
INTRODUCTION
Myeloid cells as pivotal effectors of innate immune reaction are important regulators of adaptive immunity [1]. It has been known that the differentiation and activation of myeloid cells involve quantitative regulation of essential transcription factors, including PU.1, interferon regulatory transcription factor (IRF) family, CCAAT-enhancer-binding proteins (C/EBPs) and runt-related transcription factor 1 (RUNX1) [2–3]. Dysregulation of these key transcription factors induce and affect blood cancer development as well as innate immune response [4]. Therefore, disorder of myeloid differentiation is a typical feature of acute myeloid leukemia (AML) [5]. Monocytes involved in myeloid lineage commitment are derived from pluripotent hematopoietic stem cells, and can differentiate into macrophages or dendritic cells depending on external stimuli [6–7]. Monocytes identify ongoing viral or bacterial infections, and then infiltrate the infected tissue where they differentiate into the appropriate effector cell type [8].
E26 transformation-specific (Ets) family which is a large group of transcription factors was transduced for the first time by leukemia virus, E26 [9]. PU.1 is a member of the ETS-family of transcription factors, has ETS domain which can recognize and interact ‘GGAA’ and ‘GGAT’ DNA motif, respectively [10]. The cellular function of PU.1 as a transcription factor is mainly involved in myeloid and B cell lineage development [11]. Therefore, PU.1 expression is tightly regulated in multiple hematopoietic lineages [12]. Disruption of PU.1 in mice led to lack macrophages, neutrophils, and B and T cells [13–14]. PU.1 also plays a key role as tumor suppressor for B cell malignancies [15] and classical Hodgkin lymphoma cells [16]. Indeed, an excess of PU.1 expression was found to block differentiation during development of myeloid and erythroid lineages by interacting with the transcription factors GATA-1 and GATA-2 [17–18].
IL-32 was characterized as a proinflammatory cytokine because it was expressed in the lesions of patients with rheumatoid arthritis [19]. Six isoforms of IL-32, generated by alternative splicing of the IL-32 mRNA, were previously shown to exist. Recently, however, three additional isoforms, IL-32η, IL-32θ, and IL-32s, were characterized [20]. IL-32γ, which can be spliced into IL-32β by post-translational modification, is the most highly expressed of the isoforms, as observed in inflammatory diseases and cancers [21]. As a result, IL-32β, rather than other isoforms, is commonly detected at higher levels in various immortalized cell lines [22–23]. IL-32α and IL-32γ can induce expression of other proinflammatory cytokines, including IL-6, IL-8 [24–26]. Although IL-32 is a crucial component of the immune response, previous studies on the functions of IL-32 have primarily concentrated on four of the isoforms: IL-32α, IL-32β, IL-32δ, and IL-32γ [27–28]. IL-32θ is an isoform that was recently identified in lipopolysaccharide (LPS)-differentiated dendritic cells, purified from the human periphery [20]. As a result, the function of this isoform has yet to be characterized.
Recent studies indicated that IL-32 modulates the differentiation of monocytic cells and regulates the production of inflammatory cytokines. Here, we demonstrate that the newly discovered isoform, IL-32θ, suppresses monocyte differentiation by regulating the expression of the PU.1 transcription factor.
RESULTS
Elucidation of the IL-32θ and IL-32β coding sequences
IL-32β is abundantly expressed in various tissues. It is also highly expressed in cancerous tissues and regions where inflammation is present. Conversely, expression of IL-32θ has been detected in dendritic cells derived from human peripheral blood monocytic cells (PBMCs). As a result, the functions of this isotype are thought to be limited. Because monocytes express IL-32β endogenously, we chose monocytes to compare the functions of IL-32θ to IL-32β in a monocytic line. We first analyzed the coding sequences of each isoform and determined that the IL-32θ mRNA lacks exon 6 (GenBank, accession number FJ985780), which is present in that of IL-32β (Figure 1A). The differences of both IL-32 isoforms are distinguished from alternative mRNA splicing after transcription of IL-32 mRNA [21]. The IL-32 coding sequences were then cloned into mammalian expression vectors and used to establish THP-1 myelomonocytic cell lines stably expressing the IL-32 isoforms. THP-1 cells were chosen because they have been used previously in a monocyte-to-macrophage differentiation model [33–35]. The resulting strains THP-1/IL-32θ and THP-1/IL-32β were subjected to RT-PCR and immunoblot analyses to assess IL-32 production in the presence or absence of phorbol 12-myristate 13-acetate (PMA) treatment, which has been used to induce monocyte differentiation into macrophage-like cells. Interestingly, the expression levels of both IL-32 isoforms were dramatically increased in the populations treated with 30 nM of PMA for 3 days compared to the untreated populations (Figure 1B and 1C).
PMA-induced differentiation of THP-1 monocytes into macrophages is regulated by IL-32θ
Upon injury, undifferentiated monocytes, which are generally localized within the blood, infiltrate the wound site and differentiate into macrophages or dendritic cells [36]. As a result, the specific morphological changes that occur during differentiation of a monocyte into macrophage-like cell, are crucial to the acute innate immune response. We previously reported that the IL-32α isoform inhibited both PMA-induced morphological changes and CD18/PU.1 expression in human monocytes [31]. This finding was evidenced on the interrelationship between IL-32 and monocytic differentiation because PU.1 plays a crucial role in myeloid lineage development [37]. Thus, we predicted that other isoforms may influence the differentiation of monocytes. To address this possibility, differentiation was first assessed by examining the morphology of THP-1/wt, THP-1/IL-32θ, and THP-1/IL-32β cells and quantifying adherence of differentiated cells to culture plates after stimulation with 30 nM PMA. From these analyses, we determined that the level of differentiation in the THP-1/IL-32θ population was less than 50% of that observed in the THP-1/wt and THP-1/IL-32β cells after PMA stimulation (Figure 2A and 2B). In addition, we examined the ability of these cell lines to adhere to vascular endothelium, using cultured HUVEC endothelial cells. Similar to results observed in the culture dishes, adhesion of THP-1/IL-32θ cells to HUVEC cells was significantly reduced compared to the adhesion of THP-1/wt and THP-1/IL-32β cells (Figure 2C). To then assess the relevance of IL-32θ expression in another cell line, we transiently transfected HL-60 mononuclear cells with the IL-32θ expression vector and observed any morphological changes that occurred upon PMA treatment. Compared to the wild type control, morphological changes were partially impaired in the HL-60/IL-32θ cell population (Figure 2D). Furthermore, the amount of PMA treated HL-60/IL-32θ cells that adhered to a culture dish was significantly lesser than the amount of adherent wild type cells (Figure 2E). These findings indicate that increased levels of intracellular IL-32θ, but not IL-32β, attenuate the differentiation of monocytes into macrophage after PMA stimulation.
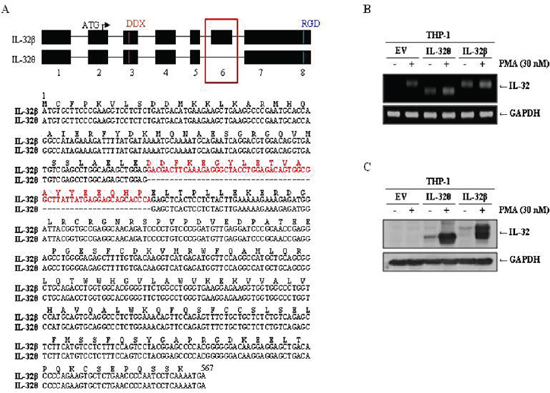
Figure 1: Comparison of the coding sequences and expression levels of IL-32θ and IL-32β. (A) The schematic diagram of the coding sequences of IL-32θ and IL-32β. The difference of exon 6 between IL-32β and IL-32θ was indicated with red box and presented with amino acid and mRNA sequence. We established THP-1 cell lines expressing IL-32β or IL-32θ. Cells were treated with 30 nM PMA for 72 h and expression levels were measured by RT-PCR using IL-32-specific primers (B) and by Western blot using an IL-32-specific antibody (C). GAPDH was used as a control in both experiments, and IL-32 expression was normalized to GAPDH expression.
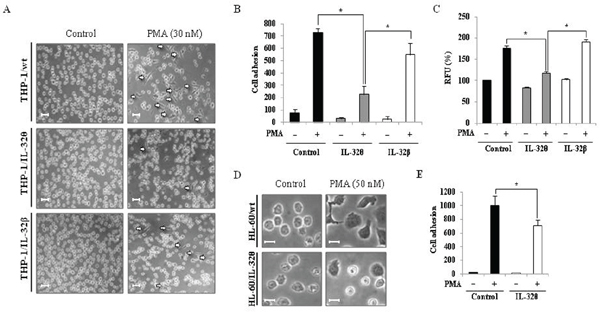
Figure 2: Effects of IL-32θ and IL-32β expression on cell morphology and adhesion capacity in THP-1 cell lines. THP-1/IL-32θ and THP-1/IL-32β cells were stimulated with 30 nM PMA for 72 h and (A) morphological changes were assessed by phase-contrast microscopy (100×) and (B) by quantifying cell adherence to culture dishes. Scale bars represent 20 μm. (C) Under identical conditions, cell adhesion capacity was measured using the leukocyte-endothelium adhesion assay kit and HUVEC endothelial cells. Treatment with 50 nM of PMA for 72 h was optimal for inducing differentiation of HL-60 cells. HL-60 cells were transfected with 1 μg pcDNA3.1+ empty vector or pcDNA 3.1+-6 × Myc-IL-32θ and incubated overnight. After PMA treatment for 72 h, HL-60 cell morphologies were examined by phase-contrast microscopy (200×) (D) and cell adherence to culture dishes was quantified (E). Scale bars represent 10 μm. Data are presented as mean ± standard error of mean (n = 3). *p < 0.05. THP-1/IL-32θ cells versus THP-1/wt or THP-1/IL-32β cells, after PMA treatment.
IL-32θ attenuates expression of monocyte/macrophage differentiation markers
To further investigate whether expression of IL-32θ inhibits differentiation of monocytes into macrophages, we measured the expression of the macrophage-specific differentiation markers CD11b, CD18, and CD36 by qRT-PCR analysis. Expression of the macrophage-1 antigen (Mac-1, CD11b/CD18) was dramatically reduced in THP-1/IL-32θ and HL-60/IL-32θ cells compared to wild type cells (Figure 3A, 3B, 3D and 3E). These expression patterns were similar to those reported for IL-32α [31]. In addition, CD36, which is highly expressed during differentiation into macrophages, was suppressed in the IL-32θ cell lines (Figure 3C and 3F). The expression patterns of the differentiation markers were erratic in the THP-1/IL-32β cells. To confirm the expression levels of the cell surface markers and detect differentiation into macrophages, we performed FACS analysis using marker-specific primary antibodies and FITC-conjugated secondary antibodies. The numbers of CD18 and CD36 positive cells (gated in M2) were greater in the PMA-treated THP-1/wt population than in the non-treated cells. However, expression of these markers was nearly identical in the treated and untreated THP-1/IL-32θ populations (Figure 3G and 3H). These findings suggest that intracellular IL-32θ inhibits expression of macrophage specific markers during PMA-induced monocyte differentiation into macrophages. Conversely, IL-32β appears to be irrelevant to this process.
Ectopic expression of IL-32θ decreases PMA-induced monocyte differentiation
To confirm whether IL-32θ inhibits monocytic differentiation, wild type THP-1 cells were transfected with an IL-32θ expressing vector. Expression of IL-32θ by the transfected cells was confirmed by RT-PCR (Figure 4A). Transfected THP-1 cells were stimulated with 30 nM of PMA and morphological changes were observed. The number of differentiated cells was reduced by IL-32θ expression in a transfection dose-dependent manner (Figure 4B). To further assess the effect of intracellular IL-32θ on the expression of macrophage-specific markers, the mRNA levels of CD11b, CD18, and CD36 were measured in cells transiently expressing IL-32θ. Consistent with results obtained from the stably expressing cell lines, the expression levels of all three macrophage-specific markers were decreased in the cells transfected with the IL-32θ construct compared to wild type (Figure 4C–4E). These data supported the conclusion that inhibition of monocytic differentiation was due to the intracellular IL-32θ expression.
Expression of IL-32θ or IL-32β does not affect PMA-induced cell cycle arrest in G0/G1 phase
PMA-induced differentiation of monocytes into macrophage is accompanied by cell cycle arrest [38]. Therefore, we quantified the number of viable THP-1/wt, THP-1/IL-32θ, and THP-1/IL-32β cells after PMA treatment and compared them to untreated populations. In the wild type and IL-32β-expressing cells, PMA treatment resulted in a significant reduction in the number of viable cells after 24 h, compared to the untreated control group. Interestingly, similar results were obtained in the IL-32θ expressing cells (Figure 5A), even though our results demonstrated that monocytic differentiation was not induced in this population (Figure 2). These results indicate that cell proliferation was halted in each of the three cell types after PMA treatment. To determine the phase of the cell cycle at which proliferation was interrupted, treated cells were stained with propidium iodide (PI), and populations were quantified by FACs analysis. As depicted in Figure 5B and 5C, the populations from each cell line were concentrated in the G0/G1 phase. Furthermore, immunoblot analyses detected decreased levels of the cell cycle regulatory proteins, cyclin D and E, and increased expression of p27, which regulates cell cycle arrest associated with cyclin E, in each of the PMA-treaded populations, in comparison to the untreated control groups (Figure 5D). These findings indicate that while IL-32θ and IL-32β were not the cause of the cell cycle arrest after PMA treatment, IL-32θ may regulate cell proliferation regardless of PMA treatment.
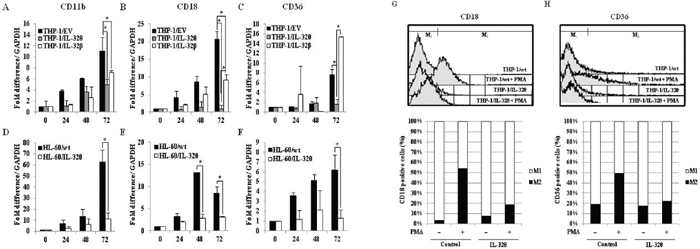
Figure 3: Expression of macrophage-specific cell surface markers in IL-32θ- and IL-32β-expressing cells after PMA stimulation. THP-1/wt, THP-1/IL-32θ, and IL-32β cell lines were treated with 30 nM PMA for 72 h and CD11b (A) CD18 (B) and CD36 (C) mRNA expression levels were measured by qRT-PCR. HL-60 cells were transfected with 1 μg IL-32θ expression vector and incubated overnight. Cells were then treated with 50 nM PMA for 72 h and CD11b (D), CD18 (E), and CD36 (F) mRNA expression levels were measured by qRT-PCR. Under identical conditions, THP-1/wt and THP-1/IL-32θ cells were harvested and fixed with 100% acetone. Cells were then labeled with marker-specific primary antibodies and FITC-conjugated secondary antibodies. The expression levels of CD18 (G) and CD36 (H) were measured by FACs analysis. Data are presented as mean ± standard error of mean (n = 3). *p < 0.05. THP-1/IL-32θ cells versus THP-1/wt or THP-1/IL-32β cells, after PMA treatment.
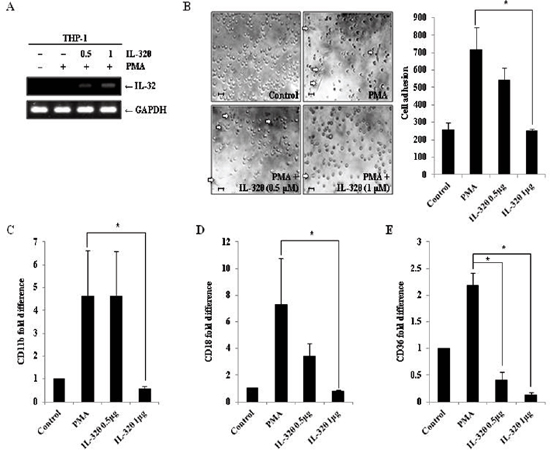
Figure 4: Ectopic expression of IL-32θ inhibits differentiation of THP-1 cells. (A) THP-1 cells were transfected with empty pcDNA3.1+ vector or the indicated amount of pcDNA3.1+-IL-32θ-6 × Myc and incubated overnight. Cells were then treated with 30 nM PMA for 72 h and RT-PCR was used to confirm IL-32 expression in the transfected cells. (B) The morphologies of cells ectopically expressing IL-32θ were assessed by phase-contrast microscopy (100×). Scale bars represent 10 μm. After transfection and PMA stimulation, undifferentiated cells (non-adhering cells) were washed out of the plates with PBS and adherent, differentiated cells were fixed, stained, and quantified. To measure expression of the macrophage-specific markers CD11b (C), CD18 (D), and CD36 (E), cells were treated and harvested, as previously described, and qRT-PCR was performed using marker-specific primers. Data are presented as mean ± standard error of mean (n = 3). *p < 0.05. THP-1 cells versus THP-1 cells with ectopic expression of IL-32θ, after PMA treatment.
IL-32θ attenuates expression of PU.1, a transcription factor necessary for differentiation of monocytes into macrophages
The regulatory role of PU.1 during myeloid lineage development has been widely studied [39]. Using qRT-PCR and immunoblot approaches, we found that expression of PU.1 was suppressed in THP-1/IL-32θ cells compared to the THP-1/wt and THP-1/IL-32β cells after PMA treatment (Figure 6A and 6B). To assess whether PU.1 could rescue the observed monocyte differentiation defect, PU.1 was transiently expressed in THP-1/IL-32θ cells, in which endogenous PU.1 expression is inhibited by IL-32θ. For these experiments, the dose-dependent effects of PU.1 were also assessed by transfecting THP-1/IL-32θ cells with 0.1, 0.5, or 1 μg of pcDNA-3.1+-PU.1-HA (Figure 6C). Interestingly, the morphology and adhesion levels of cells transfected with 1 μg of the PU.1 vector were similar to those observed in the wild type macrophage-like population (Figure 6D). Furthermore, the expression levels of the macrophage-specific markers, CD11b, CD18, CD36, were increased in cells transiently expressing PU.1, in a dose-dependent manner (Figure 6E, 6G, 6F). These findings indicate that PU.1 is a key regulatory component that can restore differentiation in THP-1 by inhibiting the effects of IL-32θ.
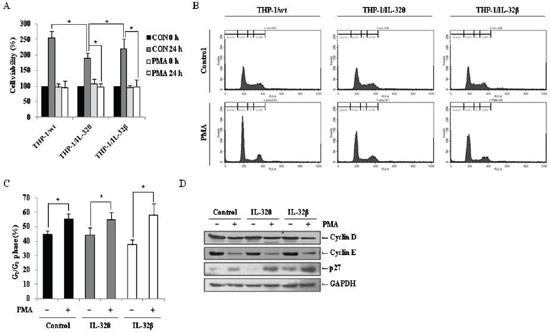
Figure 5: Effects of IL-32θ and IL-32β on PMA-mediated cell cycle arrest in THP-1 cells. (A) THP-1/Wt, THP-1/IL-32θ, and THP-1/IL-32β cells were synchronized by starvation overnight, and then seeded onto 96-well plates and treated with 30 nM PMA in complete medium for 72 h. Cell proliferation was measured using an MTS assay. (B) To determine the cell cycle phase at which proliferation of IL-32θ and IL-32β cells was halted, cells were stained with propidium iodide (PI) containing RNase A, and the population of each cell cycle phase was measured by flow cytometry. (C) The G0/G1 cell cycle phase distribution is summarized in the bar graphs. Y-axes are the proportion of counted events in an indicated phase. Data are presented as mean ± standard error of mean (n = 3). *p < 0.05. Untreated cells versus 30 nM PMA treated cells in each cell lines. (D) Expression levels of the G0/G1 phase-related factors cyclin D, cyclin E, and p27 were assessed by Western blot analysis, using specific antibodies. GAPDH was used as a loading control.
C/EBPα expression leads to additive effect with PU.1 on the restoration of monocytic differentiation
C/EBPα is a member of the Ets transcription factor family and is a key regulator during myeloid lineage development [40]. Although IL-32θ had no effect on the expression C/EBPα (data not shown), we predicted that increased expression of C/EBPα could support PU.1-mediated cell differentiation. To address this possibility, THP-1/IL-32θ cells were transfected with 1 μg of the C/EBPα and/or PU.1-expressing vectors, and transfection efficiency was confirmed by RT-PCR and Western blot analyses (Figure 7A). Morphological changes were monitored and differentiated cells were again quantified by enumerating adherent cells in culture dishes. The number of differentiated cells was significantly increased by co-transfection with PU.1 and C/EBPα compared to transfection with PU.1 or C/EBPα alone (Figure 7B). The expression levels of CD11b, CD18, and CD36 were also significantly increased in cells co-transfected with PU.1 and C/EBPα (Figure 7C–7E). Meanwhile, transfection with C/EBPα alone, resulted in a significant increase in CD18 expression levels. These findings indicate that overexpression of both PU.1 and C/EBPα resulted in a synergistic effect in the restoration of monocytic differentiation in IL-32θ-expressing cells.
DISCUSSION
Myeloid cell differentiation is tightly controlled by regulating cytokines and transcription factors because blockade of myeloid lineage development causes blood cancer and immune diseases [41]. Granulocyte-macrophage colony-stimulating factor (GM-CSF) and several interleukins are linked closely to regulate lineage commitment [42–44]. However, most studies on IL-32 have focused on the proinflammatory functions during the innate immune response. Recently, however, the role of IL-32 in apoptosis and metastasis has been investigated. In addition, IL-32 expression has been identified as a marker of gastric cancer [45–48]. Since its initial discovery, studies on relationship between IL-32 expression and cell differentiation are quite rare, but have been steadily published. IL-32 induces the differentiation of monocytes into macrophages through induction of thymic stromal lymphopoietin [49]. Furthermore, it has been reported that IL-32 is a potent regulator of osteoclastogenesis in vitro and exhibits a synergistic effect with IL-17 on differentiation of osteoclasts [50–51]. In dendritic cells, it was also known that maturation is mediated by IL-32γ-induced expression of IL-12 and IL-6 [24]. Although IL-32 was shown to enhance cellular differentiation of monocytes into macrophages, intracellular expression of IL-32α repressed differentiation of THP-1 cells by inhibiting PU.1 expression in a STAT3-dependent manner [31]. The roles of other IL-32 isoforms in cell differentiation are unclear, and it seems that the role in myeloid differentiation depends on each isoforms of IL-32. We previously reported that IL-32θ expression attenuates phosphorylation of PU.1, resulting in a reduction of IL-1β production [29]. PU.1, a member of the Ets family of transcription factors, regulates expression of macrophage-specific genes, including CD11b [52], CD18 [53], and the glycoprotein pDP4 [54], and is essential for monocyte differentiation [55–57]. We, therefore, could expect that IL-32θ may decrease PU.1 expression because PU.1 performs auto-regulatory functions, via binding to a distal enhancer on the PU.1 promoter region [58]. Similar to PU.1, C/EBPα is a transcription factor that contributes to the differentiation of monocytes into macrophage [59]. C/EBPα also binds to the distal enhancer of PU.1, thereby inducing PU.1 expression [60]. Additionally, C/EBPα forms heterodimers with AP-1, which induces PU.1 promoter activity more potently than C/EBPα homodimers or AP-1 alone [61].
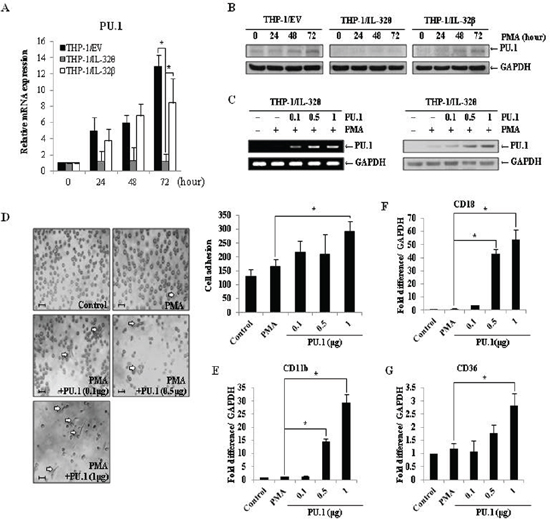
Figure 6: Expression of PU.1 rescues the IL-32θ-induced differentiation defect. (A) THP-1/wt, THP-1/IL-32θ and THP-1/IL-32β cells were treated with 30 nM PMA for 72 h. Expression levels of PU.1 were measured by qRT-PCR, using PU.1-specific primers. Data are presented as mean ± standard error of mean (n = 3). *p < 0.05. THP-1/IL-32θ cells versus THP-1/wt or THP-1/IL-32β cells after PMA treatment. (B) PU.1 expression was confirmed by Western blot. Cells were treated and harvested as described above, and protein was detected using an α-PU.1 antibody. Detection of GAPDH was used as an internal control. To assess PU.1-mediated restoration of monocyte differentiation, THP-1/IL-32θ cells were transfected with the indicated amount of empty pcDNA3.1+ vector or the indicated amount of pCDNA 3.1+-PU.1-HA. (C) Cells were treated with 30 nM PMA for 72 h, and the expression levels of PU.1 were confirmed by RT-PCR and Western blot analyses. (D) The morphology of transfected cells was observed by phase-contrast microscopy (200×). Scale bars represent 20 μm. Undifferentiated cells were washed out twice with PBS and adherent cells were stained with Diff-Quick solution. Adhesion levels were evaluated by quantifying the number of stained cells from three randomly selected fields. To measure expression levels of CD11b (E), CD18 (F), and CD36 (G), cells were treated and harvested as described above, and qRT-PCR was used for analysis. Data are presented as mean ± standard error of mean (n = 3). *p < 0.05. THP-1/IL-32θ cells versus cells transfected with indicated dose of PU.1, after PMA treatment.
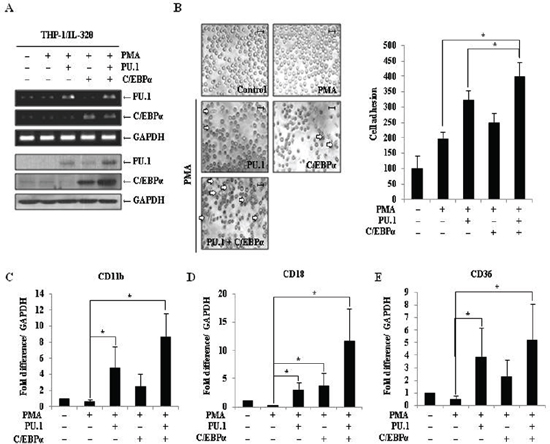
Figure 7: Effect of PU.1 and/or C/EBPα expression on monocyte differentiation in THP-1/IL-32θ cells. (A) THP-1/IL-32θ cells were transfected with 1 μg of pcDNA3.1+ empty vector, pcDNA3.1+-PU.1 and/or C/EBPα expression vectors and incubated overnight. Cells were stimulated with 30 nM PMA for 72 h, and transfection efficiency was confirmed by RT-PCR and Western blot analyses. (B) Morphological changes were assessed by phase-contrast microscopy (100 ×). Scale bars represent 20 μm. After washing out non-adherent (undifferentiated) cells with PBS, the adherent cells were stained with Diff-Quick solution. At least 150 stained cells were counted in three randomly selected fields. To assess CD11b (C), CD18 (D), and CD36 (E) expression levels, cells were prepared as described above, and mRNA levels were measured by qRT-PCR. Data are presented as mean ± standard error of mean (n = 3). *p < 0.05. THP-1/IL-32θ cells versus cells transfected with PU.1 and/or C/EBPα, after PMA treatment.
We, therefore, designed a model for the comparative analysis of monocytic differentiations between IL-32β and IL-32θ in THP-1 cell lines, and predicted that IL-32θ would be involved in regulating monocyte differentiation and that PU.1 and C/EBPα are key components in this regulatory process. In the present study, expression of IL-32θ was found to negatively regulate PMA-mediated differentiation of a monocyte cell line into macrophages compared with IL-32β. IL-32θ expression suppressed morphological changes and the adhesion capability of THP-1 cells to culture plates and to vascular endothelial cells. IL-32θ also inhibited expression of the macrophage markers, CD11b, CD18, and CD36, in THP-1/IL-32θ cells, even after PMA stimulation. Additionally, PU.1 expression levels were decreased in THP-1/IL-32θ cells compared to the wild type and THP-1/IL-32β population. In THP-1/IL-32θ cells, however, overexpression of PU.1 and/or C/EBPα rescued the observed differentiation defect after PMA treatment. Together, these data indicate that IL-32θ is a potent inhibitor of monocytic differentiation and that this inhibition occurs due to a reduction in PU.1 expression. There have been steady attempts to treat myeloid leukemia, called ‘differentiation therapy’ [62–63]. The IL-32θ may be a potent therapeutic target for myeloid differentiation-mediated diseases. In further studies, it will be crucial to examine the effects of IL-32θ expression in IL-32θ transgenic mice or in primary myeloid cells from leukemia patients.
MATERIALS AND METHODS
Cell culture and the generation of a cell line stably expressing IL-32β and IL-32θ
The human monocytic cell lines THP-1 and HL-60 were cultured in RPMI 1640 (HyClone, Logan, UT) medium, supplemented with 10% heat inactivated fetal bovine serum, 2 mM l-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin (HyClone, Logan, UT). In order to induce differentiation, cells were treated with phorbol 12-myristate 13-acetate (PMA) (St. Louis, MO). The cell line stably expressing IL-32θ and the mock control cell line were previously established and described [29]. To establish constitutive expression of IL-32β, THP-1 cells were transfected with the pcDNA3.1+6xMyc-IL-32β vector, using the Neon™ transfection system (Invitrogen, Carlsbad, CA). Cells were incubated with G-418 (700 μg/mL) and resistant cells were screened for 3 weeks, and expanded clones were acquired by serial dilution.
Cell morphology and cell adhesion assays
THP-1 cells stably expressing IL-32β and HL-60 cells transfected with IL-32θ were adhered to the bottom of culture wells by treatment with 30 nM and 50 nM of PMA, respectively, for an indicated time, and morphological changes were assessed by phase contrast microscopy at 100× and 200×. Cells were visualized using the Reastain Quick-Diff kit (Reagena, Toivala, Finland). The counting method used was described previously [30]. Briefly, after fixation and staining, a minimum of 150 cells/field were counted in three or more randomly selected fields. To assess the adhesion capability of the cell lines to vascular endothelium, we followed the manufacturer’s instructions for the CytoSelect™ Leukocyte-Endothelium Adhesion Assay kit (Cell Biolabs, San Diego, CA, USA), by using HUVEC endothelial cells.
Construction of expression vectors
We previously identified the IL-32θ isoform in human dendritic cells differentiated by treatment with lipopolysaccharide [20]. To create IL-32θ and PU.1 expression vectors, 6x-myc and HA tags were first inserted into the mammalian expression vector pcDNA 3.1+, generating pcDNA 3.1+/6 × Myc and pcDNA 3.1+/HA. The sequences of IL-32θ and PU.1 were amplified by RT-PCR, digested using EcoRI and XhoI restriction enzymes, and ligated into their respective vectors, generating pcDNA 3.1+/6 × Myc-IL-32θ and pcDNA 3.1+/HA-PU.1 [29, 31]. The C/EBPα-encoding sequence was subcloned into the pcDNA 3.1+-5 × Flag vector as previously reported [26].
MTS assay
To examine the effects of IL-32θ and IL-32β expression on cell proliferation, cell viability was evaluated in THP-1 cells by using the CellTiter 96® AQueous One Solution Assay (Promega, Madison, WI, USA). Cells were seeded in 96-well plates, treated with 30 nM of PMA, and incubated for 72 h. Untreated cells were used as controls. AQueous One solution, containing 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) and phenazine methosulfate (PMS), an electron coupling reagent, was diluted 1:5 in free medium, and 100-μL aliquots of the reagent were added to each well. After 30 min of incubation, the absorbance at 492 nm was measured using an Apollo LB 9110 microplate reader (Berthold Technologies GmbH, Bad Wildbad, Germany).
Propidium iodide staining
Approximately 1.5 × 105 cells/well were plated in 6-well plates and treated with 30 nM of PMA for 72 h. Cells were then washed twice with PBS and fixed by incubation with 70% ethanol at −20°C. The fixed cells were washed with PBS prior to staining with PBS containing 50 μg/mL PI and 100 μg/mL RNase A, for 30 min in the absence of light. The percentage of PI-stained cells in each cell cycle phase was determined using a FACSCalibur flow cytometer and analyzed with CellQuest Pro software (BD Biosciences, San Jose, CA, USA).
Quantitative real-time polymerase chain reaction (qRT-PCR) and reverse transcription polymerase chain reaction (RT-PCR) analyses
After PMA treatment, total RNA was extracted from each cell line by using the RNA-BLUE™ total RNA extraction kit (iNtRon Biotechnology, Seoul, Korea), according to the manufacturer’s protocol. The cDNA products were prepared using M-MuLV reverse transcriptase (New England Biolabs, Beverly, MA, USA). The mRNA expression levels of the macrophage-specific cell surface markers CD11b, CD18, and CD36 and the transcription factor PU.1 were detected by qRT-PCR, using a relative quantification protocol in a Chromo 4 Real-Time PCR system (Bio-Rad, Hercules, CA, USA) with the SensiFAST™ SYBR® No-ROX Kit (Bioline, Taunton, MA, USA). The CD11b, CD18, and CD36 primers sequences were as follows: CD11b, 5′-TTC CAA GAG AAC GCA AGG GG-3′ (sense) and 5′-TAG TCG CAC TGG TAG AGG CT-3′ (anti-sense); CD18, 5′-TGC TGA TCG GCA TTC TCC TGC TGG TCA TCT-3′ (sense) and 5′-CAC TGG GAC TTG AGC TTC TCC TTC TCA AAG-3′ (anti-sense); CD36, 5′-CTG GCT GTG TTT GGA GGT AT-3′ (sense) and 5′-TCT GTG CCT GTT TTA ACCCA-3′ (anti-sense); PU.1, 5′-CTG GCT GTG TTT GGA GGT AT-3′ (sense) and 5′-TCT GTG CCT GTT TTA ACC CA-3′ (anti-sense). For confirmation of transient transfection, RT-PCR was performed using IL-32-, PU.1-, and C/EBPα-specific primers. The primer sequences were as follows: IL-32, 5′-CTG GCT GTG TTT GGA GGT AT-3′ (sense) and 5′-TCT GTG CCT GTT TTA ACC CA-3′ (anti-sense); PU.1, 5′-ATG TTA CAG GCG TGC AAA ATG-3′ (sense) and 5′-TGC TTG GAC GAG AAC TGG AA-3′ (anti-sense); C/EBPα, 5′-ACG AGA CGT CCA TCG ACA TC-3′ (sense) and 5′-CAG TGC GCG ATC TGG AAC TG-3′ (anti-sense); GAPDH, 5′-GGC TGC TTT TAA CTC TGG TA-3′ (sense) and 5′-TGG AAG ATG GTG ATG GGA TT-3′ (anti-sense). GAPDH was used as an internal control.
Flow cytometry analyses
To determine the effect of IL-32θ on the expression of the macrophage surface markers CD18 and CD36, untreated THP-1 cells and cells stimulated with PMA were assessed by flow cytometry. Cells were stained with saturated concentration of anti-CD36 (Nordic-MUbio, Susteren, Netherland) or anti-CD18 (Millipore, Ma, USA) mAbs for 1 h in PBS containing 1% BSA. Cells were then washed and incubated with a FITC-conjugated goat anti-mouse antibody for 1 h. The expression levels of CD18 and CD36 were measured using a FACS Calibur flow cytometer and analyzed with CellQuest Pro software.
Western blot analyses
Cells were harvested and lysed with 50 mM HEPES (pH 7.5), 150 mM NaCl, 5% glycerol, 20 mM β-glycerophosphate, 1% Nonidet P-40, 0.5% Triton X-100, 1 mM EDTA, and 1 mM EGTA. Western blotting was performed using primary antibodies specific for cell cycle analysis; cyclin D, cyclin E, p27 from Santa Cruz Biotechnology (Santa Cruz, CA, USA), and anti-C/EBPα antibody and HRP-conjugated secondary antibodies from Millipore (Billerica, MA, USA). KU32–52, a monoclonal anti-IL-32 antibody, was produced as previously reported [32].
Statistical analysis
Quantitative data presented in figures represent the mean ± SEM of results from at least 3 independent experiments. Statistical significance for multiple groups was assessed using one-way ANOVA, followed by Tukey’s HSD tests. *p < 0.05 was considered statistically significant.
ACKNOWLEDGMENTS
This research was supported by the basic program (2012R1A2A2A 02008751) of the National Research Foundation of Korea (NRF).
CONFLICTS OF INTEREST
The authors declare that no financial or other conflicts of interest exist in relation to the content of this article.
REFERENCES
1. Alvarez-Errico D, Vento-Tormo R, Sieweke M, Ballestar E. Epigenetic control of myeloid cell differentiation, identity and function. Nat Rev Immunol. 2014; 15:7–17.
2. Rosenbauer F, Tenen DG. Transcription factors in myeloid development: balancing differentiation with transformation. Nat Rev Immunol. 2007; 7:105–117.
3. Huber R, Pietsch D, Gunther J, Welz B, Vogt N, Brand K. Regulation of monocyte differentiation by specific signaling modules and associated transcription factor networks. Cell Mol Life Sci. 2014; 71:63–92.
4. Charrad RS, Li Y, Delpech B, Balitrand N, Clay D, Jasmin C, Chomienne C, Smadja-Joffe F. Ligation of the CD44 adhesion molecule reverses blockage of differentiation in human acute myeloid leukemia. Nat Med. 1999; 5:669–676.
5. Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, Sultan C. Proposed revised criteria for the classification of acute myeloid leukemia. A report of the French-American-British Cooperative Group. Ann Intern Med. 1985; 103:620–625.
6. Auffray C, Sieweke MH, Geissmann F. Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Annu Rev Immunol. 2009; 27:669–692.
7. Chow A, Brown BD, Merad M. Studying the mononuclear phagocyte system in the molecular age. Nat Rev Immunol. 2011; 11:788–798.
8. Yasaka T, Mantich NM, Boxer LA, Baehner RL. Functions of human monocyte and lymphocyte subsets obtained by countercurrent centrifugal elutriation: differing functional capacities of human monocyte subsets. J Immunol. 1981; 127:1515–1518.
9. Nunn MF, Seeburg PH, Moscovici C, Duesberg PH. Tripartite structure of the avian erythroblastosis virus E26 transforming gene. Nature. 1983; 306:391–395.
10. Pio F, Assa-Munt N, Yguerabide J, Maki RA. Mutants of ETS domain PU.1 and GGAA/T recognition: free energies and kinetics. Protein Sci. 1999; 8:2098–2109.
11. Klemsz MJ, McKercher SR, Celada A, Van Beveren C, Maki RA. The macrophage and B cell-specific transcription factor PU.1 is related to the ets oncogene. Cell. 1990; 61:113–124.
12. Akashi K, Traver D, Miyamoto T, Weissman IL. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 2000; 404:193–197.
13. McKercher SR, Torbett BE, Anderson KL, Henkel GW, Vestal DJ, Baribault H, Klemsz M, Feeney AJ, Wu GE, Paige CJ, Maki RA. Targeted disruption of the PU.1 gene results in multiple hematopoietic abnormalities. EMBO J. 1996; 15:5647–5658.
14. Scott EW, Simon MC, Anastasi J, Singh H. Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science. 1994; 265:1573–1577.
15. Okuno Y, Yuki H. PU.1 is a tumor suppressor for B cell malignancies. Oncotarget. 2012; 3:1495–1496.
16. Yuki H, Ueno S, Tatetsu H, Niiro H, Iino T, Endo S, Kawano Y, Komohara Y, Takeya M, Hata H, Okada S, Watanabe T, Akashi K, Mitsuya H, Okuno Y. PU.1 is a potent tumor suppressor in classical Hodgkin lymphoma cells. Blood. 2013; 121:962–970.
17. Rekhtman N, Radparvar F, Evans T, Skoultchi AI. Direct interaction of hematopoietic transcription factors PU.1 and GATA-1: functional antagonism in erythroid cells. Genes Dev. 1999; 13:1398–1411.
18. Zhang P, Behre G, Pan J, Iwama A, Wara-Aswapati N, Radomska HS, Auron PE, Tenen DG, Sun Z. Negative cross-talk between hematopoietic regulators: GATA proteins repress PU.1. Proc Natl Acad Sci U S A. 1999; 96:8705–8710.
19. Joosten LA, Netea MG, Kim SH, Yoon DY, Oppers-Walgreen B, Radstake TR, Barrera P, van de Loo FA, Dinarello CA, van den Berg WB. IL-32, a proinflammatory cytokine in rheumatoid arthritis. Proc Natl Acad Sci U S A. 2006; 103:3298–3303.
20. Kang JW, Park YS, Lee DH, Kim MS, Bak Y, Ham SY, Park SH, Kim H, Ahn JH, Hong JT, Yoon DY. Interaction network mapping among IL-32 isoforms. Biochimie. 2014; 101:248–251.
21. Heinhuis B, Koenders MI, van de Loo FA, Netea MG, van den Berg WB, Joosten LA. Inflammation-dependent secretion and splicing of IL-32{gamma} in rheumatoid arthritis. Proc Natl Acad Sci U S A. 2011; 108:4962–4967.
22. Kang JW, Choi SC, Cho MC, Kim HJ, Kim JH, Lim JS, Kim SH, Han JY, Yoon DY. A proinflammatory cytokine interleukin-32beta promotes the production of an anti-inflammatory cytokine interleukin-10. Immunology. 2009; 128:e532–540.
23. Qu Y, Taylor JL, Bose A, Storkus WJ. Therapeutic effectiveness of intratumorally delivered dendritic cells engineered to express the pro-inflammatory cytokine, interleukin (IL)-32. Cancer Gene Ther. 2011; 18:663–673.
24. Jung MY, Son MH, Kim SH, Cho D, Kim TS. IL-32gamma induces the maturation of dendritic cells with Th1- and Th17-polarizing ability through enhanced IL-12 and IL-6 production. J Immunol. 2011; 186:6848–6859.
25. Kim YG, Lee CK, Kim SH, Cho WS, Mun SH, Yoo B. Interleukin-32gamma enhances the production of IL-6 and IL-8 in fibroblast-like synoviocytes via Erk1/2 activation. J Clin Immunol. 2010; 30:260–267.
26. Kang JW, Park YS, Lee DH, Kim JH, Kim MS, Bak Y, Hong J, Yoon DY. Intracellular interaction of interleukin (IL)-32alpha with protein kinase Cepsilon (PKCepsilon) and STAT3 protein augments IL-6 production in THP-1 promonocytic cells. J Biol Chem. 2012; 287:35556–35564.
27. Choi JD, Bae SY, Hong JW, Azam T, Dinarello CA, Her E, Choi WS, Kim BK, Lee CK, Yoon DY, Kim SJ, Kim SH. Identification of the most active interleukin-32 isoform. Immunology. 2009; 126:535–542.
28. Kim SH, Han SY, Azam T, Yoon DY, Dinarello CA. Interleukin-32: a cytokine and inducer of TNFalpha. Immunity. 2005; 22:131–142.
29. Kim MS, Kang JW, Lee DH, Bak Y, Park YS, Song YS, Ham SY, Oh DK, Hong J, Yoon DY. IL-32theta negatively regulates IL-1beta production through its interaction with PKCdelta and the inhibition of PU.1 phosphorylation. FEBS Lett. 2014; 588:2822–2829.
30. Urbinati C, Nicoli S, Giacca M, David G, Fiorentini S, Caruso A, Alfano M, Cassetta L, Presta M, Rusnati M. HIV-1 Tat and heparan sulfate proteoglycan interaction: a novel mechanism of lymphocyte adhesion and migration across the endothelium. Blood. 2009; 114:3335–3342.
31. Kang JW, Park YS, Kim MS, Lee DH, Bak Y, Ham SY, Song YS, Hong JT, Yoon DY. IL-32alpha down-regulates beta2 integrin (CD18) expression by suppressing PU.1 expression in myeloid cells. Cell Signal. 2014; 26:1514–1522.
32. Kim KH, Shim JH, Seo EH, Cho MC, Kang JW, Kim SH, Yu DY, Song EY, Lee HG, Sohn JH, Kim J, Dinarello CA, Yoon DY. Interleukin-32 monoclonal antibodies for immunohistochemistry, Western blotting, and ELISA. J Immunol Methods. 2008; 333:38–50.
33. Murray JL, Mehta K, Lopez-Berestein G. Induction of adenosine deaminase and 5’ nucleotidase activity in cultured human blood monocytes and monocytic leukemia (THP-1) cells by differentiating agents. J Leukoc Biol. 1988; 44:205–211.
34. Auwerx JH, Chait A, Wolfbauer G, Deeb SS. Loss of copper-zinc superoxide dismutase gene expression in differentiated cells of myelo-monocytic origin. Blood. 1989; 74:1807–1810.
35. Abrink M, Gobl AE, Huang R, Nilsson K, Hellman L. Human cell lines U-937, THP-1 and Mono Mac 6 represent relatively immature cells of the monocyte-macrophage cell lineage. Leukemia. 1994; 8:1579–1584.
36. Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, Pittet MJ. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007; 117:195–205.
37. Nerlov C, Graf T. PU.1 induces myeloid lineage commitment in multipotent hematopoietic progenitors. Genes Dev. 1998; 12:2403–2412.
38. Rovera G, Ferrero D, Pagliardi GL, Vartikar J, Pessano S, Bottero L, Abraham S, Lebman D. Induction of differentiation of human myeloid leukemias by phorbol diesters: phenotypic changes and mode of action. Ann N Y Acad Sci. 1982; 397:211–220.
39. Scott EW, Fisher RC, Olson MC, Kehrli EW, Simon MC, Singh H. PU.1 functions in a cell-autonomous manner to control the differentiation of multipotential lymphoid-myeloid progenitors. Immunity. 1997; 6:437–447.
40. Radomska HS, Huettner CS, Zhang P, Cheng T, Scadden DT, Tenen DG. CCAAT/enhancer binding protein alpha is a regulatory switch sufficient for induction of granulocytic development from bipotential myeloid progenitors. Mol Cell Biol. 1998; 18:4301–4314.
41. Iwasaki H, Akashi K. Myeloid lineage commitment from the hematopoietic stem cell. Immunity. 2007; 26:726–740.
42. Evans CA, Pierce A, Winter SA, Spooncer E, Heyworth CM, Whetton AD. Activation of granulocyte-macrophage colony-stimulating factor and interleukin-3 receptor subunits in a multipotential hematopoietic progenitor cell line leads to differential effects on development. Blood. 1999; 94:1504–1514.
43. Just U, Stocking C, Spooncer E, Dexter TM, Ostertag W. Expression of the GM-CSF gene after retroviral transfer in hematopoietic stem cell lines induces synchronous granulocyte-macrophage differentiation. Cell. 1991; 64:1163–1173.
44. Reddy VA, Iwama A, Iotzova G, Schulz M, Elsasser A, Vangala RK, Tenen DG, Hiddemann W, Behre G. Granulocyte inducer C/EBPalpha inactivates the myeloid master regulator PU.1: possible role in lineage commitment decisions. Blood. 2002; 100:483–490.
45. Ishigami S, Arigami T, Uchikado Y, Setoyama T, Kita Y, Sasaki K, Okumura H, Kurahara H, Kijima Y, Harada A, Ueno S, Natsugoe S. IL-32 expression is an independent prognostic marker for gastric cancer. Med Oncol. 2013; 30:472.
46. Sakitani K, Hirata Y, Hayakawa Y, Serizawa T, Nakata W, Takahashi R, Kinoshita H, Sakamoto K, Nakagawa H, Akanuma M, Yoshida H, Maeda S, Koike K. Role of interleukin-32 in Helicobacter pylori-induced gastric inflammation. Infect Immun. 2012; 80:3795–3803.
47. Kang YH, Park MY, Yoon DY, Han SR, Lee CI, Ji NY, Myung PK, Lee HG, Kim JW, Yeom YI, Jang YJ, Ahn DK, Song EY. Dysregulation of overexpressed IL-32alpha in hepatocellular carcinoma suppresses cell growth and induces apoptosis through inactivation of NF-kappaB and Bcl-2. Cancer Lett. 2012; 318:226–233.
48. Tsai CY, Wang CS, Tsai MM, Chi HC, Cheng WL, Tseng YH, Chen CY, Lin CD, Wu JI, Wang LH, Lin KH. Interleukin-32 increases human gastric cancer cell invasion associated with tumor progression and metastasis. Clin Cancer Res. 2014; 20:2276–2288.
49. Jeong HJ, Nam SY, Oh HA, Han NR, Kim YS, Moon PD, Shin SY, Kim MH, Kim HM. Interleukin-32-induced thymic stromal lymphopoietin plays a critical role in macrophage differentiation through the activation of caspase-1 in vitro. Arthritis Res Ther. 2012; 14:R259.
50. Moon YM, Yoon BY, Her YM, Oh HJ, Lee JS, Kim KW, Lee SY, Woo YJ, Park KS, Park SH, Kim HY, Cho ML. IL-32 and IL-17 interact and have the potential to aggravate osteoclastogenesis in rheumatoid arthritis. Arthritis Res Ther. 2012; 14:R246.
51. Mabilleau G, Sabokbar A. Interleukin-32 promotes osteoclast differentiation but not osteoclast activation. PLoS One. 2009; 4:e4173.
52. Pahl HL, Scheibe RJ, Zhang DE, Chen HM, Galson DL, Maki RA, Tenen DG. The proto-oncogene PU.1 regulates expression of the myeloid-specific CD11b promoter. J Biol Chem. 1993; 268:5014–5020.
53. Rosmarin AG, Caprio D, Levy R, Simkevich C. CD18 promoter requires PU.1 transcription factor for myeloid activity. Proc Natl Acad Sci U S A. 1995; 92:801–805.
54. Rosenbauer F, Wagner K, Zhang P, Knobeloch KP, Iwama A, Tenen DG. pDP4, a novel glycoprotein secreted by mature granulocytes, is regulated by transcription factor PU.1. Blood. 2004; 103:4294–4301.
55. Terry RL, Miller SD. Molecular control of monocyte development. Cell Immunol. 2014; 219:16–21.
56. Jego G, Lanneau D, De Thonel A, Berthenet K, Hazoume A, Droin N, Hamman A, Girodon F, Bellaye PS, Wettstein G, Jacquel A, Duplomb L, Le Mouel A, Papanayotou C, Christians E, Bonniaud P, et al. Dual regulation of SPI1/PU.1 transcription factor by heat shock factor 1 (HSF1) during macrophage differentiation of monocytes. Leukemia. 2014; 28:1676–1686.
57. de la Rica L, Rodriguez-Ubreva J, Garcia M, Islam AB, Urquiza JM, Hernando H, Christensen J, Helin K, Gomez-Vaquero C, Ballestar E. PU.1 target genes undergo Tet2-coupled demethylation and DNMT3b-mediated methylation in monocyte-to-osteoclast differentiation. Genome Biol. 2013; 14:R99.
58. Okuno Y, Huang G, Rosenbauer F, Evans EK, Radomska HS, Iwasaki H, Akashi K, Moreau-Gachelin F, Li Y, Zhang P, Gottgens B, Tenen DG. Potential autoregulation of transcription factor PU.1 by an upstream regulatory element. Mol Cell Biol. 2005; 25:2832–2845.
59. Wang D, D’Costa J, Civin CI, Friedman AD. C/EBPalpha directs monocytic commitment of primary myeloid progenitors. Blood. 2006; 108:1223–1229.
60. Yeamans C, Wang D, Paz-Priel I, Torbett BE, Tenen DG, Friedman AD. C/EBPalpha binds and activates the PU.1 distal enhancer to induce monocyte lineage commitment. Blood. 2007; 110:3136–3142.
61. Cai DH, Wang D, Keefer J, Yeamans C, Hensley K, Friedman AD. C/EBP alpha:AP-1 leucine zipper heterodimers bind novel DNA elements, activate the PU.1 promoter and direct monocyte lineage commitment more potently than C/EBP alpha homodimers or AP-1. Oncogene. 2008; 27:2772–2779.
62. Nowak D, Stewart D, Koeffler HP. Differentiation therapy of leukemia: 3 decades of development. Blood. 2009; 113:3655–3665.
63. Sell S. Leukemia: stem cells, maturation arrest, and differentiation therapy. Stem Cell Rev. 2005; 1:197–205.