, Sankila A, Nordling S, Lundin J, Rannikko A, Orešič M, Kallioniemi O and Iljin K. Phospholipase PLA2G7, associated with aggressive prostate cancer, promotes prostate cancer cell migration and invasion and is inhibited by statins. Oncotarget. 2011; 2(12):1176-1190.
8. Mira E C-RL, Tardáguila M, Azcoitia I, González-Martín A, Almonacid L, Casas J, Fabriás G and Mañes S. A lovastatin-elicited genetic program inhibits M2 macrophage polarization and enhances T cell infiltration into spontaneous mouse mammary tumors. Oncotarget. 2013; 4(12):2288-2301.
9. Demierre MF, Higgins PD, Gruber SB, Hawk E and Lippman SM. Statins and cancer prevention. Nat Rev Cancer. 2005; 5(12):930-942.
10. Osmak M. Statins and cancer: current and future prospects. Cancer Lett. 2012; 324(1):1-12.
11. Liao JK and Laufs U. Pleiotropic effects of statins. Annu Rev Pharmacol Toxicol. 2005; 45:89-118.
12. Hengst L, Dulic V, Slingerland JM, Lees E and Reed SI. A cell cycle-regulated inhibitor of cyclin-dependent kinases. Proc Natl Acad Sci U S A. 1994; 91(12):5291-5295.
13. Gray-Bablin J, Rao S and Keyomarsi K. Lovastatin induction of cyclin-dependent kinase inhibitors in human breast cells occurs in a cell cycle-independent fashion. Cancer Res. 1997; 57(4):604-609.
14. Danesh FR, Sadeghi MM, Amro N, Philips C, Zeng L, Lin S, Sahai A and Kanwar YS. 3-Hydroxy-3-methylglutaryl CoA reductase inhibitors prevent high glucose-induced proliferation of mesangial cells via modulation of Rho GTPase/ p21 signaling pathway: Implications for diabetic nephropathy. Proc Natl Acad Sci U S A. 2002; 99(12):8301-8305.
15. Hengst L and Reed SI. Translational control of p27Kip1 accumulation during the cell cycle. Science. 1996; 271(5257):1861-1864.
16. Carrano AC, Eytan E, Hershko A and Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol. 1999; 1(4):193-199.
17. Tsvetkov LM, Yeh KH, Lee SJ, Sun H and Zhang H. p27(Kip1) ubiquitination and degradation is regulated by the SCF(Skp2) complex through phosphorylated Thr187 in p27. Curr Biol. 1999; 9(12):661-664.
18. Sutterluty H, Chatelain E, Marti A, Wirbelauer C, Senften M, Muller U and Krek W. p45SKP2 promotes p27Kip1 degradation and induces S phase in quiescent cells. Nat Cell Biol. 1999; 1(4):207-214.
19. Kossatz U, Dietrich N, Zender L, Buer J, Manns MP and Malek NP. Skp2-dependent degradation of p27kip1 is essential for cell cycle progression. Genes Dev. 2004; 18(21):2602-2607.
20. Bornstein G, Bloom J, Sitry-Shevah D, Nakayama K, Pagano M and Hershko A. Role of the SCFSkp2 ubiquitin ligase in the degradation of p21Cip1 in S phase. J Biol Chem. 2003; 278(28):25752-25757.
21. Lu Z and Hunter T. Ubiquitylation and proteasomal degradation of the p21(Cip1), p27(Kip1) and p57(Kip2) CDK inhibitors. Cell Cycle. 2010; 9(12):2342-2352.
22. Rao S, Porter DC, Chen X, Herliczek T, Lowe M and Keyomarsi K. Lovastatin-mediated G1 arrest is through inhibition of the proteasome, independent of hydroxymethyl glutaryl-CoA reductase. Proc Natl Acad Sci U S A. 1999; 96(14):7797-7802.
23. Efuet ET and Keyomarsi K. Farnesyl and geranylgeranyl transferase inhibitors induce G1 arrest by targeting the proteasome. Cancer Res. 2006; 66(2):1040-1051.
24. Niknejad N, Morley M and Dimitroulakos J. Activation of the integrated stress response regulates lovastatin-induced apoptosis. J Biol Chem. 2007; 282(41):29748-29756.
25. Wojcik C, Bury M, Stoklosa T, Giermasz A, Feleszko W, Mlynarczuk I, Pleban E, Basak G, Omura S and Jakobisiak M. Lovastatin and simvastatin are modulators of the proteasome. Int J Biochem Cell Biol. 2000; 32(9):957-965.
26. Grimmler M, Wang Y, Mund T, Cilensek Z, Keidel EM, Waddell MB, Jakel H, Kullmann M, Kriwacki RW and Hengst L. Cdk-inhibitory activity and stability of p27Kip1 are directly regulated by oncogenic tyrosine kinases. Cell. 2007; 128(2):269-280.
27. Gstaiger M, Jordan R, Lim M, Catzavelos C, Mestan J, Slingerland J and Krek W. Skp2 is oncogenic and overexpressed in human cancers. Proc Natl Acad Sci U S A. 2001; 98(9):5043-5048.
28. Hershko DD. Oncogenic properties and prognostic implications of the ubiquitin ligase Skp2 in cancer. Cancer. 2008; 112(7):1415-1424.
29. Frescas D and Pagano M. Deregulated proteolysis by the F-box proteins SKP2 and beta-TrCP: tipping the scales of cancer. Nat Rev Cancer. 2008; 8(6):438-449.
30. Wirbelauer C, Sutterluty H, Blondel M, Gstaiger M, Peter M, Reymond F and Krek W. The F-box protein Skp2 is a ubiquitylation target of a Cul1-based core ubiquitin ligase complex: evidence for a role of Cul1 in the suppression of Skp2 expression in quiescent fibroblasts. EMBO J. 2000; 19(20):5362-5375.
31. Wei W, Ayad NG, Wan Y, Zhang GJ, Kirschner MW and Kaelin WG, Jr. Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature. 2004; 428(6979):194-198.
32. Bashir T, Dorrello NV, Amador V, Guardavaccaro D and Pagano M. Control of the SCF(Skp2-Cks1) ubiquitin ligase by the APC/C(Cdh1) ubiquitin ligase. Nature. 2004; 428(6979):190-193.
33. Kamura T, Hara T, Kotoshiba S, Yada M, Ishida N, Imaki H, Hatakeyama S, Nakayama K and Nakayama KI. Degradation of p57Kip2 mediated by SCFSkp2-dependent ubiquitylation. Proc Natl Acad Sci U S A. 2003; 100(18):10231-10236.
34. Zhang H, Kobayashi R, Galaktionov K and Beach D. p19Skp1 and p45Skp2 are essential elements of the cyclin A-CDK2 S phase kinase. Cell. 1995; 82(6):915-925.
35. Berndt N, Hamilton AD and Sebti SM. Targeting protein prenylation for cancer therapy. Nat Rev Cancer. 2011; 11(11):775-791.
36. Schulman BA, Carrano AC, Jeffrey PD, Bowen Z, Kinnucan ER, Finnin MS, Elledge SJ, Harper JW, Pagano M and Pavletich NP. Insights into SCF ubiquitin ligases from the structure of the Skp1-Skp2 complex. Nature. 2000; 408(6810):381-386.
37. Ecker K and Hengst L. Skp2: caught in the Akt. Nat Cell Biol. 2009; 11(4):377-379.
38. Bai C, Sen P, Hofmann K, Ma L, Goebl M, Harper JW and Elledge SJ. SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell. 1996; 86(2):263-274.
39. Ji P, Goldin L, Ren H, Sun D, Guardavaccaro D, Pagano M and Zhu L. Skp2 contains a novel cyclin A binding domain that directly protects cyclin A from inhibition by p27Kip1. J Biol Chem. 2006; 281(33):24058-24069.
40. Xu S, Abbasian M, Patel P, Jensen-Pergakes K, Lombardo CR, Cathers BE, Xie W, Mercurio F, Pagano M, Giegel D and Cox S. Substrate recognition and ubiquitination of SCFSkp2/Cks1 ubiquitin-protein isopeptide ligase. J Biol Chem. 2007; 282(21):15462-15470.
41. Rao S, Lowe M, Herliczek TW and Keyomarsi K. Lovastatin mediated G1 arrest in normal and tumor breast cells is through inhibition of CDK2 activity and redistribution of p21 and p27, independent of p53. Oncogene. 1998; 17(18):2393-2402.
42. Tang BK and Kalow W. Variable activation of lovastatin by hydrolytic enzymes in human plasma and liver. 4. Eur J Clin Pharmacol. 1995; 47(5):449-451.
43. Sjogren AK, Andersson KM, Liu M, Cutts BA, Karlsson C, Wahlstrom AM, Dalin M, Weinbaum C, Casey PJ, Tarkowski A, Swolin B, Young SG and Bergo MO. GGTase-I deficiency reduces tumor formation and improves survival in mice with K-RAS-induced lung cancer. J Clin Invest. 2007; 117(5):1294-1304.
44. Hirai A, Nakamura S, Noguchi Y, Yasuda T, Kitagawa M, Tatsuno I, Oeda T, Tahara K, Terano T, Narumiya S, Kohn LD and Saito Y. Geranylgeranylated rho small GTPase(s) are essential for the degradation of p27Kip1 and facilitate the progression from G1 to S phase in growth-stimulated rat FRTL-5 cells. J Biol Chem. 1997; 272(1):13-16.
45. Laufs U, Marra D, Node K and Liao JK. 3-Hydroxy-3-methylglutaryl-CoA reductase inhibitors attenuate vascular smooth muscle proliferation by preventing rho GTPase-induced down-regulation of p27(Kip1). J Biol Chem. 1999; 274(31):21926-21931.
46. Hu W, Bellone CJ and Baldassare JJ. RhoA stimulates p27(Kip) degradation through its regulation of cyclin E/CDK2 activity. J Biol Chem. 1999; 274(6):3396-3401.
47. Kazi A, Carie A, Blaskovich MA, Bucher C, Thai V, Moulder S, Peng H, Carrico D, Pusateri E, Pledger WJ, Berndt N, Hamilton A and Sebti SM. Blockade of protein geranylgeranylation inhibits Cdk2-dependent p27Kip1 phosphorylation on Thr187 and accumulates p27Kip1 in the nucleus: implications for breast cancer therapy. Mol Cell Biol. 2009; 29(8):2254-2263.
48. Bond M, Wu YJ, Sala-Newby GB and Newby AC. Rho GTPase, Rac1, regulates Skp2 levels, vascular smooth muscle cell proliferation, and intima formation in vitro and in vivo. Cardiovasc Res. 2008; 80(2):290-298.
49. Aktories K, Wilde C and Vogelsgesang M. Rho-modifying C3-like ADP-ribosyltransferases. Rev Physiol Biochem Pharmacol. 2004; 152:1-22.
50. Fukushima H, Ogura K, Wan L, Lu Y, Li V, Gao D, Liu P, Lau AW, Wu T, Kirschner MW, Inuzuka H and Wei W. SCF-mediated Cdh1 degradation defines a negative feedback system that coordinates cell-cycle progression. Cell Rep. 2013; 4(4):803-816.
51. Sigl R, Wandke C, Rauch V, Kirk J, Hunt T and Geley S. Loss of the mammalian APC/C activator FZR1 shortens G1 and lengthens S phase but has little effect on exit from mitosis. J Cell Sci. 2009; 122(Pt 22):4208-4217.
52. Kullmann MK, Grubbauer C, Goetsch K, Jakel H, Podmirseg SR, Trockenbacher A, Ploner C, Cato AC, Weiss C, Kofler R and Hengst L. The p27-Skp2 axis mediates glucocorticoid-induced cell cycle arrest in T-lymphoma cells. Cell Cycle. 2013; 12(16):2625-2635.
53. Barisic M, Sohm B, Mikolcevic P, Wandke C, Rauch V, Ringer T, Hess M, Bonn G and Geley S. Spindly/CCDC99 is required for efficient chromosome congression and mitotic checkpoint regulation. Mol Biol Cell. 2010; 21(12):1968-1981.