
INTRODUCTION
Altered glucose metabolism is a key feature that distinguishes cancer cells from normal cells. Most cancer cells consume higher amounts of glucose and subsequently produce much more lactate than normal cells, even in the presence of ample O2. This phenomenon is known as the Warburg effect [1]. Since Otto Warburg made this important observation in 1924 [2], many researchers have attempted to elucidate the underlying molecular mechanisms in cancer cells. Hypoxia-inducible factor 1 (HIF-1), Myc, p53, Ras, Akt, Src, pyruvate kinase (PK) M2, and lactate dehydrogenase A (LDHA) have been implicated in the Warburg effect [3-8]. We recently discovered that the glycolytic enzyme PKM2 promotes the Warburg effect by serving as a transcriptional coactivator for HIF-1 in cancer cells [9]. This research perspective will discuss these recent findings regarding physical and functional interactions of HIF-1 and PKM2.
HIF-1 AND METABOLIC REPROGRAMMING IN CANCER CELLS
HIF-1 is a heterodimeric transcription factor, consisting of an O2-regulated HIF-1α subunit and a constitutively expressed HIF-1β subunit [10, 11]. HIF-1 is a master regulator of transcriptional responses to reduced O2 availability (hypoxia), which is found in the majority of advanced human cancers [12, 13]. In well-oxygenated human cells, HIF-1α is hydroxylated at proline-402 and/or proline-564 by the prolyl hydroxylase domain proteins, PHD1-3 [14]. PHD2 is primarily responsible for regulating basal HIF-1α levels in cancer cells [15]. Prolyl-hydroxylated HIF-1α is bound by the von Hippel-Lindau (VHL) tumor suppressor protein, which is the substrate recognition component of an E3 ubiquitin-ligase complex, leading to HIF-1α protein degradation by the 26S proteasome [16]. Under hypoxic conditions, HIF-1α prolyl hydroxylation is inhibited, thereby stabilizing HIF-1α protein [17]. HIF-1α protein levels are also increased in normoxic cancer cells with loss of function of certain tumor suppressors, most notably VHL in the clear cell type of renal cell carcinoma [16, 18, 19]. HIF-2α is a paralog of HIF-1α that is also O2-regulated, dimerizes with HIF-1β, and transactivates a group of target genes that partially overlaps the battery of genes regulated by HIF-1 [20, 21].
Activation of HIF-1 commonly occurs in many cancer types and is a driving force regulating many steps in cancer progression [18, 22]. HIF-1 activates the transcription of genes encoding proteins that mediate angiogenesis, invasion, metastasis, and the shift from oxidative to glycolytic metabolism [12, 18, 19, 22, 23]. By activating the transcription of genes encoding glucose transporters and glycolytic enzymes, HIF-1 enhances glucose uptake and glycolysis in cells [23-27]. HIF-1 also controls expression of LDHA and pyruvate dehydrogenase kinase 1 (PDK1) [25, 26, 28, 29]. LDHA catalyzes the conversion of pyruvate to lactate (Figure 1), thereby decreasing mitochondrial utilization of pyruvate as a substrate for pyruvate dehydrogenase (PDH), which converts pyruvate to acetyl coenzyme A (AcCoA). PDK1 phosphorylates the catalytic subunit of PDH, leading to its inactivation, which shunts pyruvate away from the mitochondria. HIF-1 activation shifts the balance of metabolism from oxidative phosphorylation toward glycolysis and mediates the Warburg effect in VHL-null renal carcinoma cells [30].
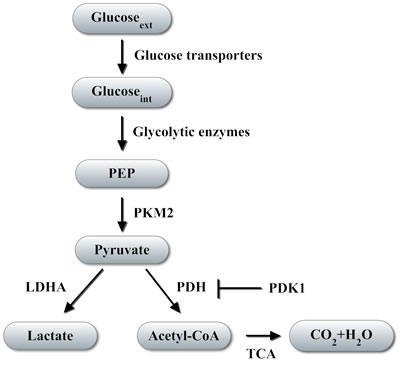
Figure 1: Regulation of glucose metabolism by HIF-1. HIF-1 controls transcription of genes encoding glucose transporters, which transport glucose from the extracellular (ext) to the intracellular (int) milieu, and glycolytic enzymes, which convert glucose to lactate as glycolytic end-product or acetyl coenzyme A (Acetyl-CoA) that is metabolized in the tricarboxylic acid cycle (TCA). Pyruvate kinase M2 converts phosphoenolpyruvate (PEP) into pyruvate, which is upstream of the decision point for glycolytic vs oxidative metabolism. Arrow indicates direction of glucose metabolism; blocked line indicates inhibition.
REGULATION OF PKM2 EXPRESSION IN CANCER CELLS
PK catalyzes the conversion of phosphoenolpyruvate to pyruvate (Figure 1) and is composed of M1-/M2-type and L-/R-type isoforms, which are encoded by PKM2 and PKLR genes, respectively [31]. Tissue-specific promoters control expression of PKL, which is expressed in liver and kidney, and PKR, which is expressed in erythrocytes. PKM1 and PKM2 are the alternatively spliced products of the PKM2 primary RNA transcript with PKM1 and PKM2 mRNA containing sequences encoded by exon 9 or exon 10, respectively [32]. PKM1 is expressed in muscle and brain, whereas PKM2 is expressed in the embryo and in cancer cells. The transcription factors Sp1 and Sp3 bind to a GC-rich element in the promoter of the human PKM2 gene [33]. Sp1 constitutively activates transcription of PKM2, whereas Sp3 functions as a transcriptional repressor that dissociates from the PKM2 gene under hypoxic conditions.
We recently identified a hypoxia response element (HRE) within the first intron of the human PKM2 gene [9]. Heterodimer complexes of HIF-1β with HIF-1α, but not HIF-2α, bound to the PKM2 HRE and increased the activity of a luciferase reporter gene driven by the PKM2 HRE in hypoxic HeLa cells. Mutation of the HIF-1 binding site in the PKM2 HRE or knockdown of HIF-1α protein expression suppressed reporter gene activity. Hypoxia induced the expression of PKM1 and PKM2 mRNA in wild-type, but not in HIF-1α-knockout, mouse embryo fibroblasts [9].
PKM2 expression in cancer cells is also regulated by microRNAs (miRs). miR-326 matches two regions in the 3’-untranslated region (UTR) of PKM2 mRNA and transfection of miR-326 precursor decreased PKM2 3’-UTR-luciferase reporter activity and PKM2 protein levels in glioma cells [34]. miR-133a and miR-133b are also implicated in PKM2 expression. PKM2 overexpression is associated with downregulation of miR-133a and miR-133b, whereas transfection of miR-133a or miR-133b precursors inhibited PKM2 expression in tongue squamous cell carcinoma cells [35]. The significance of this mutual antagonism between miR-133a/b and PKM2 has not been determined.
Recent studies revealed the molecular mechanism underlying PKM2 mRNA splicing. Heterogeneous nuclear ribonucleoproteins (hnRNP) I, A1, and A2 bind to RNA sequences encoded by exon 9 and inhibit PKM1-specific mRNA splicing [36, 37]. The c-Myc oncoprotein regulates transcription of hnRNPI, hnRNPA1 and hnRNPA2, resulting in preferential PKM2 isoform expression in cancer cells overexpressing c-Myc [36]. Mammalian target of rapamycin (mTOR), a serine/threonine protein kinase that regulates cell growth, cell survival, and protein synthesis, also stimulates PKM2 expression through activation of HIF-1 and c-Myc [38]. Thus, activation of transcription factors and kinases, and downregulation of microRNAs results in high expression of PKM2 in cancer cells. However, analysis of human tumor and non-tumor tissues from kidney, liver, lung, and thyroid using an absolute quantification approach by mass spectrometry revealed that PKM2 protein expression is predominant in both human tumor tissues and tissue-matched normal controls, suggesting that no switch from PKM1 to PKM2 is required for tumor development [39]. These findings challenge the conclusion, which was based on the analysis of cancer cell lines, that PKM2 overexpression is a hallmark of cancer cells [3]. The proteomic study found that total PKM expression (PKM1 + PKM2) is increased 3-fold in tumor tissue compared to normal tissue [39]. HIF-1α is overexpressed in solid tumors due to intratumoral hypoxia, genetic alterations, or both [12, 18, 22, 23], and thus may be a predominant regulator that contributes to elevated levels of PKM2 in human tumor tissues.
PKM2 AND THE WARBURG EFFECT IN CANCER CELLS
Christofk et al. demonstrated that PKM2 expression was associated with increased glucose uptake and lactate production, but decreased O2 consumption in cancer cells [3]. Genetic manipulation of cancer cells that switched them from PKM2 to PKM1 expression reversed the Warburg effect, suggesting that high expression of PKM2 is required for aerobic glycolysis in cancer cells. Moreover, expression of PKM2, but not PKM1, induced tumor xenograft growth in nude mice [3]. The binding of tyrosine-phosphorylated peptides to PKM2 at lysine-433 was found to inhibit PKM2 enzymatic activity through release of the allosteric activator fructose-1,6-bisphosphate (FBP) and to promote cell growth and glycolytic metabolism in cancer cells [40]. Tyrosine kinases play critical roles in cell growth, cell metabolism, and angiogenesis in cancer [41, 42]. Hitosugi et al. reported that fibroblast growth factor receptor type 1 (FGFR1) phosphorylated PKM2 at tyrosine residues-83, -105, -148, -175, -370, and -390 in murine Ba/F3 hematopoietic cells [43]. Phosphorylation of PKM2 at tyrosine-105 induced FBP release from active tetrameric PKM2, promoted formation of less active dimeric PKM2, and subsequently decreased PKM2 enzymatic activity. In contrast, the phosphorylation of PKM2 at other tyrosine residues caused by FGFR1 failed to regulate PKM2 activity. However, it remained unclear how alterations in PKM2 activity could determine whether the product of the PKM2 reaction, pyruvate, was converted to lactate or to AcCoA (Figure 1).
We recently delineated a molecular mechanism by which PKM2 mediates the Warburg effect in cancer cells [9]. PKM2 was found to interact with HIF-1α in the nucleus and to function as a transcriptional coactivator in HeLa cervical carcinoma and Hep3B hepatoblastoma cells. PKM2 increased HIF-1 binding to HREs at target genes, recruitment of coactivator p300, histone acetylation, and subsequent transactivation of HIF-1 target genes including SLC2A1 (which encodes glucose transporter 1), LDHA, and PDK1 in HeLa and Hep3B cells. PKM2-stimulated expression of HIF-1 target genes promotes the shift from oxidative phosphorylation to glycolytic metabolism. PKM2 also binds to HIF-2α and promotes HIF-2-mediated transactivation in cancer cells [9]. In addition to its effects on genes encoding metabolic enzymes, PKM2 stimulates HIF-1- and HIF-2-mediated expression of the VEGF gene (which encodes vascular endothelial growth factor), thereby promoting angiogenesis. Thus, PKM2 may play a far broader role in promoting cancer progression than was previously appreciated (Figure 2).
Figure 2: PKM2 contributes to metabolic reprogramming and cancer progression by serving as a PHD3-dependent coactivator for HIF-1. Prolyl hydroxylation of PKM2 by PHD3 promotes the interaction of PKM2 with HIF-1α, thereby stabilizing HIF-1 binding to the HRE of target genes, recruitment of coactivator p300, histone acetylation, and subsequent transcription of HIF-1 target genes, which encode proteins that are involved in metabolic reprogramming, angiogenesis, and many other critical aspects of cancer progression.
The enzymatic activity of PKM2 is not required for HIF-1 transactivation [9]. Interestingly, PKM2 is prolyl hydroxylated in the PKM2-specific domain encoded by exon 10, and prolyl hydroxylation of PKM2 is required for HIF-1-mediated transactivation in cancer cells. PHD3 catalyzes hydroxylation of PKM2 and PHD3 knockdown reduced expression of the HIF-1 target genes SLC2A1, LDHA, and PDK1, and reversed the Warburg effect in VHL-null RCC4 renal carcinoma cells [9]. PKM2, but not PKM1, is prolyl hydroxylated and PKM2, but not PKM1, interacts with HIF-1α, thus providing a molecular basis for the selective effect of PKM2 on HIF-1-mediated transactivation and the Warburg effect in cancer cells [9].
Recently, the interaction of PHD3 with PKM2 was reported to increase the formation of dimeric PKM2, which has decreasd activity compared to the tetrameric form of the enzyme [44]. Chen et al. concluded that the hydroxylase activity of PHD3 was not required for its effect on PKM2 oligomerization/enzyme activity because mutant PHD3 (R205K) behaved similarly to wild-type PHD3. However, we found that mutation of arginine-205 alone was not sufficient to inactivate the hydroxylase activity of PHD3 (W.L. and G.L.S., unpublished) and thus it would be interesting to repeat these experiments using the PHD3 (H135A/D137A) mutant that lacks hydroxylase activity [9]. PHD3 is encoded by a HIF-1 target gene and increased PHD3 mRNA and protein expression is induced by HIF-1 under hypoxic conditions [45], which compensates for the reduced hydroxylase activity [9]. Thus, PHD3 and PKM2 exert a positive feedback loop in cancer cells that amplifies HIF-1 activity, which may play a major role in driving metabolic reprogramming, angiogenesis, and other critical aspects of cancer progression (Figure 3).
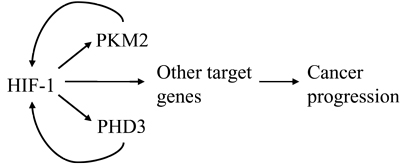
Figure 3: PKM2 and PHD3 are components of a positive feedback loop that amplifies HIF-1 transcriptional activity. HIF-1 activates transcription of genes encoding PKM2 and PHD3, which interact with HIF-1α to stimulate transactivation of HIF-1 target genes that promote cancer progression.
UNANSWERED QUESTIONS, FUTURE DIRECTIONS
Does PKM2 have other functions in the nucleus that promote cancer progression? PKM2 also binds to the transcription factor Oct-4 and enhances Oct-4-dependent gene transcription [46]. Oct-4 is a key mediator of pluripotency in embryonic stem cells [47] and induced pluripotent stem cells [48]. Oct-4 is also expressed in human breast cancer stem cells [49]. Hoshino et al. also found that nuclear translocation of PKM2 is induced by interleukin-3 and stimulates cell proliferation [50], although the nuclear target of PKM2 was not identified. It is likely that the stimulation of cell proliferation by PKM2 is independent of its regulation of HIF-1α transactivation.
Post-translational modification by prolyl hydroxylation and tyrosine phosphorylation regulate PKM2 activity as a transcriptional coactivator and glycolytic enzyme, respectively. PKM2 is also subjected to sumoylation [51] and lysine acetylation [52] and further studies are required to determine whether these post-translational modifications also regulate the role of PKM2 as a HIF-1 coactivator.
Several compounds have been shown to inhibit PKM2 enzymatic activity [53, 54]. However, those inhibitors may not suppress HIF-1 transactivation in cancer cells because the enzymatic activity of PKM2 is not required for its coactivator functions [9]. Combination therapy with HIF inhibitors [12, 18, 19] may prove to be more efficacious.
ACKNOWLEDGMENTS
We thank Ryan Chang for assistance with figure preparation. This work was supported, in part, by contracts N01-HV28180 and HHS-N268201000032C from NHLBI and an International Cooperative Research Project award (ERATO-ICORP Gas Biology Project) from the Japan Science and Technology Agency. G.L.S. is the C. Michael Armstrong Professor at the Johns Hopkins University School of Medicine.
REFERENCES
1. Warburg O. On the origin of cancer cells. Science. 1956; 123:309-314.
2. Warburg O, Posener K, Negelein E. Über den stoffwechsel der carcinomzelle. Biochem Zeitschr. 1924; 152:309-344.
3. Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008; 452:230-233.
4. Semenza GL. HIF-1 mediates the Warburg effect in clear cell renal carcinoma. J Bioenerg Biomembr. 2007; 39:231-234.
5. Dang CV, Kim JW, Gao P, Yustein J. The interplay between MYC and HIF in cancer. Nat Rev Cancer. 2008; 8:51-56.
6. Hsu PP, Sabatini DM. Cancer cell metabolism: Warburg and beyond. Cell. 2008; 134:703-707.
7. Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A. 2010; 107:2037-2042.
8. Levine AJ, Puzio-Kuter AM. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science. 2010; 330:1340-1344.
9. Luo W, Hu H, Chang R, Zhong J, Knabel M, O’Meally R, Cole RN, Pandey A, Semenza GL. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. 2011; 145:732-744.
10. Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992; 12:5447-5454.
11. Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995; 92:5510-5514.
12. Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003; 3:721-732.
13. Vaupel P, Mayer A, Hockel M. Tumor hypoxia and malignant progression. Methods Enzymol. 2004; 381:335-354.
14. Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian YM, Masson N, Hamilton DL, Jaakkola P, Barstead R, Hodgkin J et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001; 107:43-54.
15. Berra E, Benizri E, Ginouves A, Volmat V, Roux D, Pouyssegur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1α in normoxia. EMBO J. 2003; 22:4082-4090.
16. Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999; 399:271-275.
17. Kaelin WG, Jr., Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008; 30:393-402.
18. Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene. 2010; 29:625-634.
19. Melillo G. Targeting hypoxia cell signaling for cancer therapy. Cancer Metastasis Rev. 2007; 26:341-352.
20. Ema M, Taya S, Yokotani N, Sogawa K, Matsuda Y, Fujii-Kuriyama Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1α regulates the VEGF expression and is potentially involved in lung and vascular development. Proc Natl Acad Sci U S A. 1997; 94:4273-4278.
21. Löfstedt T, Fredlund E, Holmquist-Mengelbier L, Pietras A, Ovenberger M, Poellinger L, Påhlman S. Hypoxia inducible factor-2α in cancer. Cell Cycle. 2007; 6:919-926.
22. Harris AL. Hypoxia--a key regulatory factor in tumor growth. Nat Rev Cancer. 2002; 2:38-47.
23. Semenza GL. HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev. 2010; 20:51-56.
24. Semenza GL, Roth PH, Fang HM, Wang GL. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J Biol Chem. 1994; 269:23757-23763.
25. Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, Giallongo A. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem. 1996; 271:32529-32537.
26. Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY, Semenza GL. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1α. Genes Dev. 1998; 12:149-162.
27. Seagroves TN, Ryan HE, Lu H, Wouters BG, Knapp M, Thibault P, Laderoute K, Johnson RS. Transcription factor HIF-1 is a necessary mediator of the pasteur effect in mammalian cells. Mol Cell Biol. 2001; 21:3436-3444.
28. Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006; 3:187-197.
29. Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006; 3:177-185.
30. Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI, Dang CV, Semenza GL. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell. 2007; 11:407-420.
31. Mazurek S, Boschek CB, Hugo F, Eigenbrodt E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin Cancer Biol. 2005; 15:300-308.
32. Noguchi T, Inoue H, Tanaka T. The M1- and M2-type isozymes of rat pyruvate kinase are produced from the same gene by alternative RNA splicing. J Biol Chem. 1986; 261:13807-13812.
33. Discher DJ, Bishopric NH, Wu X, Peterson CA, Webster KA. Hypoxia regulates β-enolase and pyruvate kinase-M promoters by modulating Sp1/Sp3 binding to a conserved GC element. J Biol Chem. 1998; 273:26087-26093.
34. Kefas B, Comeau L, Erdle N, Montgomery E, Amos S, Purow B. Pyruvate kinase M2 is a target of the tumor-suppressive microRNA-326 and regulates the survival of glioma cells. Neuro Oncol. 2010; 12:1102-1112.
35. Wong TS, Liu XB, Chung-Wai Ho A, Po-Wing Yuen A, Wai-Man Ng R, Ignace Wei W. Identification of pyruvate kinase type M2 as potential oncoprotein in squamous cell carcinoma of tongue through microRNA profiling. Int J Cancer. 2008; 123:251-257.
36. David CJ, Chen M, Assanah M, Canoll P, Manley JL. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature. 2010; 463:364-368.
37. Clower CV, Chatterjee D, Wang Z, Cantley LC, Vander Heiden MG, Krainer AR. The alternative splicing repressors hnRNP A1/A2 and PTB influence pyruvate kinase isoform expression and cell metabolism. Proc Natl Acad Sci U S A. 2010; 107:1894-1899.
38. Sun Q, Chen X, Ma J, Peng H, Wang F, Zha X, Wang Y, Jing Y, Yang H, Chen R, Chang L, Zhang Y, Goto J, Onda H, Chen T, Wang MR et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc Natl Acad Sci U S A. 2011; 108:4129-4134.
39. Bluemlein K, Gruning NM, Feichtinger RG, Lehrach H, Kofler B, Ralser M. No evidence for a shift in pyruvate kinase PKM1 to PKM2 expression during tumorigenesis. Oncotarget. 2011; 2:393-400.
40. Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature. 2008; 452:181-186.
41. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646-674.
42. Hunter T. Tyrosine phosphorylation: thirty years and counting. Curr Opin Cell Biol. 2009; 21:140-146.
43. Hitosugi T, Kang S, Vander Heiden MG, Chung TW, Elf S, Lythgoe K, Dong S, Lonial S, Wang X, Chen GZ, Xie J, Gu TL, Polakiewicz RD, Roesel JL, Boggon TJ, Khuri FR et al. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci Signal. 2009; 2:ra73.
44. Chen N, Rinner O, Czernik D, Nytko KJ, Zheng D, Stiehl DP, Zamboni N, Gstaiger M, Frei C. The oxygen sensor PHD3 limits glycolysis under hypoxia via direct binding to pyruvate kinase. Cell Res. 2011; 21:983-986.
45. Pescador N, Cuevas Y, Naranjo S, Alcaide M, Villar D, Landazuri MO, Del Peso L. Identification of a functional hypoxia-responsive element that regulates the expression of the egl nine homologue 3 (egln3/phd3) gene. Biochem J. 2005; 390:189-197.
46. Lee J, Kim HK, Han YM, Kim J. Pyruvate kinase isozyme type M2 (PKM2) interacts and cooperates with Oct-4 in regulating transcription. Int J Biochem Cell Biol. 2008; 40:1043-1054.
47. Pesce M, Scholer HR. Oct-4: gatekeeper in the beginnings of mammalian development. Stem Cells. 2001; 19:271-278.
48. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006; 126:663-676.
49. Trosko JE. From adult stem cells to cancer stem cells: Oct-4 Gene, cell-cell communication, and hormones during tumor promotion. Ann N Y Acad Sci. 2006; 1089:36-58.
50. Hoshino A, Hirst JA, Fujii H. Regulation of cell proliferation by interleukin-3-induced nuclear translocation of pyruvate kinase. J Biol Chem. 2007; 282:17706-17711.
51. Spoden GA, Morandell D, Ehehalt D, Fiedler M, Jansen-Durr P, Hermann M, Zwerschke W. The SUMO-E3 ligase PIAS3 targets pyruvate kinase M2. J Cell Biochem. 2009; 107:293-302.
52. Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, Li Y, Shi J, An W, Hancock SM, He F, Qin L et al. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010; 327:1000-1004.
53. Vander Heiden MG, Christofk HR, Schuman E, Subtelny AO, Sharfi H, Harlow EE, Xian J, Cantley LC. Identification of small molecule inhibitors of pyruvate kinase M2. Biochem Pharmacol. 2010; 79:1118-1124.
54. Chen J, Xie J, Jiang Z, Wang B, Wang Y, Hu X. Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene. 2011.