
INTRODUCTION
Malignant epithelial salivary gland tumors (SGTs) represent a heterogeneous group of tumors, including the major subtypes of adenoid cystic carcinomas (ACC), mucoepidermoid carcinomas (MEC), and adenocarcinomas (ADC) [1]. Each of these malignant SGTs has no effective systemic treatment for patients who present or recur with unresectable disease and, until recently, there was little insight into the molecular basis for malignant salivary gland tumorigenesis. In 2003, the CRTC1-MAML2 fusion oncogene was isolated from a recurrent t(11;19) translocation in MEC [2]. This led to the discovery of a new CREB co-activator gene CRTC1 that is regulated by the Peutz-Jegher LKB1 kinase proposing a direct link between anabolic metabolism and salivary gland tumorigenesis [3-6]. MEC research efforts were facilitated by the availability of human tumor cell lines that allowed positional mapping of chromosomal breakpoints to isolate the etiologic fusion oncogene. In contrast, molecular characterization of a recurrent t(6;9) rearrangement in ACC was delayed due to lack of validated ACC tumor cell lines [7-10] until 2009 when the MYB-NFIB fusion oncogene was isolated using short-term cultures of ACC surgical biopsies [11]. ACC is a particularly aggressive subtype of SGT that frequently recurs with incurable metastatic disease many years after the initial surgical resection. Therefore, the identification of a recurrent MYB fusion event in ACC tumorigenesis provides an important clue to pursue new therapeutic strategies. MYB is a nuclear transcription factor that plays an essential role in the development and homeostasis of hematopoiesis [12-15]. In addition, MYB has been suggested to play a role in the development and homeostasis of selected glandular tissues including colon [16, 17] and breast [18]. In human cancer, tandem duplications of the MYB gene were detected in a subset of human T cell acute lymphoblastic leukemia (T-ALL) [19] and over-expression of MYB is associated with leukemia, breast and colorectal cancers [20-24]. Ectopic expression of MYB, however, has shown limited in vitro transforming activity that is largely restricted to hematopoietic animal model systems. Therefore, the identification of a recurrent MYB:NFIB translocation in ACC offered a new opportunity to study MYB biology in a defined epithelial human cancer model system.
The terminal NFIB exon, containing a small conserved open reading frame, is a recurrent translocation 3’ gene fusion partner in salivary gland ACC [11], pleomorphic adenomas (PA) [25] and lipoma [26]. Gründer et al.[25] indicated that this sequence resembled the CTD (C-terminal domain) of the largest RNA polymerase II subunit that can generate a physical bridge between the RNA transcription and pre-mRNA processing complexes through CTD-tail protein-protein interactions [27]. The functional role of NFIB in human cancer is unclear. For example, activation of HMGA2 by a reciprocal translocation with NFIB was proposed to arise by deletion of regulatory HMGA2 miRNA let-7 binding sites within 3’ untranslated (UT) sequences [28]. However, the detection of variant HMGA2 translocation breakpoints downstream of these consensus let-7 sites did not support this hypothesis [29]. MYB is also known to undergo 3’UT regulation in hematopoietic cell lineages and disruption of miRNA binding by the chromosomal translocation in ACC was recently proposed as a critical event for MYB activation [11]. Therefore, it is important to study both the functional properties of the novel MYB:NFIB fusion product as well as the biology and gene expression patterns in ACC samples that are both MYB fusion-positive and negative.
While MYB rearrangements were detected by FISH in approximately 50% of cases, there was an overall low rate of somatic mutations suggesting candidate cooperating driver mutations [30-32]. These data confirmed the importance of MYB activation but raised questions regarding downstream MYB signaling events in ACC tumorigenesis that might identify new therapeutic targets for patients with advanced disease.
To address these uncertainties, we have now performed global mRNA and miRNA expression analysis for a large collection of well-annotated malignant SGT and their matched normal tissues and integrated this data with somatic mutational analyses. We also performed functional assays to determine the transforming activity of activated MYB by either 3’ truncation or by fusion with terminal NFIB exon(s). We have detected a distinct ACC mRNA signature that includes over-expression of a network of extracellular matrix (ECM) components that arise from both MYB-dependent and -independent signals. These data suggests a cooperating role between MYB and RUNX1 /NOTCH1 signaling and proposes new therapeutic targets for this lethal disease.
RESULTS
ACC mRNA signature is independent of MYB rearrangement and enriched for cell membrane and extracellular matrix targets
To define a diagnostic adenoid cystic cancer (ACC) gene signature and to identify new therapeutic targets, we performed global mRNA and miRNA expression array analysis for 12 ACC tumors and matched adjacent normal samples. We also compared the ACC signatures to 14 mucoepidermoid cancers (MEC) and 11 salivary adenocarcinomas (ADC). All samples showed high RNA integrity (RIN >8) and passed quality controls described in Methods. We first performed principle component analysis (Figure 1A), both unsupervised (Supplementary Figure 1) and supervised (Figure 1B) clustering analysis for tumor and matched normal tissues to detect a unique mRNA signature that easily separates ACC from normal tissues. We detected 1160 genes that were differentially expressed in ACC as compared to matched normal tissues by using paired t-tests (Figure 1B, FDR=0.05, see Supplementary Table 1 for complete gene list). In addition, we were able to detect a secondary cluster across normal tissues that suggested a minor signature contribution from the specific tissue of origin (major vs minor salivary glands, Supplementary Figure 1). A striking finding was a distinct ACC mRNA signature regardless of tissue of origin that suggests a core mutational signaling pathway for this disease. This unique ACC mRNA signature was also markedly distinct from MEC and ADC mRNA signatures (data not shown). Although a recurrent MYB-NFIB fusion transcript is detected in many ACC samples, we and others have detected fusion-negative tumors in 20-40% of ACC samples. To test if MYB fusion-negative ACC tumors exhibit a different downstream gene expression profile, we initially pre-selected 6 fusion-positive and 6 fusion-negative ACC tumor samples scored by RT-PCR. As shown in Figure 1, there were no significant differences between these samples (Figure 1, panels A and B, Supplementary Figure 2, ANOVA FDR=0.05). We mined the 1160 genes with at least 2-fold change in ACC for predicted subcellular localization and functional properties using Fisher’s t-test. We noted an enrichment of downstream gene targets that localized to the cell membrane, extracellular space, and extracellular matrix (Figure 1C). As expected, using the IPA analysis we also noted a marked enrichment in genes that were known MYB target genes or MYB interaction genes (Figure 1D). We further confirmed this observation by merging our dataset with the ChIP-Seq public MYB target genes list obtained using highly specific MYB antibodies [33], and showing that approximately 50% of the ACC signature were candidate MYB-related genes.
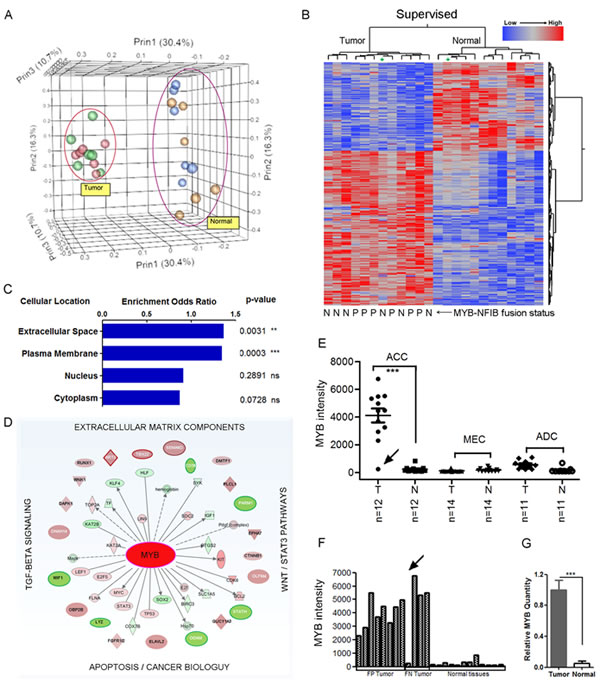
Figure 1: A unique mRNA gene signature distinguishes salivary gland adenoid cystic cancer (ACC) from matched normal salivary gland tissue. A) Principle component analysis (PCA) for all salivary gland adenoid cystic cancer (ACC) and matched normal samples. Red and green spheres indicate fusion positive and negative tumors respectively. Blue and brown indicate their matched normal tissues respectively. B) Supervised hierarchical-ward clustering analysis using 1160 mRNA probe sets that were significantly expressed in ACC (two sides paired t-test, FDR=0.05; see Supplementary Table 1 for complete gene list). Star depicts tumor sample with negligible MYB expression and matched normal tissue. MYB-NFIB fusion positive samples (P) and fusion negative samples (N). C) Gene localization and molecular enrichment analysis using Fisher’s t-test (***, paired t-test, two side p-value<0.0001; **, paired t-test, two side p-value<0.001; ns, paired t-test, two side p-value>=0.05). Gene localization was defined by Ingenuity IPA. D) MYB-related network in ACC. The inner layer: MYB-interaction genes defined by Ingenuity IPA. The outer layer, MYB regulating genes defined by published ChIP-Seq data [33]. E) Average intensities of MYB probe sets for three subtypes of salivary gland tumors and their matched normal samples (***, paired t-test, two side p-value<0.0001). F) MYB average intensity in ACC for each sample. Arrow depicts sample with low MYB. G) qRT-PCR validation of relative MYB expression levels in ACC tumors (***, two sample t-test, two side p-value<0.0001; error bars are S.E.M. of 12 samples).
Analysis of ACC tumors with negligible MYB expression identifies MYB-dependent and independent target genes
An unbiased inspection of global ACC gene expression identified MYB as the top differentially expressed gene as compared to either matched normal tissue or to other SGT subtypes such as MEC and ADC (Figure 1E, 1F, Supplementary Table 1). We confirmed this by independent qRT-PCR in the same set of ACC samples (Figure 1G). We also noted other previously identified ACC activation markers, such as c-KIT [34] and fibroblast growth factor receptor 1 (FGFR1) [35] that were up-regulated 5-fold and 4-fold respectively (Supplementary Table 1). The detection of MYB as the top differentially activated gene validates the accuracy of our data collection and methodology. However, we also noted a unique ACC sample with negligible MYB expression indistinguishable from basal levels in normal tissue, which, remarkably, exhibited an identical ACC mRNA gene signature (see asterix in Figure 1B and arrow in Figures 1E, 1F).
The detection of a similar ACC mRNA gene signature in both MYB fusion-positive and fusion-negative tumors as well as a sample with low MYB expression suggested several alternative mechanisms: i) presence of subtle variant MYB fusion events that were not detected in our samples with RT-PCR. ii) alternative, non-fusion somatic events that activate MYB, or iii) activation of an alternative gene pathway that mimics activated MYB signaling. To address the first possibility, we re-analyzed the relative expression levels of MYB exons using detailed exon array expression data. This microarray platform allows for multiple probes covering most MYB exons and we are able to generate ‘exon-plot’ images to examine exon-specific expression except for MYB exon 10 that was not present in ACC tumors. As expected, we were able to infer the approximate MYB breakpoints by noting relative drops in average probe set intensities immediately after the breakpoints in validated MYB-NFIB fusion positive samples. In contrast, true fusion-negative samples showed consistent probe intensities across the whole gene. With this ‘exon-plot’ approach, we identified two misdiagnosed ‘fusion negative’ tumors (Figure 2B, asterix), which were then validated for MYB rearrangements by FISH (data not shown). After these corrections, however, we were still unable to detect significant differences in downstream mRNA gene signature between MYB fusion-positive and the remaining fusion-negative tumors (Supplementary Figure 2). It is possible that cryptic MYB 3’ rearrangements may have escaped detection of the exon array probes and FISH analyses or, alternatively, these data may suggest that MYB activation is not necessarily dependent on 3’ structural rearrangement to drive ACC downstream signaling pathway. Inspection of the MYB 3’ terminal exon array data also allowed us to infer co-expression of the reciprocal and/or remaining wildtype (wt) MYB allele in fusion-positive samples. For example, we could infer the absent expression of both reciprocal and wildtype (wt) MYB alleles in at least 4 ACC samples (Figure 2B) suggesting a minor role for these alleles in ACC tumorigenesis.
As noted in Figure 1, we detected a unique ACC tumor sample with negligible MYB expression that unexpectedly retained the diagnostic ACC mRNA signature. We pursued this observation as a tool to search for potential MYB-dependent and MYB independent targets genes in ACC tumorigenesis. To validate this observation, we first searched the public gene array database for additional ACC cases and detected 3 independent ACC tumors with negligible MYB expression (GSE28996 [10]). These MYB-low ACC tumor samples were tested at different times on different array platforms but still showed an identical ACC downstream gene expression patterns indicating both MYB-dependent and independent gene transcripts (Figure 3A-C) that were enriched for extracellular matrix products (Figure 3C).
We then integrated our new ACC gene signature with data from three recent whole exome ACC sequencing studies [30-32] and identified RUNX1 and NOTCH1 as the only overlapping genes targeted for potential gain of function mutations (missense or C-terminal truncated protein) and associated with elevated gene expression (Figure 3D). It is also notable that the clustering of MYB, NOTCH1, RUNX1, and FBXW7 mutations seen in this uncommon epithelial tumor ACC overlaps with frequent somatic mutations seen in human acute lymphocytic leukemia patients [36]. Approximately 30% of the top-scoring gene loci in ACC encode components of the extracellular matrix including the HAPLN1/VCAN binding partners (Figure 3A-C). We also noted that RUNX1 (aka AML1/CBFA/PEBP2) cooperates with MYB to activate enhancer elements for many eukaryotic genes [37-40] and has conserved binding sites upstream of the human HAPLN1 promoter (Figure 3E) necessary for HAPLN1 expression [41, 42]. These data suggest that activated RUNX1 could participate as a co-activator in ACC for the VCAN/HAPLN1 complex. Although these extracellular components can be upregulated in other tumor lineages, we noted that the top 16 differentially expressed ECM genes on the basis of fold-induction were specifically observed in ACC but not in the other two SGT subtypes of MEC and ADC (Figure 4).
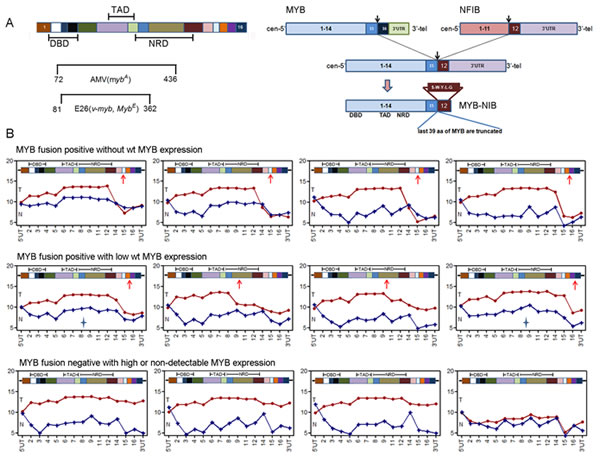
Figure 2: Exon array analysis is a sensitive tool to identify MYB C-terminal rearrangement status. A) Illustrations of MYB protein structure and MYB-NFIB fusion events in ACC. DBD, DNA binding domain; TAD, transcriptional activation domain; NRD, negative regulation domain. B) MYB exon intensity plot for 12 ACC tumor samples identifies variable expression of MYB C-terminal exons and validates MYB fusion status. Red line, MYB exon intensities in ACC tumor; Blue line, MYB exon intensities in matched normal tissue; Star depicts two false fusion-positive samples validated by FISH.
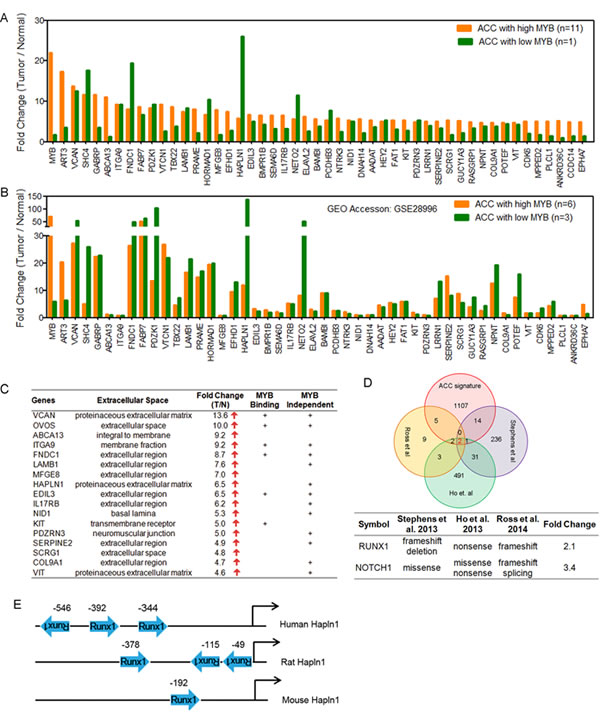
Figure 3: Integrative analysis identifies both MYB-dependent and MYB-independent genes in ACC. A) Differentially expressed genes with the highest fold change compared to normal in high or low MYB ACC tumors indicate extracellular gene activation. B) The same analysis as panel A was performed on ACC gene expression array data (Affy_HG133) obtained from public GEO database (accession: GSE28996). C) Top scoring genes that are components of extracellular matrix (ECM). MYB binding genes were defined by published ChIP-Seq data. MYB independent targets were defined as genes that were activated in both high and low MYB ACC tumors. D) Integrative analysis of ACC gene signature to recently published ACC mutational sequencing data identifies potential roles of RUNX1 in ACC tumorigenesis. E) Regulation of extracellular gene HAPLN1 by RUNX1.
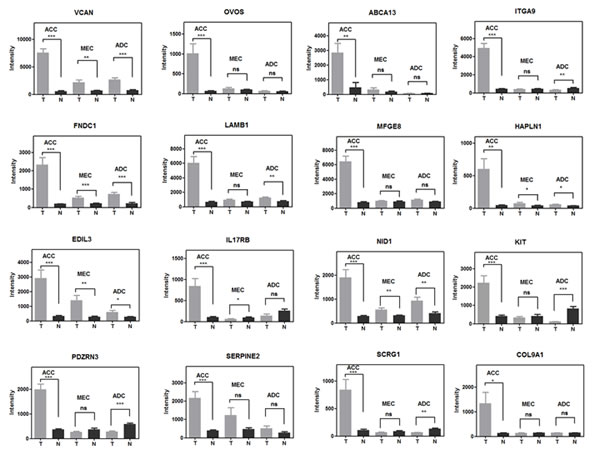
Figure 4: Comparison of the differential ECM activation in three subtypes of salivary gland tumors and their matched normal samples. ***, t-test, two side p-value<0.0001; **, p-value<0.001; *, p-value<0.05; ns, p-value >=0.05. Error bars are S.E.M. of 12 replicates of ACC, 14 replicates of MEC and 11 replicates of ADC.
Global miRNA signature distinguishes ACC from normal salivary gland tissues
Since MYB expression can be regulated by miRNA binding, deletion of these 3’ UT binding sites were proposed as a primary mechanism for MYB activation in ACC tumors. Therefore, to test this hypothesis, we performed global unbiased miRNA microarray analyses for the same collection of ACC and adjacent normal tissue samples. We tested a total of 847 probes with each probe representing a distinct human mature miRNA species. We detected a unique ACC miRNA profile distinct from matched normal samples (Figure 5A, see Supplementary Table 2 for complete list) and distinct from MEC and ADC tumors (data not shown). However, we again noted no significant difference in the miRNA signature between MYB fusion positive samples with deleted 3’UT sequences and fusion-negative ACC samples that are predicted to retain these miRNA binding sites. These data do not exclude a role for miRNA regulation of MYB since the MYB inhibitory hsa-miR-150 was down-regulated in all ACC tumors (Figure 5A). However, they argue against miRNA binding sites as a specific target for the recurrent t(6;9) chromosomal translocation. We also integrated the miRNA data with our global mRNA gene expression data and noted potential regulatory interconnections including patterns of ECM components gene expression (Figure 5B). For example, the HAPLN1 inhibitory hsa-miR-29 was down-regulated 2.5 fold (Figure 5B).
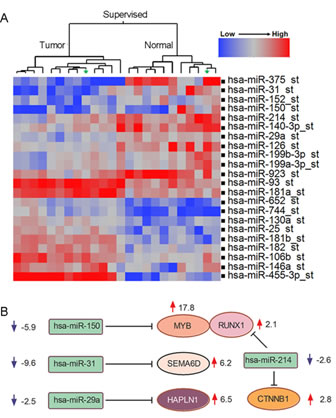
Figure 5: Distinct miRNA expression in ACC tumorigenesis. A) Supervised hierarchical ward clustering for ACC using 22 mature miRNA seed probe sets that are significantly expressed (two sides paired t-test, FDR=0.05). B) Regulatory miRNA target predictions (TargetScan, Pictar) suggest potential roles of miRNAs in regulation of extracellular genes in ACC tumorigenesis.
Forced expression of MYB-NFIB alters human salivary gland cell morphology and adhesion in vitro
To study functional roles of MYB and MYB-NFIB in ACC tumorigenesis, we generated expression vectors including full-length MYB with intact 5’ and 3’ UT (MYB wt); MYB with only 5’UT (MYB no 3’UTR), truncated MYB with a stop codon inserted at the most common translocation breakpoint in exon 15 (Trunc MYB), and MYB-NFIB fusion (MYB-NFIB) (Figure 6A). We obtained normal human salivary gland cells that were immortalized with SV40 to select clones that morphologically represent ductal cells (DC) [43] for transient and stable transfection with the MYB constructs. We observed lower MYB protein levels after either transient or stable expression of cDNA constructs that retained 3’ UT regions (Figure 6A). We also detected a modest, but statistically significant prolongation of MYB protein half life in the MYB-NFIB fusion product as compared to the truncated or wt MYB protein (Figure 6B). Interestingly, we detected clusters of loosely attached oncospheres as well as enhanced colony growth in soft agar of the DC cells transfected with the fusion MYB-NFIB product as compared to other MYB constructs (Figure 6C). We also noted that these cells were more loosely attached to the plastic dishes in vitro which we measured by counting viable cells in the supernatant at day 4 after initial plating and then reseeding these cells onto fresh dishes to monitor for growth by crystal violet staining (Figure 6D). A similar morphological appearance was detected when MYB-NFIB was transfected into primary mouse salivary gland cells (Supplementary Figure 3). These data, as well as the ACC mRNA gene expression signature identified in Figure 1, suggests cell membrane and extracellular candidate genes as a focus for new therapeutic targets in ACC. We selected VCAN from our data since this target was the highest expressed ECM gene target in our expression studies and VCAN (aka CSPG2) belongs to a family of versican proteoglycan genes with broad cellular functions including cell morphogenesis, adhesion, migration, angiogenesis, inflammation and growth factor regulation. VCAN was also previously shown to suppress tumor cell growth in mesothelioma, osteosarcoma, prostate, and other tumor cell lineages suggesting it may be a promising therapeutic target for preclinical development [44]. Since there are no validated ACC tumor cell lines [7-10] we selected a short term culture of a human ACC biopsy collected through an IRB approved protocol and treated cells with either control or 2 different lentiviral VCAN shRNAs. We detected marked growth inhibition of ACC tumor cells in vitro (Figure 6E) and also reduced nude mouse xenograft tumor growth in vivo (Figure 6F) with both VCAN shRNAs.
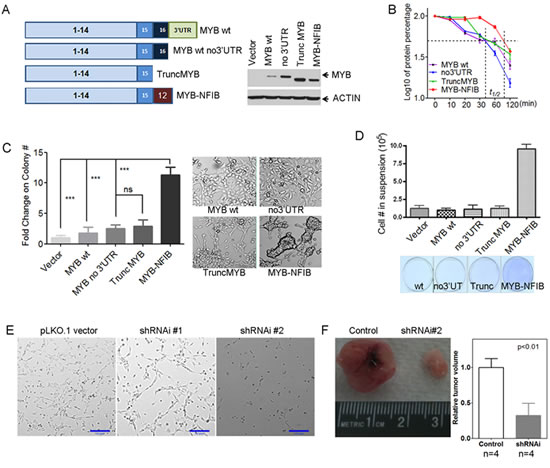
Figure 6: Functional analysis of MYB fusion protein. A) Illustration of MYB cDNA clones tested fragments tested and their expression efficiency in human immortalized salivary ductal cells (DC). MYB-NFIB protein showed increased steady-state protein and B) prolonged half-life in human immortalized myoepithelial cells (MC). C) Forced expression of MYB-NFIB enhanced the anchorage independent colony formation of human salivary DC cells. ***, t-test, two side p-value<0.0001; ns, p-value >=0.05. Error bars are S.E.M. of 3 replicates for each group. D) Decreased attachment and increased viability of suspension cells after ectopic MYB-NFIB expression. Error bars are S.E.M. of 3 replicates for each group. E) ACC tumor cell growth inhibition after shRNAi depletion of VCAN, and F) decreased xenograft tumor growth ability in NOD.SCID mice (two sample t-test, error bars are S.E.M. for 4 replicates).
DISCUSSION
ACC is one of the most common malignant SGT subtypes with no curative strategy for unresectable disease. In addition, lethal metastatic recurrences can present many years after the initial surgical resection that has further stimulated an urgent search for new therapies. Cytotoxic chemotherapy has not been effective for advanced disease and clinical trials testing imatinib to target KIT over-expression [45] or lapatinib for HER activation [46] have been unsuccessful for ACC; Although activation of FGFR has been proposed as another candidate target for ACC that is currently under clinical investigation; more work is needed to define the core cancer signaling pathways and to identify new cancer treatment targets. A critical clue towards this goal was the detection of a recurrent MYB-NFIB fusion transcript in a subset of ACC tumors, however there are numerous obstacles preventing the direct targeting of a nuclear transcription factors, such as MYB, with pharmacological or biological agents. To address this, we have now pursued a global nonbiased search for new ACC-specific mRNA and miRNA signatures that would identify candidates genes localized on the cell membrane or extracellular space that would be more accessible for therapeutic interventions.
In fact, we discovered that 30% of the top ranking genes measured by fold overexpression were localized within the extracellular space and this pattern appeared unique for ACC as compared to other SGT subtypes such as mucoepidermoid carcinomas and adenocarcinomas. Unexpectedly, we also detected an ACC sample with low MYB expression that still retained this global ACC gene expression signature. We validated this observation by identifying 3 additional ACC tumor samples from the public microarray databases with low MYB levels suggesting that approximately 5-10% of ACC samples may have this alternate phenotype with negligible MYB levels. Although MYB activation defines ACC tumorigenesis and MYB overexpression has been associated with a worse clinical outcome in patients with ACC [11, 47-49], there may be heterogeneity of MYB staining within tumor samples. For example, Costa et al (2014) showed that highly transformed area of ACC tends to have lower MYB expression although not statistically significant (p=0.123, Wilcoxon test), and they also observed an absence of MYB expression in 1 case (out of 8 ACC cases) [50]. We hypothesize that ACC tumors with negligible MYB expression may have acquired an alternate somatic alteration in a gene target that mimics activated MYB signaling.
To pursue this possibility we performed an integrated analysis of gene expression and somatic mutational data and these data proposed a role for MYB, NOTCH1, and RUNX1 in regulating a subset of key ACC target genes. RUNX1, a MYB transcriptional co-factor, may be a specific target for missense or C-terminal truncated activating mutations in ACC that cooperate to deregulate MYB transcriptional activation [51-55]. For example, MYB is required for both RUNX1 transcriptional activity [54, 56], for the ability of a chimeric RUNX1 fusion oncogene to transform hematopoietic cells [54, 57, 58], and RUNX1 can cooperates with MYB to activate ectopic targets through novel MYB binding sites in cancers [53]. Interestingly, we discovered that two of the most highly expressed ACC target genes, VCAN and HAPLN1, can bind within a predicted ECM complex in ACC tumor cells and, in the case of HAPLN1, appear to be regulated by RUNX1 activation [41, 42]. In addition to the ability of RUNX/NOTCH to cooperate with MYB for promoter activity [59], others have noted that either activated NOTCH or aberrant accumulation of VCAN can induce the same heart valve phenotype in rodent models suggesting another connection between NOTCH/RUNX/MYB and ECM regulations [60]. VCAN/HAPLN1 binding in the ECM is predicted to activate a broad range of signaling pathways including the epidermal growth factor receptor (EGFR), TGF-β, insulin-like growth factor receptor (IGFIR) and others [61, 62]. The pro-tumorigenic role of VCAN/HAPLN1 complex has been tested in several other tumor types, such as malignant pleural mesothelioma [63] and breast and prostate cancers and increased tumor-specific VCAN expression levels have been reported in ACC and several other tumor types [64-69]. For example, VCAN was found to be 29-fold up-regulation in human ACC tumors comparing to normal tissues [69] and increased VCAN protein staining was detected in mouse xenograft tumor models for human ACCs [10]. Although stromal VCAN expression was associated with a poor prognosis in oral squamous cell carcinoma [70] and was associated with high rate of tumor recurrence and more advanced disease in non-small cell lung cancers [71], our data does not yet confirm a direct link between VCAN overexpression and many of the clinicopathological and prognostic features in ACC. We have now demonstrated tumor growth inhibition in vitro and in vivo with short term ACC cell cultures following depletion of VCAN and the generation of new validated immortalized ACC tumor cell lines and animal models will allow preclinical testing of these new promising extracellular targets for development of new treatment strategies.
METHODS
RNA extraction, quality control and quantification
A total of 74 salivary gland tumors including 24 salivary gland adenoid cystic cancer, 28 mucoepiderrmoid cancer and 22 adenocarcinoma samples were excised from human patients under approved IRB protocols from University of Texas MD Anderson Cancer Center. MYB-NFIB fusion positive tumors were preselected by RT-PCR. Total RNAs were extracted and evaluated using NanodropTM 8000 (ThermoFisher Scientific, MA) and the Agilent 2100 Bioanalyzer with RNA 6000 Nano Labchip® (Agilent Technologies, CA).
Microarray assay and data analysis
Microarray assays were performed by following manufacturer’s instructions (GeneChip ®_human gene 1.0_ST and miRNA_GeneChip_1.0; Affymetrix, CA). Data were processed through the robust multichip average algorithm and quantile normalization methods followed by log2 transformation [72, 73]. Processed data were analyzed using SAS JMP® Genomics (SAS Institute, NC). Both baseline filter (intensity >=50) and variation filter (|fold-change|>=2) were applied before mRNA differential expression analysis. Both baseline filter (intensity >=800) and variation filter (|fold-change|>=2) were applied for microRNA chip analysis. Hierarchical cluster analysis and the principle component analysis were performed for all samples using SAS (SAS Institute, NC). Paired t-tests were performed to compare tumor samples to their matched normal tissues and Hochberg-Benjamini FDR was set at 0.05 [74]. The mixed ANOVA analysis was performed for the comparison between MYB-NFIB fusion-positive and fusion-negative tumors. All data were deposited in the NCBI GEO database (GSE59702). MYB interaction genes were extracted from IPA interactions (Ingenuity IPA, Redwood City, CA). MYB target gene was downloaded from public ChIP-Seq data that listed all genes having significant MYB binding sites in their promoters [33]. Gene enrichment analysis was performed using Fisher’s test.
Plasmid construction, transfection, cell culture and shRNAi knock-down
MYB fragments were amplified from commercial MYB cDNA clone (Open Biosystems, AL, Cat MHS1010-9206139) and inserted into the pAcGFP1N1 expression plasmid using NotI and KpnI restriction sites and the terminal NFIB exon with or without NFIB 3’UT sequences was introduced by PCR-mediated cloning. All constructs were validated by sequencing. For stable clone selection, 50 ng/ml Geneticin® selective antibiotics (Gibco®, Cat #11811-098) was introduced after 48 hours of transfection. The pLKO.1 lentiviral VCAN-shRNA constructs were obtained from Open Biosystems (Huntsville, AL). The lentiviral procedures applied for shRNAi were performed as described in [75]. The pBABE-puro-hTERT plasmid was purchased from Addgene (MA, plasmid1771).
NIH/3T3 cells were cultured in DMEM (Sigma) supplemented with 10% FBS and 1% pen/strep (Gibco®). HEK293T cells were cultured in high glucose DMEM medium supplemented with 10% FBS and 1% pen/strep (Gibco®). RK3E cells were cultured in RPMI 1640 (Sigma) with 10% FBS and 1% pen/strep (Gibco®). Normal human immortalized salivary gland cells [76] were generous gift from Dr Stephen Hsu at Medical College of Georgia and cells were cultured with the Keratinocyte-SFM medium with L-Glutamine, EGF and BPE (Gibco®, Cat17005-075). Defined Trypsin Inhibitor was also purchased from Invitrogen (CatR-007). The primary normal salivary gland cell culture for wt FVB mice (The Jackson Laboratory, Jacksonville, FL) was performed following procedures described in [76]. Cells were observed under Leica microscope (Leica microsystems, IL).
Quantitative RT-PCR
Reverse transcription and amplification of cDNA was performed in the 7900 HT Fast (Applied Biosystems) system by following manufacture instruction using High-Capacity cDNA Reverse Transcription Kits (Applied Biosystems®, Cat4368814) and TaqMan Fast Universal PCR Master Mix (Applied Biosystems®, Cat4367846). All gene expression probes were commercially purchased (Applied Biosystems®). Relative expression of target gene was calculated in comparison to 18s rRNA values. The quantitative miRNA RT-PCR analysis was performed by following procedures as described in [77].
Western-blot, and antibodies
Cells pellet was lysed in RIPA buffer (Boston Bioproducts) and subjected to SDS- polyacrylamide tris-glycine gel separation (Invitrogen). And the membrane transfer was performed using iBlot® 7 Minute Blotting System (Invitrogen) following the manufacturer’s instructions. Membrane was blocked with 3% BSA overnight at 4°C and then incubated with primary antibodies for 1 hour at room temperature. Antibody against N-terminus MYB was purchased from Abcam (EP769Y). After washed with PBS, the membrane was incubated with a horseradish peroxidase-conjugated secondary antibody for 45 minutes. Bands were visualized with Chemiluminescence (Pierce).
Cycloheximide blocking assay
The protein half life was determined by cycloheximide blocking assay. Cells were seeded into 100mm dish and subjected to inhibition of protein synthesis with 100 µg/ml cycloheximide (Sigma) for 10 minutes, 20 minutes, 30 minutes, 60 minutes, 120 minutes or no treatment as control. Cycloheximide treated cells were then harvested and subjected to SDS-PAGE gel separation and western-blot analysis for retained protein. The relative protein quantity were measured using ImageJ [78].
Soft-agar assay for anchorage independent colony formation
A total of 1x105 trypsinized cells were subjected to soft-agar assay as described in [79]. Generally, top agar was prepared with 0.8% Difco Agar and 0.2% Difco peptone and then mixed with 2x Media (Gibco). Base agar was prepared with 1.6% Difco Agar and 0.4% Difco peptone and then mixed with 2x Media (Gibco). Triplicates were set for each cell construct. Agar plates were then put into incubator (Thermo Scientific) at 37°C and 5% CO2 for 3 weeks. And cells were feed once every week. Colonies were counted manually under10x microscope (Leica microsystems, IL).
Self-suspension viable cell growth
A total of 1x106 cells were seeded into 6-well plates in day one, and then viable cells in the culture supernatant were counted with TrypanBlue (Gibco) staining at day 4. Unstained detached cells were washed twice with PBS, reseeded to 6-well plates for growth and then stained with crystal violet for cell density observation after 4 days.
Xenograft tumor model and primary cell culture
A total of 1×106 cells were washed twice with PBS and then subjected to subcutaneous flank injection into NOD.SCID mice (The Jackson Laboratory, Jacksonville, FL). Freshly prepared human ACC tumor obtained under IRB-approved tissue protocol was also subjected to xenograft tumor growth and short term cell culture. Xenograft tumor volumes were calculated as: volume = (width)2 x length/2. The primary tumor cell culture was performed using methods as described in [43, 76]. The media used for primary tumor cell culture includes F12K (Sigma) 1:1 mixture with DMEM (Sigma) supplemented with 5-20% FBS (Gibco) and ACL4 medium supplemented with 5% FBS.
ACKNOWLEDGEMENTS
The study is supported by the NIH National Institute of Dental and Craniofacial Research and the NIH Office of Rare Diseases Research grant number U01DE019765, the Head and Neck SPORE program grant number P50 CA097007, the Kenneth D. Muller Professorship, the NCI CA-16672 grant, the Gatorade funding UF College of Medicine Department of Medicine and the National Institute Of Dental & Craniofacial Research of the National Institutes of Health R01DE023641. The authors would also like to thank Dr Stephen Hsu, Medical College of Georgia, GA, who generously provided the human immortalized normal salivary gland cells.
REFERENCES
1. L Barnes JE, P Reichart, D Sidransky (2005). World Health Organization International Classification of Tumors. Pathology and Genetics of Head and Neck Tumors. Lyon: IARC Press).
2. Tonon G, Modi S, Wu L, Kubo A, Coxon AB, Komiya T, O’Neil K, Stover K, El-Naggar A, Griffin JD, Kirsch IR and Kaye FJ. t(11;19)(q21;p13) translocation in mucoepidermoid carcinoma creates a novel fusion product that disrupts a Notch signaling pathway. Nat Genet. 2003; 33(2):208-213.
3. Tirado Y, Williams MD, Hanna EY, Kaye FJ, Batsakis JG and El-Naggar AK. CRTC1/MAML2 fusion transcript in high grade mucoepidermoid carcinomas of salivary and thyroid glands and Warthin’s tumors: implications for histogenesis and biologic behavior. Genes Chromosomes Cancer. 2007; 46(7):708-715.
4. Komiya T, Coxon A, Park Y, Chen WD, Zajac-Kaye M, Meltzer P, Karpova T and Kaye FJ. Enhanced activity of the CREB co-activator Crtc1 in LKB1 null lung cancer. Oncogene. 29(11):1672-1680.
5. Coxon A, Rozenblum E, Park YS, Joshi N, Tsurutani J, Dennis PA, Kirsch IR and Kaye FJ. Mect1-Maml2 fusion oncogene linked to the aberrant activation of cyclic AMP/CREB regulated genes. Cancer Res. 2005; 65(16):7137-7144.
6. Cao C, Gao R, Zhang M, Amelio AL, Fallahi M, Chen Z, Gu Y, Hu C, Welsh EA, Engel BE, Haura EB, Cress WD, Wu L, Zajac-Kaye M and Kaye FJ. Role of LKB1-CRTC1 on Glycosylated COX-2 and response to COX-2 Inhibition in lung cancer. J Natl Cancer Inst. 2015; 107(1):11.
7. Choi S, Sano D, Cheung M, Zhao M, Jasser SA, Ryan AJ, Mao L, Chen WT, El-Naggar AK and Myers JN. Vandetanib inhibits growth of adenoid cystic carcinoma in an orthotopic nude mouse model. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008; 14(16):5081-5089.
8. Phuchareon J, Ohta Y, Woo JM, Eisele DW and Tetsu O. Genetic profiling reveals cross-contamination and misidentification of 6 adenoid cystic carcinoma cell lines: ACC2, ACC3, ACCM, ACCNS, ACCS and CAC2. PloS one. 2009; 4(6):e6040.
9. El-Naggar AK, Kaye FJ and Stenman G. The 3rd Salivary Gland Tumor Workshop meeting report. Head & neck. 2013; 35(8):1057-1060.
10. Moskaluk CA, Baras AS, Mancuso SA, Fan H, Davidson RJ, Dirks DC, Golden WL and Frierson HF, Jr. Development and characterization of xenograft model systems for adenoid cystic carcinoma. Laboratory investigation; a journal of technical methods and pathology. 2011; 91(10):1480-1490.
11. Persson M, Andren Y, Mark J, Horlings HM, Persson F and Stenman G. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proceedings of the National Academy of Sciences of the United States of America. 2009; 106(44):18740-18744.
12. Golay J, Basilico L, Loffarelli L, Songia S, Broccoli V and Introna M. Regulation of hematopoietic cell proliferation and differentiation by the myb oncogene family of transcription factors. International journal of clinical & laboratory research. 1996; 26(1):24-32.
13. Oh IH and Reddy EP. The myb gene family in cell growth, differentiation and apoptosis. Oncogene. 1999; 18(19):3017-3033.
14. Lieu YK and Reddy EP. Conditional c-myb knockout in adult hematopoietic stem cells leads to loss of self-renewal due to impaired proliferation and accelerated differentiation. Proceedings of the National Academy of Sciences of the United States of America. 2009; 106(51):21689-21694.
15. Klempnauer K-H. (2004). Target Genes Of V-Myb And C-Myb. Myb Transcription Factors: Their Role in Growth, Differentiation and Disease.
16. Thompson MA, Rosenthal MA, Ellis SL, Friend AJ, Zorbas MI, Whitehead RH and Ramsay RG. c-Myb down-regulation is associated with human colon cell differentiation, apoptosis, and decreased Bcl-2 expression. Cancer Res. 1998; 58(22):5168-5175.
17. Malaterre J, Carpinelli M, Ernst M, Alexander W, Cooke M, Sutton S, Dworkin S, Heath JK, Frampton J, McArthur G, Clevers H, Hilton D, Mantamadiotis T and Ramsay RG. c-Myb is required for progenitor cell homeostasis in colonic crypts. Proceedings of the National Academy of Sciences of the United States of America. 2007; 104(10):3829-3834.
18. Drabsch Y, Robert RG and Gonda TJ. MYB suppresses differentiation and apoptosis of human breast cancer cells. Breast cancer research : BCR. 2010; 12(4):R55.
19. O’Neil J, Tchinda J, Gutierrez A, Moreau L, Maser RS, Wong KK, Li W, McKenna K, Liu XS, Feng B, Neuberg D, Silverman L, DeAngelo DJ, Kutok JL, Rothstein R, DePinho RA, et al. Alu elements mediate MYB gene tandem duplication in human T-ALL. The Journal of experimental medicine. 2007; 204(13):3059-3066.
20. Gonda TJ and Metcalf D. Expression of myb, myc and fos proto-oncogenes during the differentiation of a murine myeloid leukaemia. Nature. 1984; 310(5974):249-251.
21. Shen-Ong GL. The myb oncogene. Biochimica et biophysica acta. 1990; 1032(1):39-52.
22. Lipsick JS and Wang DM. Transformation by v-Myb. Oncogene. 1999; 18(19):3047-3055.
23. Ramsay RG, Barton AL and Gonda TJ. Targeting c-Myb expression in human disease. Expert opinion on therapeutic targets. 2003; 7(2):235-248.
24. Ramsay RG and Gonda TJ. MYB function in normal and cancer cells. Nature reviews. 2008; 8(7):523-534.
25. Geurts JM, Schoenmakers EF, Roijer E, Astrom AK, Stenman G and van de Ven WJ. Identification of NFIB as recurrent translocation partner gene of HMGIC in pleomorphic adenomas. Oncogene. 1998; 16(7):865-872.
26. Nilsson M, Panagopoulos I, Mertens F and Mandahl N. Fusion of the HMGA2 and NFIB genes in lipoma. Virchows Arch. 2005; 447(5):855-858.
27. Grunder A, Qian F, Ebel TT, Mincheva A, Lichter P, Kruse U and Sippel AE. Genomic organization, splice products and mouse chromosomal localization of genes for transcription factor Nuclear Factor One. Gene. 2003; 304:171-181.
28. Mayr C, Hemann MT and Bartel DP. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science. 2007; 315(5818):1576-1579.
29. Wang X, Hulshizer RL, Erickson-Johnson MR, Flynn HC, Jenkins RB, Lloyd RV and Oliveira AM. Identification of novel HMGA2 fusion sequences in lipoma: evidence that deletion of let-7 miRNA consensus binding site 1 in the HMGA2 3’ UTR is not critical for HMGA2 transcriptional upregulation. Genes Chromosomes Cancer. 2009; 48(8):673-678.
30. Ross JS, Wang K, Rand JV, Sheehan CE, Jennings TA, Al-Rohil RN, Otto GA, Curran JC, Palmer G, Downing SR, Yelensky R, Lipson D, Balasubramanian S, Garcia L, Mahoney K, Ali SM, et al. Comprehensive genomic profiling of relapsed and metastatic adenoid cystic carcinomas by next-generation sequencing reveals potential new routes to targeted therapies. The American journal of surgical pathology. 2014; 38(2):235-238.
31. Stephens PJ, Davies HR, Mitani Y, Van Loo P, Shlien A, Tarpey PS, Papaemmanuil E, Cheverton A, Bignell GR, Butler AP, Gamble J, Gamble S, Hardy C, Hinton J, Jia M, Jayakumar A, et al. Whole exome sequencing of adenoid cystic carcinoma. The Journal of clinical investigation. 2013; 123(7):2965-2968.
32. Ho AS, Kannan K, Roy DM, Morris LG, Ganly I, Katabi N, Ramaswami D, Walsh LA, Eng S, Huse JT, Zhang J, Dolgalev I, Huberman K, Heguy A, Viale A, Drobnjak M, et al. The mutational landscape of adenoid cystic carcinoma. Nat Genet. 2013; 45(7):791-798.
33. Quintana AM, Liu F, O’Rourke JP and Ness SA. Identification and regulation of c-Myb target genes in MCF-7 cells. BMC cancer. 2011; 11:30.
34. Holst VA, Marshall CE, Moskaluk CA and Frierson HF, Jr. KIT protein expression and analysis of c-kit gene mutation in adenoid cystic carcinoma. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 1999; 12(10):956-960.
35. Ding L, Zhu SR, Xie SX and Wu XB. [Effects of basic fibroblast growth factor on the proliferation of human salivary adenoid cystic carcinoma cell line ACC-2 and extracellular signal-regulated kinase, Cyclin D1, p2waf/cip1 signaling pathway]. Hua xi kou qiang yi xue za zhi = Huaxi kouqiang yixue zazhi = West China journal of stomatology. 2008; 26(2):118-120.
36. O’Neil J and Look AT. Mechanisms of transcription factor deregulation in lymphoid cell transformation. Oncogene. 2007; 26(47):6838-6849.
37. Britos-Bray M and Friedman AD. Core binding factor cannot synergistically activate the myeloperoxidase proximal enhancer in immature myeloid cells without c-Myb. Molecular and cellular biology. 1997; 17(9):5127-5135.
38. Mertz JA, Kobayashi R and Dudley JP. ALY is a common coactivator of RUNX1 and c-Myb on the type B leukemogenic virus enhancer. Journal of virology. 2007; 81(7):3503-3513.
39. Hernandez-Munain C and Krangel MS. c-Myb and core-binding factor/PEBP2 display functional synergy but bind independently to adjacent sites in the T-cell receptor delta enhancer. Molecular and cellular biology. 1995; 15(6):3090-3099.
40. Martensson A, Xie XQ, Persson C, Holm M, Grundstrom T and Martensson IL. PEBP2 and c-myb sites crucial for lambda5 core enhancer activity in pre-B cells. European journal of immunology. 2001; 31(11):3165-3174.
41. Liu J, Park ES, Curry TE, Jr. and Jo M. Periovulatory expression of hyaluronan and proteoglycan link protein 1 (Hapln1) in the rat ovary: hormonal regulation and potential function. Molecular endocrinology (Baltimore, Md. 24(6):1203-1217.
42. Jo M and Curry TE, Jr. Luteinizing hormone-induced RUNX1 regulates the expression of genes in granulosa cells of rat periovulatory follicles. Molecular endocrinology (Baltimore, Md. 2006; 20(9):2156-2172.
43. Liu X, Ory V, Chapman S, Yuan H, Albanese C, Kallakury B, Timofeeva OA, Nealon C, Dakic A, Simic V, Haddad BR, Rhim JS, Dritschilo A, Riegel A, McBride A and Schlegel R. ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. The American journal of pathology. 2012; 180(2):599-607.
44. Ricciardelli C, Sakko AJ, Ween MP, Russell DL and Horsfall DJ. The biological role and regulation of versican levels in cancer. Cancer metastasis reviews. 2009; 28(1-2):233-245.
45. Pfeffer MR, Talmi Y, Catane R, Symon Z, Yosepovitch A and Levitt M. A phase II study of Imatinib for advanced adenoid cystic carcinoma of head and neck salivary glands. Oral oncology. 2007; 43(1):33-36.
46. Agulnik M, Cohen EW, Cohen RB, Chen EX, Vokes EE, Hotte SJ, Winquist E, Laurie S, Hayes DN, Dancey JE, Brown S, Pond GR, Lorimer I, Daneshmand M, Ho J, Tsao MS, et al. Phase II study of lapatinib in recurrent or metastatic epidermal growth factor receptor and/or erbB2 expressing adenoid cystic carcinoma and non adenoid cystic carcinoma malignant tumors of the salivary glands. J Clin Oncol. 2007; 25(25):3978-3984.
47. Mitani Y, Li J, Rao PH, Zhao YJ, Bell D, Lippman SM, Weber RS, Caulin C and El-Naggar AK. Comprehensive analysis of the MYB-NFIB gene fusion in salivary adenoid cystic carcinoma: Incidence, variability, and clinicopathologic significance. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010; 16(19):4722-4731.
48. Mitani Y, Rao PH, Futreal PA, Roberts DB, Stephens PJ, Zhao YJ, Zhang L, Mitani M, Weber RS, Lippman SM, Caulin C and El-Naggar AK. Novel chromosomal rearrangements and break points at the t(6;9) in salivary adenoid cystic carcinoma: association with MYB-NFIB chimeric fusion, MYB expression, and clinical outcome. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011; 17(22):7003-7014.
49. Brill LB, 2nd, Kanner WA, Fehr A, Andren Y, Moskaluk CA, Loning T, Stenman G and Frierson HF, Jr. Analysis of MYB expression and MYB-NFIB gene fusions in adenoid cystic carcinoma and other salivary neoplasms. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2011; 24(9):1169-1176.
50. Costa AF, Altemani A, Garcia-Inclan C, Fresno F, Suarez C, Llorente JL and Hermsen M. Analysis of MYB oncogene in transformed adenoid cystic carcinomas reveals distinct pathways of tumor progression. Laboratory investigation; a journal of technical methods and pathology. 2014; 94(6):692-702.
51. Friedman AD. Runx1, c-Myb, and C/EBPalpha couple differentiation to proliferation or growth arrest during hematopoiesis. Journal of cellular biochemistry. 2002; 86(4):624-629.
52. Carabana J, Ortigoza E and Krangel MS. Regulation of the murine Ddelta2 promoter by upstream stimulatory factor 1, Runx1, and c-Myb. Journal of immunology. 2005; 174(7):4144-4152.
53. Mansour MR, Abraham BJ, Anders L, Berezovskaya A, Gutierrez A, Durbin AD, Etchin J, Lawton L, Sallan SE, Silverman LB, Loh ML, Hunger SP, Sanda T, Young RA, Look AT. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science. 2014.
54. Burns CE, Galloway JL, Smith AC, Keefe MD, Cashman TJ, Paik EJ, Mayhall EA, Amsterdam AH and Zon LI. A genetic screen in zebrafish defines a hierarchical network of pathways required for hematopoietic stem cell emergence. Blood. 2009; 113(23):5776-5782.
55. Rothenberg EV. Regulatory factors for initial T lymphocyte lineage specification. Current opinion in hematology. 2007; 14(4):322-329.
56. Zaiman AL and Lenz J. Transcriptional activation of a retrovirus enhancer by CBF (AML1) requires a second factor: evidence for cooperativity with c-Myb. Journal of virology. 1996; 70(8):5618-5629.
57. Pattabiraman DR, McGirr C, Shakhbazov K, Barbier V, Krishnan K, Mukhopadhyay P, Hawthorne P, Trezise A, Ding J, Grimmond SM, Papathanasiou P, Alexander WS, Perkins AC, Levesque JP, Winkler IG and Gonda TJ. Interaction of c-Myb with p300 is required for the induction of acute myeloid leukemia (AML) by human AML oncogenes. Blood. 2014; 123(17):2682-2690.
58. Bresciani E, Carrington B, Wincovitch S, Jones M, Gore AV, Weinstein BM, Sood R, Liu PP. CBFβ and RUNX1 are required at 2 different steps during the development of hematopoietic stem cells in zebrafish. Blood. 2014; 124(1):9.
59. Del Real MM and Rothenberg EV. Architecture of a lymphomyeloid developmental switch controlled by PU.1, Notch and Gata3. Development. 2013; 140(6):1207-1219.
60. Dupuis LE, Osinska H, Weinstein MB, Hinton RB and Kern CB. Insufficient versican cleavage and Smad2 phosphorylation results in bicuspid aortic and pulmonary valves. Journal of molecular and cellular cardiology. 2013; 60:50-59.
61. Goldoni S and Iozzo RV. Tumor microenvironment: Modulation by decorin and related molecules harboring leucine-rich tandem motifs. International journal of cancer Journal international du cancer. 2008; 123(11):2473-2479.
62. Schaefer L and Schaefer RM. Proteoglycans: from structural compounds to signaling molecules. Cell and tissue research. 2010; 339(1):237-246.
63. Ivanova AV, Goparaju CM, Ivanov SV, Nonaka D, Cruz C, Beck A, Lonardo F, Wali A and Pass HI. Protumorigenic role of HAPLN1 and its IgV domain in malignant pleural mesothelioma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009; 15(8):2602-2611.
64. Touab M, Villena J, Barranco C, Arumi-Uria M and Bassols A. Versican is differentially expressed in human melanoma and may play a role in tumor development. The American journal of pathology. 2002; 160(2):549-557.
65. Yeung TL, Leung CS, Wong KK, Samimi G, Thompson MS, Liu J, Zaid TM, Ghosh S, Birrer MJ and Mok SC. TGF-beta modulates ovarian cancer invasion by upregulating CAF-derived versican in the tumor microenvironment. Cancer Res. 2013; 73(16):5016-5028.
66. Kischel P, Waltregny D, Dumont B, Turtoi A, Greffe Y, Kirsch S, De Pauw E and Castronovo V. Versican overexpression in human breast cancer lesions: known and new isoforms for stromal tumor targeting. International journal of cancer Journal international du cancer. 2010; 126(3):640-650.
67. Gorter A, Zijlmans HJ, van Gent H, Trimbos JB, Fleuren GJ and Jordanova ES. Versican expression is associated with tumor-infiltrating CD8-positive T cells and infiltration depth in cervical cancer. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2010; 23(12):1605-1615.
68. Miquel-Serra L, Serra M, Hernandez D, Domenzain C, Docampo MJ, Rabanal RM, de Torres I, Wight TN, Fabra A and Bassols A. V3 versican isoform expression has a dual role in human melanoma tumor growth and metastasis. Laboratory investigation; a journal of technical methods and pathology. 2006; 86(9):889-901.
69. Frierson HF, Jr., El-Naggar AK, Welsh JB, Sapinoso LM, Su AI, Cheng J, Saku T, Moskaluk CA and Hampton GM. Large scale molecular analysis identifies genes with altered expression in salivary adenoid cystic carcinoma. The American journal of pathology. 2002; 161(4):1315-1323.
70. Pratheepa L PR, Herald J Sherlin, Anuja N, Priya Premkumar. Expression of Emerging Novel Tumor markers in Oral Squamous cell carcinoma and their Clinical and Pathological correlation to determine the Prognosis and Usefulness as a Therapeutic target – A Systematic Review. Journal of Natural Sciences Research. 2012; 2(1).
71. Pirinen R, Leinonen T, Bohm J, Johansson R, Ropponen K, Kumpulainen E and Kosma VM. Versican in nonsmall cell lung cancer: relation to hyaluronan, clinicopathologic factors, and prognosis. Human pathology. 2005; 36(1):44-50.
72. Barczak A, Rodriguez MW, Hanspers K, Koth LL, Tai YC, Bolstad BM, Speed TP and Erle DJ. Spotted long oligonucleotide arrays for human gene expression analysis. Genome research. 2003; 13(7):1775-1785.
73. Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B and Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic acids research. 2003; 31(4):e15.
74. Hochberg Y and Benjamini Y. More powerful procedures for multiple significance testing. Statistics in medicine. 1990; 9(7):811-818.
75. Chen Z, Chen J, Gu Y, Hu C, Li JL, Lin S, Shen H, Cao C, Gao R, Li J, Ha PK, Kaye FJ, Griffin JD and Wu L. Aberrantly activated AREG-EGFR signaling is required for the growth and survival of CRTC1-MAML2 fusion-positive mucoepidermoid carcinoma cells. Oncogene. 2013.
76. Azuma M, Tamatani T, Kasai Y and Sato M. Immortalization of normal human salivary gland cells with duct-, myoepithelial-, acinar-, or squamous phenotype by transfection with SV40 ori- mutant deoxyribonucleic acid. Laboratory investigation; a journal of technical methods and pathology. 1993; 69(1):24-42.
77. Yan Y, Salazar TE, Dominguez JM, 2nd, Nguyen DV, Li Calzi S, Bhatwadekar AD, Qi X, Busik JV, Boulton ME and Grant MB. Dicer expression exhibits a tissue-specific diurnal pattern that is lost during aging and in diabetes. PloS one. 2013; 8(11):e80029.
78. Schneider CA, Rasband WS and Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nature methods. 2012; 9(7):671-675.
79. Zajac-Kaye M and Ts’o PO. DNAase I encapsulated in liposomes can induce neoplastic transformation of Syrian hamster embryo cells in culture. Cell. 1984; 39(3 Pt 2):427-437.