
Introduction
The development of mutant BRAF and of MEK1/2 inhibitors has markedly improved treatment of advanced BRAF-mutant melanoma [1-2] showing highly significant effects on progression-free and/or overall survival in several phase III trials, in comparison to chemotherapy [3]. Nevertheless, a fraction of patients does not benefit from target-specific therapy and duration of clinical responses can be limited. Intrinsic and acquired resistance to small molecule inhibitors limit the efficacy of these therapeutic approaches [4-5], highlighting the urgent need for identification of new molecules and pathways that may contribute to melanoma growth. Several screening strategies have been proposed to identify genes with a significant role in BRAFV600Emelanoma persistence. These include identification of BRAF target genes by comparing gene expression profiles in melanomas treated with/without BRAF inhibitors [7], screening for expression of relevant molecules to be targeted by inhibitors [8-11], and comparison of gene expression profiles of BRAFV600E vs wild-type tumors [12-13]. The latter approach has the advantage of contributing to define the gene expression landscape associated with a frequently activated oncogene, but conflicting results have been published [14-19] possibly due to strong inter-tumor genetic heterogeneity. To reduce the impact of this heterogeneity, we exploited a set of melanoma clones, isolated from a metastasis containing both BRAFV600E and NRASQ61R cells [20]. The clones, previously shown to have mutually exclusive expression of activated BRAFV600E or NRASQ61R [20], were used for whole genome gene expression profiling aimed at identifying genes overexpressed in BRAFV600E cells. This approach led us to identify the semaphorin SEMA6A, a ligand of Plexin A2 and A4 [21-23], as one of the most discriminating genes, overexpressed in BRAF-mutant clones compared to NRASQ61R clones. Interestingly, another gene in the semaphorin-plexin pathway, MICAL1 (molecule interacting with CasL), that links Sema/PlexinA signaling to F-actin disassembly [24-26], showed similar differential expression in BRAF-mutants compared to NRAS-mutant clones. Although the sema/plexin/mical pathway has been thoroughly investigated for its role in repulsive guidance, recent studies point to a role of its different components in regulating cell survival and migratory functions even in neoplastic cells [27]. It has been demonstrated that semaphorine 3A regulates tumor persistence by suppressing apoptosis triggered by plexin D1 dependent receptor [28]. Among this class of molecules, Sema6A regulates vascular development in tumors and angiogenesis by modulating VEGFR2 signaling in endothelial cells [22]. Mical1 is a multi-domain flavoprotein monoxigenase binding NDR1/2 kinases, thus inhibiting both their phosphorylation by MST1 and NDR-mediated apoptosis [26, 29]. We tested whether Sema6A and Mical1 could exert a significant pro-tumoral role in BRAFV600E melanomas by contributing to regulate their growth, survival, and invasion. The results confirmed this hypothesis and suggested that different components of the sema/plexin/mical signaling cascade may represent new therapeutic targets in melanoma.
Results
BRAFV600E and NRASQ61R clones isolated from the same metastatic melanoma have distinct biological behavior and gene expression profiles
The distinct genetic makeup of BRAF-mutant and NRAS- mutant clones derived from the 665/2 cell line [20] was associated with different biological behavior in vivo. Upon intracardiac injection, BRAFV600E clones metastatized in all mice and all organs, except for spleen and testis, while only one NRASQ61R clone was able to metastasize, and only in one mouse and exclusively to the intestine and stomach (Table S1). These marked differences were reflected in the gene expression profiles. Whole genome microarray analysis of BRAFV600E and NRASQ61R clones (Fig. S1) identified SEMA6A as one of the most discriminating genes, highly expressed in the BRAFV600E clones compared to the NRASQ61R ones. Another gene belonging to the sema/plexin signaling pathway, MICAL1, was also expressed at significantly higher levels in BRAF clones (Fig. S1). Higher expression of Sema6A and Mical1, at both RNA and protein levels, in BRAFV600E clones, was confirmed by qRT-PCR and WB, respectively (Fig. 1A, and B). We also found high expression of both Sema6A and Mical1 in BRAFV600E cells derived from seven different melanoma cell lines isolated from primary tumor or from lymph node metastases (Fig. 1B). Most BRAFV600E clones and cell lines in this panel showed constitutive activation of the AKT and ERK pathway, while NRASQ61R clones showed reduced activation of AKT pathway, a finding consistent with the lower/missing expression of ErbB2/ErbB3 receptor, the strongest activator in nature of PI3K [30].
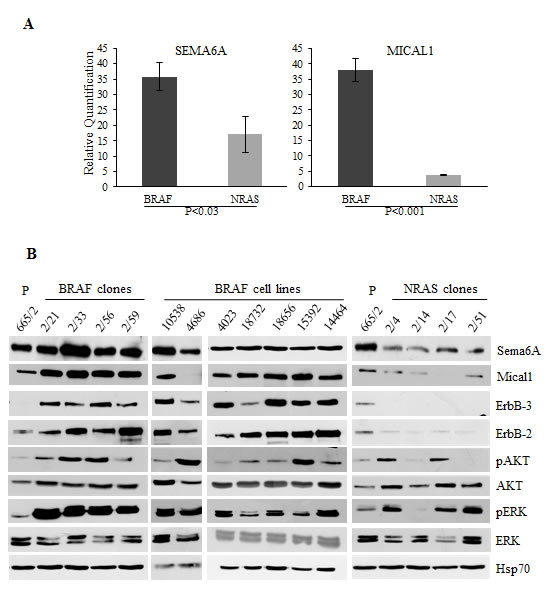
Figure 1: Sema6A and Mical1 are highly expressed in BRAFV600E compared to NRASQ61R melanoma. (A) Average expression of SEMA6A and MICAL1 mRNA examined by qRT-PCR±SD. (B) Total cell lysates from cell line 665/2, from BRAF and NRAS clones isolated from 665/2 and from seven BRAF-mutant cell lines were analyzed by WB for expression of Sema6A, Mical1, ErbB3, ErbB2, total and pAkt, total and pERK, and Hsp70.
Sema6A and Mical-1 are highly expressed in vivo in BRAFV600E tumors compared to wild type melanomas and nevi
To assess whether Sema6A and Mical-1 were preferentially expressed in BRAFV600E tumors, we analyzed their expression by qRT-PCR and immunohistochemistry (IHC) on BRAFV600E and BRAF wild type (WT) melanoma specimens derived from patients surgically treated at the Regina Elena National Cancer Institute. Both molecules were also analyzed in nevi. The results demonstrated that Sema6A and Mical-1 were significantly more expressed in BRAFV600E than in WT melanomas (P<0.02 and P<0.009, respectively) (Fig. 2A and B). In nevi, Sema6A and Mical-1 expression was comparable to the levels found in WT melanoma (Fig. S2). Representative IHC analyses with the relative internal controls are reported for both molecules in the two subtypes of melanoma (Fig. 2A and B) and in nevi (Fig.S2). These data supported our in vitro results.
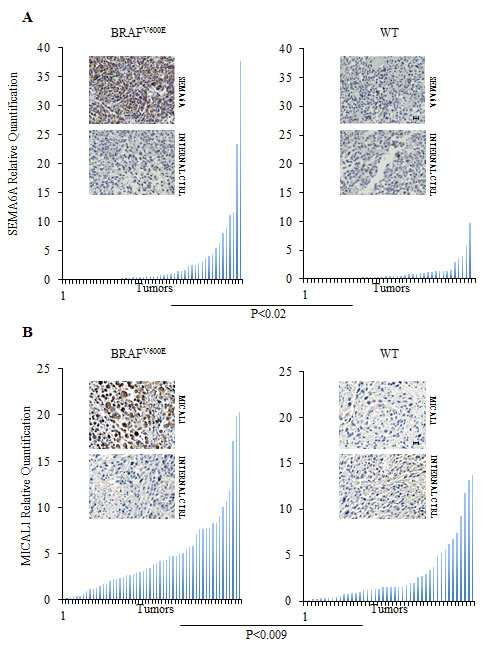
Figure 2: Sema6A and Mical1 are highly expressed in vivo in B-RAFV600E compared to WT melanomas. (A-B) Expression of SEMA6A and MICAL1 mRNA was examined by qRT-PCR on lymph node biopsies from patients carrying BRAFV600E or WT tumors (left and right graphs). Representative Sema6A and Mical1 IHC on sections derived from same samples. Secondary antibody was used as internal control. Scale bar 30 µm.
Depletion of Sema6A in BRAF-mutant melanoma cells promotes cell death
To investigate the role of Sema6A in BRAFV600E cells, we carried out siRNA experiments, with different Sema6A-specific silencing sequences (siSema6A) in three clones isolated from 665/2 cell line and in one cell line (10538) isolated from a BRAFV600E primary tumor. Sema6A depletion strongly induced PARP cleavage in clone 2/21 and reduced total PARP in the clones 2/56 and 2/59 and in the melanoma cell line 10538 (Fig. 3A). Surprisingly, Sema6A silencing inhibited ErbB3 expression and phosphorylation of AKT, and to a lesser extent, of ERK (Fig. 3A). ErbB3 down-regulation was likely post-transcriptional, secondary to p-Akt inhibition, as previously described [31], and not transcriptional, as demonstrated by qRT-PCR (Fig. 3B). The above results were confirmed by silencing of Sema6A by a second specific siSema6A sequence (Fig. S3A), and even by a third specific commercially available siSema6A (data not shown). Interestingly, PI3K and MAPK activity appeared to be regulated downstream of Sema6A, as their phosphorylation levels (Fig. 3A, and S3A) clearly correlated with Sema6A depletion, suggesting that this semaphorin can regulate major pathways supporting melanoma cell viability. To further explore this possibility, we carried out apoptosis assays by FACS analysis. Annexin-V/PI staining assays indicated that Sema6A depletion induced apoptosis, associated with caspase 3/7 activation, in BRAFV600E clones 2/56 and 2/59 and in melanoma cell line 10538, with effects already evident at 24-36h (Fig. S4). Trypan blue exclusion assays confirmed that siSema6A induced death in all cells tested compared to controls or siScr transfected cells (Fig. 3C). Taken together these results suggested that Sema6A promotes survival of BRAF-mutant melanoma cells.
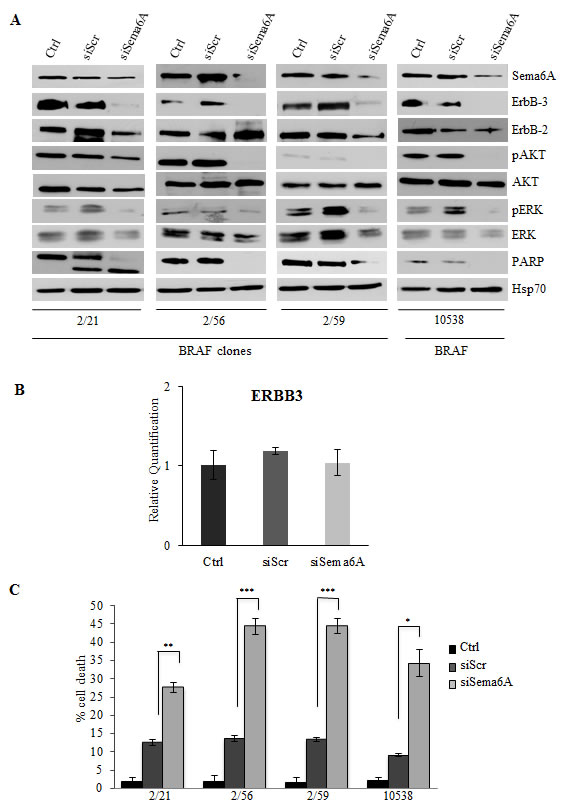
Figure 3: Interference for Sema6A induces cells death, and inhibits PI3K pathway. (A) siSema6A/BRAFV600E clones and primary melanoma cell line 10538 were analyzed by WB for expression of Sema6A, ErbB-3, ErbB-2, P-AKT, P-ERK1/2, PARP, and Hsp70. (B) Average expression of ErbB-3 mRNA examined by qRT-PCR±SD in Control, siScr, and siSema6A in clone 2/59. (C) Cell death of siSema6A/BRAFV600E cells was evaluated at 48 h post-transfection by Trypan blue exclusion from three independent experiments; bars ± S.D.
Depletion of Sema6A in BRAF-mutant melanoma cells alters the cytoskeleton and impairs anchorage-independent growth, as well as motility and invasive activities
Sema6A silencing led to down-regulation of Caspase 3 and reduction of total PARP when cells where plated on conventional cell culture dishes, but not on Fibronectin-coated dishes suggesting cell death by loss of cell adhesion (Fig. 4A). The Cell death was confirmed by TUNEL assays showing fragmentation of nuclei only in siSema6A cells plated on poli-lysine (Fig. 4B). Graphical representation of percentage of TUNEL positive cells is reported in Fig. 4C (P<0.004). Silencing of Sema6A also induced changes in the actin cytoskeleton as shown by loss of actin stress fibers consistent with impaired cell adhesion and with promotion of cell death by anoikis (Fig. 4D, and E, P<0.0001). These results were further confirmed in clones 2/56, 2/21, and in 10538 cell line (Fig. S5A and B, right panels). These changes were not observed in untreated or siScr/cells (Fig. 4D, and E, and Fig. S5A and B, left and central panels). These results suggested that Sema6A might contribute to anchorage-independent growth of BRAFV600E melanoma cells. Indeed, Sema6A depletion strongly suppressed growth of BRAFV600E cells in soft agar (Fig. 4F, and G). siSema6A cells formed very small colonies appearing later, compared to control or to siScr/cells (P<0.0001) confirming that Sema6A controls anchorage-independent growth. In agreement with above results, Sema6A depletion in BRAF mutant cells strongly reduced their capability to migrate and to invade in vitro (P≤0.001) (Fig. S6A and B, upper and lower panels).
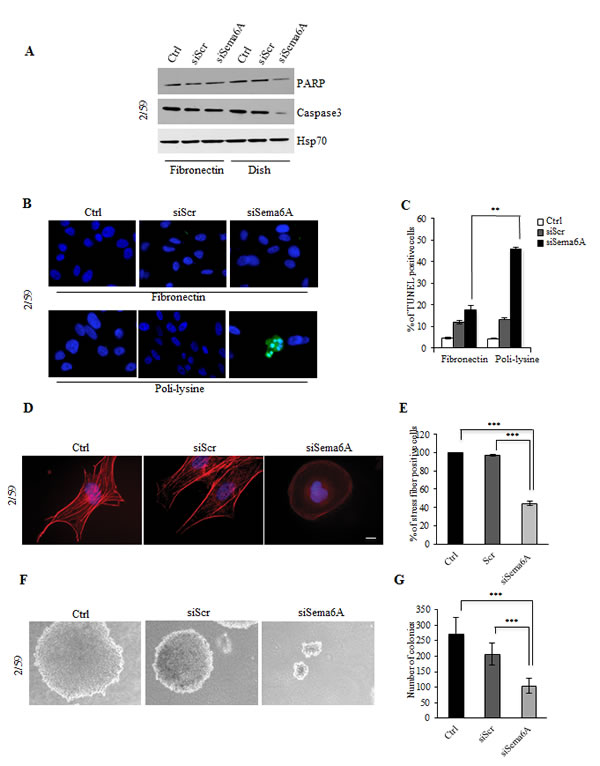
Figure 4: Interference with Sema6A expression in BRAF 2/59 cells induces cytoskeletal remodeling and inhibits anchorage-independent growth. (A) Control, siScr- or siSema6A/BRAFV600E cells were plated onto FN coated-dish or conventional culture-dish. Total cell lysates were analyzed by WB with PARP, Caspase3, and Hsp70 antibodies. (B) Tunel assay of BRAF/siSema6A cells plated on FN and/or poli-lysine. (C) Histogram reported percentage of tunel positive cells (P<0.004). (D) The cells, 48 h post-transfection, were plated on poly-l lysine coated slides, and stained with phalloidin-TRITC to show actin filaments. Scale bar is 10 µm. (E) Histogram reported percentage of stress fiber positive-cells. (F) Ctrl, siScr and siSema6A/BRAFV600E cells soft agar assay. (G) Histogram reported the number of colonies obtained in soft agar assay in Ctrl, siScr and siSema6A cells.
Ectopic expression of Sema6A in NRAS-mutant cells promotes in vitro and in vivo invasion
We then tested whether forced overexpression of Sema6A in NRASQ61R clones could promote anchorage-independent growth and invasiveness. In contrast with in vivo results on development of metastases (Table S1) and with data indicating that NRAS mutant clones yield only small colonies in soft agar [20], we found that overexpression of Sema6A in NRASQ61R melanoma clones conferred a strong invasive activity in vitro (P≤0.0001) (Fig. 5A), and capability to generate large colonies in soft agar (P≤0.02) (Fig. 5B). Furthermore, depletion of Sema6A in NRASQ61R melanoma clones down-regulated their capability to invade (P≤0.001) (Fig. S7). More importantly, by in vivo assessment for metastatic ability of NRAS-mutant cells overexpressing Sema6A, we found that several components of the skeletal system of the mice (spinal column, femurs, and upper and lower limbs) were characterized by presence of metastatic foci. Moreover, we found a significant increase, at these anatomical sites, in the fraction of tissue being interested by metastatic foci, compared to tissues from mice receiving control NRAS-mutant cells (Fig. 5C). Analysis of additional organs (Table S2) demonstrated that NRAS cells overexpresing Sema6A frequently produced metastases also in the brain, lung, pancreas and heart, compared to control cells. These data strongly support the role of Sema6A in the mechanisms that promote melanoma invasion.
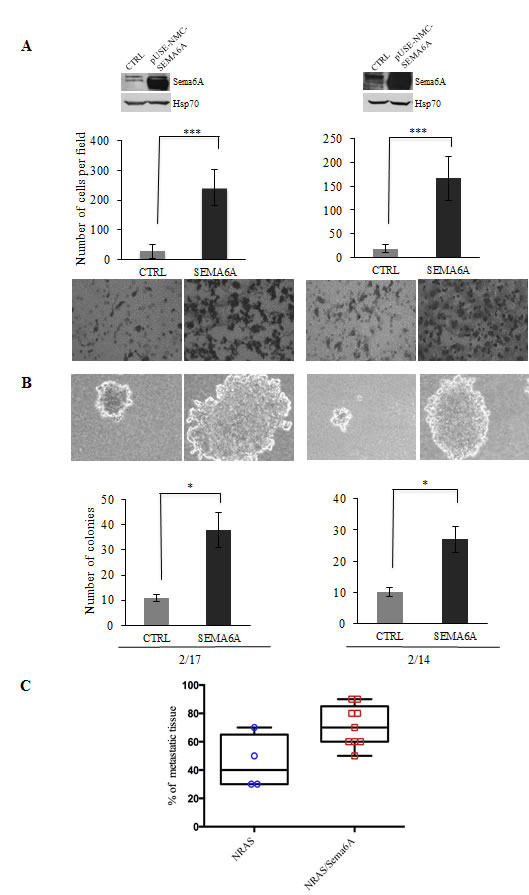
Figure 5: Sema6A controls invasion. (A) Overexpression of Sema6A by WB (upper part of the figure), and invasion assay (lower part of the figure), and (B) soft agar assay, of control and Sema6A/NRASQ61R overexpressing clones (2/14 and 2/17). (C) Box plot of in vivo skeletal tissues metastases analysis was obtained by software Prism version 6.0.
Sema6A functions in BRAF-mutant melanoma cells are independent from PlexinA2 and PlexinA4 receptors
We then looked at expression and function in melanoma cells of Plexin-A2 and Plexin-A4, the Sema6A receptors. In agreement with previous results [32], all BRAFV600E clones and cell lines expressed heterogeneous levels of Plexin-A4 (Fig. S8A, and B) compared to the level found for Sema6A (Fig. 1), while all clones and cell lines were negative for the expression of Plexin-A2 (data not shown). Both PlexinA4 and PlexinA2 were absent in NRASQ61R clones (Fig. S8A and B, and data not shown). Surprisingly, depletion of Plexin-A4 in BRAFV600E clone by siRNA did not induce cell death and did not alter the actin cytoskeleton (data not shown). Silencing of both molecules led to the expected changes already found upon siSema6A alone (Fig. S8C). Taken together these results suggest that Sema6A and Plexin-A4 have distinct roles in regulating biological functions in BRAF-mutant melanoma cells.
Mical1 depletion induces melanoma cell death by apoptosis
Mical1 is a mediator of Sema/Plexin repulsion, and acts by promoting F-actin disassembly [33-34]. Mical1 enhanced expression in BRAFV600E clones, and its known role as an effector of the Sema/Plexin axis, suggested that it could exert functions similar to those regulated by Sema6A. To address this possibility we depleted Mical1 by siRNA. Surprisingly, Mical1 silencing did not change the actin cytoskeleton organization, indicating that its role in BRAFV600E melanoma cells is not overlapping with that of Sema6A. A candidate function for Mical1 is interaction with NDR, a pro-apoptotic kinase, antagonizing MST1-induced NDR phosphorylation to trigger apoptosis [29]. Indeed, silencing of Mical1 in BRAFV600E melanoma clones (2/59, 2/56) and in 10538 melanoma cell line caused strong NDR phosphorylation between 30 h and 36 h post transfection associated with PARP cleavage (Fig. 6A). By Annexin-V/PI flow cytometry assay, we also found that Mical1 depletion in these melanoma cells increased both early (annexin-V+PI-) and late (Annexin-V+PI+) apoptosis, compared with the effects seen in cells transfected with control siRNA (siScr, Fig. 6B), suggesting that Mical-1 protects BRAFV600E melanomas from apoptosis. These results were confirmed by the use of a second siMical1 (Fig. S3B), and by a commercially available siMical1 (data not shown). By caspase-3 activation assays on the same cells, we confirmed that siMical1 induced melanoma apoptosis (Fig. 7A, B). Analysis for phosphorylation of H2B-S14, that co-localizes with Hoechst positivity in fragmented nuclei, further supported the evidence that siMical1 induces apoptosis in BRAFV600E melanoma without affecting the actin cytoskeleton (Fig. 7C, D). Taken together, these results support the notion that both Sema6A and Mical1 promote cell survival, and inhibit cell death of BRAFV600E melanomas through different pathways.
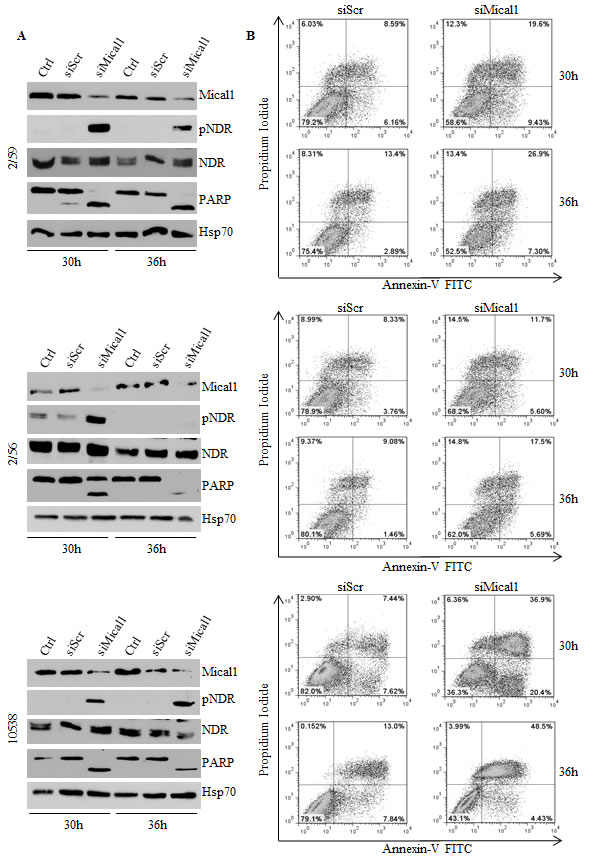
Figure 6: Silencing Mical1 induces apoptosis. (A) siMical-1/BRAFV600E cell lysates from 2/59 and 2/56 clones, and 10538 primary tumor were analyzed at the indicated time post-transfection for expression of Mical1, total and pNDR, and PARP. (B) Apoptosis was evaluated by Annexin-V/PI flow cytometric assay.
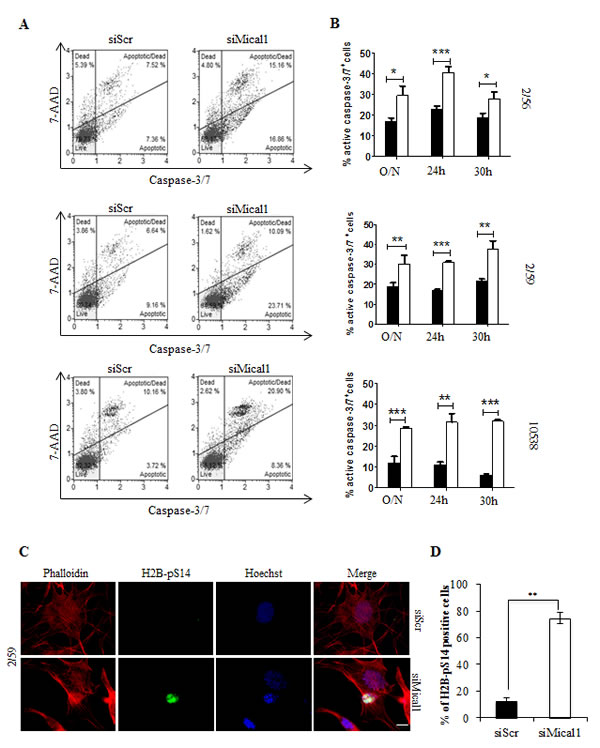
Figure 7: Silencing Mical1 induces Caspase 3/7 activation and H2B phosphorylation on Serine 14. (A) siMical-1/BRAFV600Ecells were analyzed with Muse™ Caspase-3/7 assay. (B) Caspase-3/7 activity after transfection with siScr (black bars) or siMICAL1 (white bars) is reported. (C) Cells plated on poly-l lysine coated slides, stained with phalloidin-TRITC, and anti-H2B-pS14 that co-stains with Hoechst of apoptotic nuclei of siMical-1/BRAFV600E cells. (D) Scale bar is 10 µm. Histogram reported the percentage of H2B-pS14 positive cells.
Discussion
In attempts to find molecules regulating aggressiveness of BRAFV600E melanoma cells, by whole-genome array we found that SEMA6A and MICAL1, two genes of the SEMA/PLEXIN signaling pathway, were over-expressed in BRAFV600E clones compared to NRASQ61R clones isolated from the same metastasis. In vitro and in vivo assays confirmed the selective expression of the corresponding proteins in BRAFV600E cell lines derived from primary and metastatic melanoma lesions or in BRAF mutant tumors compared to BRAF wild type tumors and nevi. Sema6A belongs to a large family of secreted or trans-membrane proteins and its receptors are PlexinA2 and PlexinA4. Sema6A/Plexin complexes were previously characterized for their role in vascular and neural development [22]. However a role of SEMA6A in cell proliferation, survival, anchorage-independent cell growth, and invasion of BRAFV600E melanoma was not previously described. We found that Sema6A-depletion in BRAFV600E clones and primary tumor cells results in cell death. Specifically, Sema6A depletion reduces Akt phosphorylation and suppresses ErbB-3 expression suggesting that its inhibition of cell proliferation could depend on loss of ErbB-2/ErbB-3 heterodimer formation. Loss of Sema6A by siRNA caused a very low reduction of ErbB-2 expression and of ERK activity suggesting that in these cells ErbB-2 homodimer may still contribute to cell proliferation. However, when the cells were forced to adhere to fibronectin (FN) we did not find cleavage of PARP and activation of caspase 3.
Previous studies have shown that depletion of Sema6A in endothelial cells reduces VEGFR2 expression and abolish its phosphorylation, thus inducing cell death. Interestingly, stimulation by VEGF did not restore cell proliferation in Sema6A-depleted endothelial cells, demonstrating that Sema6A mainly control proliferation through the VEFGR2 receptor [22]. Overall, the results of this study add further evidence for a direct role of SEMA6A as regulator of cell proliferation even in neoplastic cells.
Cells with depleted Sema6A showed loss of stress fiber and actin distribution exclusively under the cell membrane. This was associated with loss of adhesion to the substrate, reduced growth in soft agar, inhibition of chemotaxis and invasion. In agreement, there are several evidences demonstrating that semaphorins control the adhesion regulating integrin function in vascular morphogenesis and cancer progression [35-36]. In particular, it has been shown that Sema3A impairs endothelial cell adhesion and migration by inhibiting integrin activation [35]. In contrast but in agreement with our data, Sema3C promotes endothelial cell adhesion and migration by increasing integrin activity [36]. It is also been described that neuropilins interacting with numerous growth factors receptor and semaphorins promote tumor progression [37]. All these data suggest the complexity of Semaphorins functions and that there is much still to be learned about their involvement in tumor initiation, growth, and metastatization.
Our study demonstrated strong capability of BRAFV600E clones to metastazise in mice, and in agreement with previous results [20], to form high number of big colonies in soft agar compared to NRASQ61R clones. These data were further confirmed by the strong acquired capability of NRASQ61R clones, overexpressing Sema6A, to grow in soft agar and to metastasize in vitro and in vivo suggesting a specific role of Sema6A regulating tumor progression. These finding are strongly in agreement with previous study showing that Sema6A represents, in melanoma cell line M14, one of the major modulated genes in response to MAPK inhibitor PD0325901 [38] and with our data showing that Vemurafenib treatment increases Sema6A expression in vemurafenib-resistant cells but inhibits Sema6A expression in the vemurafenib-responsive cells (unpublished data).
PlexinA2 and PlexinA4, known Sema6A receptors [39-40], showed differential expression in BRAF-mutant clones and melanomas, indicating that in such tumors only Plexin-A4 acted as Sema6A receptor. We hypothesized that depletion of Plexin-A4 may recapitulate the effects of Sema6A silencing. However, PlexinA4 silencing did not induce any of the effects of Sema6A depletion, suggesting that other, as yet unidentified, receptors could be involved. This result is in agreement with the finding that Sema6A regulates angiogenesis by a mechanism that is PlexinA2- and PlexinA4-independent [22].
The other Sema/Plexin pathway-related molecule we found highly expressed in BRAFV600E cells and tumors compared to NRASQ61R cells, WT tumors and nevi, is MIical1. In vertebrates, this molecule interact with class A Plexins [41], suggesting that it could be a mediator of Sema6A signaling in melanoma cells. However, MICAL1 silencing, as well as PlexinA4, did not produce the same effects as Sema6A depletion. Specifically, we observed early and massive cell death by apoptosis in all cell lines tested suggesting that Mical1 plays a Sema6A-independent role in these tumors. These results are in agreement with the finding that Mical1 control cell survival by binding to the phosphorylation site of NDR, that in turn inhibits NDR phosphorylation-dependent apoptosis [29]. Indeed, Mical-1 depletion induced NDR phosphorylation and cell death by apoptosis.
In conclusion our results revealed that BRAFV600E melanomas express high level of Sema6A and MICAL1 whose functions are strongly involved in the mechanisms that control cell proliferation and survival. Our results strongly support the hypothesis that both molecules could represent new potentially relevant therapeutic targets in BRAFV600E melanoma as emerging from recent literature [41-42].
Methods
Cell lines and Transfection
BRAFV600E melanoma cell lines were derived from primary tumor (10538), from lymph node metastases (4686, 4023, 18732, 18656, 15392, 14464), and from s.c. 665/2 metastasis. All melanoma cell lines were isolated from surgical specimens of melanoma patients, not previously subjected to chemotherapy and admitted to Fondazione IRCCS Istituto Nazionale dei Tumori, Milan. All lesions were histologically confirmed to be cutaneous malignant melanomas [20, 43-45]. From the parental cell line 665/2 (P), which was previously shown to be a heterogeneous tumor consisting of BRAF- as well as of NRAS-mutant cells (46, 20), several BRAFV600E clones (2/21, 2/33, 2/56, 2/59) or NRASQ61R clones (2/4, 2/14, 2/17, 2/51) were established as described (46). Selection of clones for the study was based on the intention to select those with the most known and relevant phenotypic differences, as previously described [20, 46-48].
The study was conducted according to the Declaration of Helsinki Principles, and following institutional guidelines. Molecular and biological features of the cell lines and clones, are described in refs [20, 43, 46]. Methods for identification of mutations in BRAF and NRAS have been previously reported [20, 43].
BRAFV600E clones and primary tumor 10538, were depleted for Sem6A, Mical1, and PlexinA4 expression with specific siRNA (siSema6A, siMical1, siPlexinA4) or scramble (siScr) as described [49]. The siRNAs were either generated by Silencer® siRNA construction kit (Ambion, Glasgow, UK) or purchased from Origene or Life Technlogies.
Template oligonucleotides sequences were:
SEMA6A-5’AAATAGTCGCGGCGACTACTTCCTGTCTC3’
5’AAAAGTAGTCGCCGCGACTATCCTGTCTC3’
SEMA6A (2): AS: 5’- AAGCCTTGCTGCTATATTTCACCTGTCTC – 3’
S: 5’ – AATGAAATATAGCAGCAAGGCCCTGTCTC – 3’
SEMA6A (3): Life Technologies, Code N. AM16708
MICAL1-5’AAGGACGTCAAATTCATAGTTCCTGTCTC3’
5’AAAACTATGAATTTGACGTCCCCTGTCTC3’
MICAL1 (2):AS: 5’- AACTACTGGAGCGCCAAGTCACCTGTCTC – 3’
S: 5’- AATGACTTGGCGCTCCAGTAGCCTGTCTC – 3’
MICAL1(3): Origene, Code N. SR312119
PLEXINA4-5’AAGCAGCGGTCATTTGTCACACCTGTCTC 3’
5’AATGTGACAAATGACCGCTGCCCTGTCTC 3’
Scr1-5’AAGCGCAACTCTACCTCTACCTGTCTC3’
5’AATAGAGGTAGAGTTGCGCGCCCTG TCTC3’
Scr2-5’AATAGAGGTAGAGTTGCGCGCCCTGTCTC3’
5’AAGCGCGCAACTCTACCTCTACCTGTCTC3’
Antibodies
Anti-Sema6A and anti-Mical1 (SIGMA-Aldrich, MO, USA), anti-total and phospho-ser473-Akt, anti-total and phospho-ERK 1/2, and anti-PARP were from Cell Signaling (Milan, Italy), anti-ErbB-2, anti-ErbB-3, anti-Caspase 3, and anti-Hsp-70 (Oncogene, MA, USA), (Santa Cruz Biotechnology, CA, USA), and (Stressgen, NY, USA), anti-phospho-NDR(Thr444) was provided by Dr. Brian Hemmings (Friedrich Miescher Institute for Biomedical Research, Basel), anti-total NDR and anti-phospho-H2B-S14, and HRP-conjugated secondary antibodies were from (Millipore, Billerica, MA, USA), and (Bio-Rad, CA, USA). For IHC, the secondary antibody was used as internal control.
Western Blots
Clones and cell lines, before and after transfection, were lysed, analyzed by SDS-PAGE and probed (WB) with antibodies of interest and secondary HRP-conjugated antibodies [49]. Signals were detected by LuminataTM Classico Western HRP substrate (Millipore). Same amount of total protein from three independent experiments were pooled and analyzed.
In vivo experiments
CD-1 male nude (nu/nu) mice, 6–8 weeks old and weighing 22-24 g were purchased from Charles River Laboratories (Calco, Italy). The procedures involving mice were in compliance with Regina Elena National Cancer Institute animal care guidelines and with national and international directives (D.L. March 4, 2014, no. 26; directive 2010/63/EU of the European parliament and of the council; Guide for the Care and Use of Laboratory Animals, United States National Research Council, 2011).
For the intracardiac experimental metastasis model, nude mice (age 8–10 weeks) were anesthetized and injected with 2x106 cells of NRASQ61L (2/14, 2/17) or BRAFV600E (2/21, 2/33) clones, suspended in 100 μl sterile PBS, into the left ventricle of the heart by nonsurgical means. The spontaneous, pulsatile entrance of bright red oxygenated blood into the transparent needle hub indicated proper positioning of the needle. After 5 weeks, the mice were sacrificed; selected organs were excised from the mice at necropsy and were preserved in 10% formalin solution for subsequent scoring of metastasis. NRASQ61L (2/14, 2/17) and NRAS/Sema6A cells (2/14, and 2/17) were injected intracardially as above described, and after 7 weeks the mice were sacrificed and the organs were processed and analyzed as described above. Box plot of in vivo metastasis analysis was obtained by software Prism version 6.0.
Gene expression profiles
Gene expression profiles of BRAFV600E and NRASQ61R melanoma clones isolated from 665/2 cell line were assessed as described [44].Three biological replicates of each clone were analyzed. Single-color hybridization of RNAs was performed on Illumina Bead Chip HumanHT-12_v4 Microarrays (Illumina) containing more than 48,000 transcript probes. The expression profiles have been deposited in NCBI’s Gene Expression Omnibus (GEO) with GSE accession number GSE58199. Background correction, filtering of data, and quantile normalization were done with BeadStudio Illumina software. Analysis for differentially expressed genes was carried out by BRB-array Tools (Vers.4.3.0) software. Class comparison was carried out by a random-variance F-test with a nominal significance level of 0.001. Permutation P values for significant genes were computed based on 10,000 random permutations.
Quantitative RT-PCR
Total RNA from NRASQ61R and BRAFV600E clones was prepared using TRIzol® (Ambion). First-strand cDNA was synthesized with the M-MLV RT kit (Invitrogen, Glasgow, UK). Human tissue samples were obtained from the Regina Elena National Cancer Institute, after approval by the institutional ethic committee. Total RNA, derived from nevi, BRAFV600E and wild type (WT) melanoma, was isolated by PureLink FFPE kit (Invitrogen), and reverse-transcribed using PrimeScript RT reagent kit (Takara). Quantitative PCR (qPCR) was performed using SYBR Green on an ABI Prism 7500 apparatus (Applied Biosystems, Glasgow, UK) in 2 independent experiments in triplicate. The comparative threshold (∆Ct) method was used.
Primer sequences were:
SEMA6A-Fw-5’ACAATTCCTTTGTGGCACTGAA, Rev5’TCTTGAGCCGTGGAATCTGA
MICAL1-Fw-5’ATGGGCAGCCTGATGTCTCT, Rev5’GGCGCCATGCTTCTCTTG
PLEXINA4-Fw-5’TGGCTCAGGCGACCC, Rev5’GACGGAGATATTGTTGGGATG
HER3-Fw5’GCAGGATTGGTAGTGATTTTCATG,
Rev-5’TATCGCCTCATAGCCCTTTTATTC
GAPDH-Fw-5’TCCCTGAGCTGAACGGGAAG, Rev5’-GGAGGAGTGGGTGTCGCTGT
Immunohistochemistry
Formalin-fixed paraffin-embedded sections from melanoma lymph node metastases of patients treated at the Regina Elena National Cancer Institute (53 BRAFV600E melanomas, 43 BRAF WT, and 8 nevi) were analyzed as described [42]. Antigen retrieval was performed at 96°C (10 mM/L citrate buffer, pH 6) for 20 minutes. Sections were incubated with the primary antibody anti-Mical-1 1:100 (Sigma Prestige), anti-SEMA6A 1:50 (Sigma Prestige) for 30 minutes at room temperature. Immunoreactions were revealed by Bond Polymer Refine Detection Kit according to manufacturer’s procedure (Leica Biosystems) in an automated autostainer Bond III Leica Biosystems. Diaminobenzidine was used as chromogenic substrate. Microscope Nikon ECLIPSE 55i with digital camera HESP Tecnology was used. Scale bars 30 µm. The study was reviewed and approved by the ethical committee of Regina Elena National Cancer Institute, and informed consent was obtained from all patients.
Cell adhesion, Immunofluorescence, Invasion and chemotaxis assays
siSema6A, and siScr/BRAFV600E cells, 24 hours post-transfection were plated onto fibronectin (FN)-coated dishes (20 µg/ml, SIGMA-Aldrich), or culture dishes or poly-l lysine coated slides and 24 hours later analyzed by immunofluorescence. The cells incubated with Phalloidin-Tetramethylrhodamine B isothiocyanate (TRITC) were counterstained with Hoechst to highlight nuclei (SIGMA-Aldrich). For Tunel assay, siSema6A, and siScr/BRAFV600E cells, 24 hours post-transfection were plated onto FN-coated or poly-l lysine coated slides, as above described. 48 hours post-transfection TUNEL assay was performed following manufacturer’s instructions (Millipore). Microscope OLYMPUS BX53 was used to evaluate fluorescence and Tunel. Scale bars 20 µm. NRAS/Sema6A cells chemoinvasion was assessed as described [49] by overexpression of pUSENMC-Sema6A expression vector, kindly provided by Silvia Prislei (University Cattolica, Rome) [50]. Chemoinvasion or chemotaxis assays after depletion of Sema6A in BRAF and NRAS mutant cells, were performed as previously described [49]. Each assay was carried out in quadruplicate and repeated at least three times.
Soft agar assay
siSema6A, siScr/BRAFV600E, and Sema6A/NRASQ61R cells were transfected and 48 hours later cells (5×103) were plated onto 0.4% agar. After 2 weeks viable colonies were counted [42] by the software ImageJ 1.47v (NHI, USA).
Cell death, apoptosis, and caspase activity
siMical1-, siSema6A-, and siScr/BRAFV600E cells were plated at concentration of 4 × 105. After 30, and 36 hours cell vitality was evaluated by Trypan blue exclusion. Apoptosis was assessed by staining with FITC-conjugated Annexin-V and Propidium Iodide (BD Pharmingen, BD Biosciences San Diego, CA). The samples were acquired by FACSCalibur flow cytometer (BD). Enzymatic activity of caspases-3/7 was measured with Muse™ Caspase-3/7 Assay Kit and read by Muse™ Cell Analyzer (Millipore).
Statistical analysis
Data were reported as mean and standard deviation . Differences were considered statistically significant when P≤0.05. All analyses were performed using SPSS, version 17.0 (SPSS, Chicago, Illinois). Student T test was performed for the comparison of results from qRT-PCR and from all other different test (*P<0.05, **P<0.001, ***P<0.0001).
Disclosure of Potential Conflict of Interest
No potential conflicts of interest were disclosed
Acknoledgements
The authors thank M. Fanciulli, P. Zizza, S. Giordano, and L. Tamagnone for critical discussion and suggestion, E. Gaucci for technical support, and S. Bacchetti for revision of the manuscript.
Grant Support
R.F. is supported by: New Idea Award (Ministero della Salute), Filas, and Italian Association for Cancer Research (AIRC) 5x1000 (SPMCO 9979); R.M. is supported by AIRC (IG 12020).
Conflict of interest
No potential conflicts of interest were disclosed
REFERENCES
1. Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007; 445: 851-7.
2. Chapman PB, Hauschild A, Robert C, Haanen JB Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutations. N Engl J Med. 2011; 364: 2507-16.
3. Menzies AM, Long GV. Dabrafenib and Trametinib, Alone and in Combination for BRAF-Mutant Metastatic Melanoma. Clin Cancer Res. 2014; 20: 2035-43.
4. Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, Chodon T, Guo R, Johnson DB, Dahlman KB, Kelley MC, Kefford RF, Chmielowski B, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitory therapy. Cancer Discov. 2014; 4: 80-93.
5. Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, Chodon T, Nelson SF, McArthur G, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS up-regulation. Nature. 2010; 468: 973-7.
6. Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, Shi H, Arefi M, Titz B, Gabay MT, Salton M, Dahlman KB, Tadi M, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature. 2011; 480: 387-90.
7. Wangle N, Van Allen EM, Treacy DJ, Frederick DT, Cooper ZA, Taylor-Weiner A, Rosenberg M, Goetz EM, Sullivan RJ, Farlow DN, Friedrich DC, Anderka K, Perrin D, et al. Map kinase pathway alteration in BRAF-mutant melanoma patient with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 2014; 4: 61-80.
8. Shi H, Hong A, Kong X, Koya RC, Song C, Moriceau G, Hugo W, Yu CC, Ng C, Chodon T, Scolyer RA, Kefford RF, Ribas A, et al. A novel AKT1 mutant amplifies an adaptive melanoma response to BRAF inhibition. Cancer Discov. 2014; 4: 69-79.
9. Van Allen EM, Wagle M, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, Place CS, Taylor-Weiner A, Whittaker S, Kryukov GV, Hodis E, Rosenberg M, McKenna A, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014; 4: 94-109.
10. Reschke M, Mihic-Probst D, vad der Horst EH, Knyazev P, Wild PJ, Hutterer M, Meyer S, Dummer R, Moch H, Ullrich A. HER3 is a determinant for poor prognosis in melanoma. Clin Cancer Res. 2008; 14: 5188-97.
11. Chakrabarty A, Sánchez V, Kuba MG, Rinehart C, Arteaga CL. Feedback upregulation of HER3 (ErbB3) expression and activity attenuates antitumor effect of PI3K inhibitor. Proc Natl Acad Sci U S A. 2012; 109: 2718-23.
12. Greaves WO, Verma S, Patel KB, Davies MA, Barkoh BA, Galbincea JM, Yao H, Lazar AJ, Aldape KD, Medeiros LJ, Luthra R. Frequency and spectrum of BRAF mutations in a retrospective, single-institution study of 1112 cases of melanoma. J Mol Diagn. 2013; 15: 220-6.
13. Mann GJ, Pupo GM, Campain AE, Carter CD, Schramm SJ, Pianova S, Gerega SK, De Silva C, Lai K, Wilmott JS, Synnott M, Hersey P, Kefford RF. BRAF mutation, NRAS mutation, and the absence of an immune-related expressed gene profile predict poor outcome in patients with stage III melanoma. J Invst Dermatol. 2013; 133: 509-17.
14. Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, Cho KH, Aiba S, Bröcker EB, LeBoit PE, Pinkel D, Bastian BC. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005; 353: 2135–47.
15. Bloethner S, Chen B, Hemminki K, Müller-Berghaus J, Ugurel S, Schadendorf D, Kumar R. Effect of common B-RAF and N-RAS mutations on global gene expression in melanoma cell lines. Carcinogenesis. 2005; 26: 1224-32.
16. Pavey S, Johansson P, Packer L, Taylor J, Stark M, Pollock PM, Walker GJ, Boyle GM, Harper U, Cozzi SJ, Hansen K, Yudt L, Schmidt C. Microarray expression profiling in melanoma reveals a BRAF mutations signature. Oncogene. 2004; 23: 4060-7.
17. Hoek KS, Schlegel NC, Brafford P, Sucker A, Ugurel S, Kumar R, Weber BL, Nathanson KL, Phillips DJ, Herlyn M, Schadendorf D, Dummer R. Metastatic potential of melanomas defined by specific gene expression profiles with no BRAF signature. Pigment Cell Res. 2006; 19: 290-302.
18. Johansson P, Pavey S, Hayward N. Confirmation of a BRAF mutation-associated gene expression signature in melanoma. Pigment Cell Res. 2007; 20: 216-21.
19. Ellerhorst JA, Greene VR, Ekmekcioglu S, Warneke CL, Johnson MM, Cooke CP, Wang LF, Prieto VG, Gershenwald JE, Wei Q, Grimm EA. Clinical correlation of NRAS and BRAF mutations in primary human melanoma. Clin Cancer Res. 2011; 17: 229-35.
20. Sensi M, Nicolini G, Petti C, Bersani I, Lozupone F, Molla A, Vegetti C, Nonaka D, Mortarini R, Parmiani G, Fais S, Anichini A. Mutually exclusive NRASQ61R and BRAFV600E mutations at the single-cell level in the same human melanoma. Oncogene. 2006; 25: 3357-64.
21. Neufeld G, Kessler O. The semaphorins: versatile regulators of tumour progression and tumour angiogenesis. Nat Rev Cancer. 2008; 8: 632-45.
22. Segarra M, Ohnuki H, Maric D, Salvucci O, Hou X, Kumar A, Li X, Tosato G. Semaphorin 6A regulates angiogenesis by modulating VEGF signaling. Blood. 2012; 120: 4105-15.
23. Renaud J, Kerjan G, Sumita I, Zagar Y, Georget V, Kim D, Fouquet C, Suda K, Sanbo M, Suto F, Ackerman SL, Mitchell KJ, FujisawaH, et al. Plexin-A2 and its ligand, Sema6A, control nucleus-centrosome coupling in migrating granule cells. Net Neurosci. 2008; 11: 440-9.
24. Cabodi S, del Pilar Camacho-Leal M, Di Stefano P, Defilippi P. Integrin signalling adaptors: not only figurants in the cancer story. Nat Rev Cancer. 2010; 10: 858-70.
25. Terman JR, Mao T, Pastercamp RJ, Yu HH, Kolodkin AL. MICALs, a family of conserved flavoprotein oxidoreductases, function in plexin-mediated axonal repulsion. Cell. 2002; 109: 887-900.
26. Suzuki T, Nakamoto T, Ogawa S, Seo S, Matsumura T, Tachibana K, Morimoto C, Hirai H. MICAL, a novel CasL interacting molecule, associates with vimentin. J Biol Chem. 2002; 277: 14933-41.
27. Giordano S, Corso S, Conrotto P, Artigiani S, Gilestro G, Barberis D, Tamagnone L, Comoglio PM. The semaphorin 4D receptor controls invasive growth by coupling Met. Nat Cell Biol. 2002; 4: 720-4.
28. Luchino J, Hocine M, Amoureux MC, Gilbert B, Bernet A, Royet A, Treilleux I, Lécine P, Borg JP, Mehlen P, Chauvet S, Mann F. Semaphorin 3A suppress tumor cell death triggered by the Plexin D1 dependence receptor in metastatic breast cancers. Cancer Cell. 2013; 24: 673-85.
29. Zhou Y, Adolfs Y, Pijnappel WW, Fuller SJ, Van der Schors RC, Li KV, Sugden PH, Smit AB, Hergovich A, Pasterkamp RJ. MICAL-1 is a negative regulator of MST-NDR kinase signaling and apoptosis. Mol Cell Biol. 2011; 31: 3603-15.
30. Wallasch C, Weiss FU, Nierderfellner G, Jallal B, Issing W, Ullrich A. Heregulin-dependent regulation of HER2/neu oncogenic signaling by heterodimerization with HER3. EMBO J. 1995; 14: 4267-75.
31. Folgiero V, Bacheleder RE. Bon G, Sacchi A, Falcioni R, Mercurio AM. The α6β4 can regulate ErbB-3 expression: implications for α6β4 signaling and function. Cancer Res. 2007; 67: 1645-52.
32. Balakrishnan A, Penachioni JY, Lamba S, Bleeker FE, Zanon C, Rodolfo M, Vallacchi V, Scarpa A, Felicioni L, Buck M, Marchetti A, Comoglio PM, Bardelli A, et al. Molecular profiling of the ‘Plexisome’ in melanoma and pancreatic cancer. Human Mutat. 2009; 30: 1167-1174.
33. Suzuki T, Nakamoto T, Ogawa S, Seo S, Matsumura T, Tachibana K, Morimoto C, Hirai H. MICAL, a novel CasL interacting molecule, associates with vimentin. J Biol Chem. 2002; 277: 14933-41.
34. Hung RJ, Terman JR. Extracellular inhibitors, repellents, and semaphorin/plexin/Mical-mediated actin filament disassembly. Cell. 2011; 68: 415-33.
35. Serirni G, Valdembri B, Zanivan S, Morterra G, Burkhardi C, Caccavari F, Zammataro R, Primo L, Tamagnone L, Logan M, Tessier-Lavigne M, Taniguchi M, Puschel A, et al. Class 3 semaphorins control vascular morphogenesis by inhibiting integrin function. Nature. 2003; 424: 391-97.
36. Banu N, Teichman J, Dunlap-Brown M, Villegas G, Tufro A. Semaphorin 3C regulates endothelial cell function by increasing integrin activity. Faseb J. 2006; 20: 2150-52.
37. Prud’homme G, Glinka Y. Neuropilins are multifunctional coreceptors involved in tumor initiation, growth, metastasis and immunity. Oncotarget. 2012; 3: 921-939.
38. Giuffreda L, Del Bufalo D, Desideri M, Di Sanza C, Stoppacciaro A, Ricciardi MR, Chiaretti S, Tavolaro S, Benassi B, Bellacosa A, Foà R, Tafuri A, Cognetti F, et al. Therapeutic potential of PD0325901 in Melanoma. Neoplasia. 2009; 11: 720-31.
39. Suto F, Tsuboi M, Kamiya H, Mizuno H, Kiyama Y, Komai S, Shimizu M, Sanbo M, Yagi T, Hiromi Y, Chédotal A, Mitchell KJ, Manabe T, et al. Interactions between plexin-A2, plexin-A4, and semaphorin 6A control lamina-restricted projection of hippocampal mossy fibers. Neuron. 2007; 53: 535-47.
40. Huber AB, Kolodkin AL, Ginty DD, Cloutier JF. Signaling at the growth cone: ligand-receptor complexes and the control of axon growth and guidance. Ann Rev Neurosci. 2003; 26: 509-62.
41. Tamagnone L. Emerging role of semaphorins as major regulatory signals and potential therapeutic targets in cancer. Cancer Cell. 2012; 22: 145-152.
42. Worzfeld T, Offermann S. Semaphorins and plexins as therapeutic targets. Nature Rev. 2014; 13: 603-621.
43. Daniotti M, Oggionni M, Ranzani T, Vallacchi V, Campi V, Di Stasi D, Torre GD, Perrone F, Luoni C, Suardi S, Frattini M, Pilotti S, Anichini A et al. BRAF alterations are associated with complex mutational profiles in malignant melanoma. Oncogene. 2004; 23: 5968-77.
44. Tassi E, Zanon M, Vegetti C, Molla A, Bersani I, Perotti V, Pennati M, Zaffaroni N, Milella M, Ferrone S, Carlo-Stella C, Gianni AM, Mortarini M, et al. Role of Apollon in human melanoma resistance to antitumor agents that activate the intrinsic or the extrinsic apoptosis pathways. Clin Cancer Res. 2012; 18: 3316-27.
45. Perotti V, Baldassari P, Bersani I, Molla A, Vegetti C, Tassi E, Dal Col J, Dolcetti R, Anichini A, Mortarini R. NFATc2 is a potential therapeutic target in human melanoma. J Invest Dermatol. 2012; 132: 2652-60
46. Anichini A, Mazzocchi A, Fossati G, Parmiani G. Cytotoxic T lymphocyte clones from peripheral blood and from tumor site detect intratumor heterogeneity of melanoma cells. Analysis of specificity and mechanims of interaction. J Immunol. 1989; 142: 3692-701.
47. Anichini A, Mortarini R, Supino R, Parmiani G. Human melanoma cells with high susceptibility to cell-mediated lysis can be identified on the basis of ICAM-1 phenotype, VLA profile and invasive ability. Int J Cancer 1990; 46: 508-15.
48. Castelli C, Sensi M, Lupetti R, Mortarini R, Panceri P, Anichini A, Parmiani G. Expression of interleukin 1 alpha, interleukin 6, and tumor necrosis factor alpha genes in human melanoma clones is associated with that of mutated N-RAS oncogene. Cancer Res 1994; 54: 4785-90.
49. Folgiero V, Avetrani A, Bon G, Di Carlo SE, Fabi A, Nisticò C, Vici P, Melucci E, Buglioni S, Perracchio L. Sperduti I, Rosanò L, Sacchi A, et al. Induction of ErbB-3 Expression by α6β4 Integrin Contributes to Tamoxifen Resistance in ERβ1-Negative Breast Carcinomas. PLoS ONE. 2008; 3: e1592.
50. Prislei S, Mozzetti T, Filippetti F, De Donato M, Raspaglio G, Cicchillitti L, Scambia G, Ferlini C. From plasma membrane to cytoskeleton: a novel function for Semaphorine 6A. Mol Cancer Ther. 2008; 7: 233-41.