

INTRODUCTION
Prostate cancer (PCa) is the most common non-cutaneous male malignancy and the second leading cause of cancer-related deaths in males, with an estimated 233,000 new cases (27% of all new cancer cases in males) expected to occur in the United States in 2014 [1]. During prostate tumorigenesis, oncogenic signaling pathways promote the progression of androgen-dependent carcinomas to castration-resistant (hormone-refractory) prostate cancer (CRPC), the major contributing factor in PCa fatalities. The primary treatment for localized PCa is radiotherapy (external beam or brachytherapy) or complete removal of the prostate (radical prostatectomy). However, patients relapse in 30-50% of intermediate to high risk cases and neoplastic growth of aberrant prostate cells resumes either locally or systematically [2]. Relapsed patients are often subjected to androgen deprivation therapy (ADT) in which either chemical or surgical castration is performed along with anti-androgen treatment. Tumor cell proliferation despite the use of androgen deprivation (e.g. increased prostate specific antigen (PSA) levels in serum regardless of ongoing treatment), defines the presence of CRPC [3].
Histological studies of PCa tissues have identified a specific lesion of prostatic intraductal dysplasia [4], now commonly referred to as prostatic intraepithelial neoplasia (PIN). PIN is characterized by the appearance of luminal epithelial hyperplasia, enlarged nuclei and nucleoli, a reduction in basal cells while maintaining an intact basement membrane, and is generally regarded as a pre-neoplastic lesion in humans. The majority of prostate cancers are pathologically classified as adenocarcinoma and display a luminal phenotype, with the absence of staining for basal cell markers p63 and cytokeratins 5 and 14 (CK5/14) [5]. Less common histological variants of PCa include, among others, squamous cell carcinoma and small cell carcinoma [6]. While individual prognosis is related to the stage and histological grade of the index lesion (i.e. largest cancer in the prostate), PCa is multifocal in nature with focal lesions having been found to possess different genetic abnormalities from one another [7]. The presence of multiple neoplastic lesions with unique genetic compositions in PCa suggests that different cells within prostate epithelia may be vulnerable and contribute towards tumorigenesis.
In this review, we define prostate stem cells (PSCs) as the cells capable of differentiating into single (unipotent) or multiple cell types (multipotent) that form the prostate epithelium. The PCa cell-of-origin refers to the cancer-initiating cell(s) within the prostate epithelium, whether it is a stem/progenitor cell itself or its differentiated progeny which acquire mutations that re-instigate self-renewal ability and prevent post-mitotic differentiation, resulting in the emergence of cancer stem cells [8]. Prostate cancer stem cells (PCSCs) are a subset of cells within the prostate tumor that display tumor-propagating ability, demonstrate long-term self-renewal potential, and support a model in which survival from radiotherapy, chemotherapy and development of castration-resistant disease is met with an increased proportion and/or activity of PCSCs. Therefore, understanding the cell-of-origin and the role of PCSCs in prostate tumorigenesis, the signaling pathways that promote PCSC maintenance as well as their involvement in tumor progression will be necessary towards achieving novel therapeutic approaches towards combating PCa.
CELLULAR HIERARCHY IN NORMAL MURINE AND HUMAN PROSTATES
The prostate contains epithelial cells of various lineage hierarchy that compose this glandular tissue. In adult males, the prostate gland contributes to the production of secretory proteins which are present within the seminal fluid [5]. Within the prostate epithelium, three epithelial cell types exist: luminal secretory cells, basal cells and intermediate cells. Luminal cells are a differentiated androgen-dependent cell type that produce and release prostatic secretory proteins into the lumen including PSA [9], a serine (Ser) protease responsible for preventing semen coagulation during ejaculation [10]. Luminal cells are characterized by the expression of androgen receptor (AR) [11], CD24 and CD26 cell surface proteins [12], as well as CK8 and CK18 [13]. Basal cells, which are located between luminal cells and the basement membrane, express CD44, p63 and CK5/14 proteins [13-15], display low AR protein levels [11] and lack PSA expression [9]. Intermediate cells are luminally-located cells in budding acini during prostate development that express cytokeratins found in basal and luminal cells [16], and are associated with luminal differentiation of basal cells [16-18]. Dispersed among basal and luminal cells are neuroendocrine cells, a rare AR-negative cell type in the prostate epithelium [19] (Figure 1).
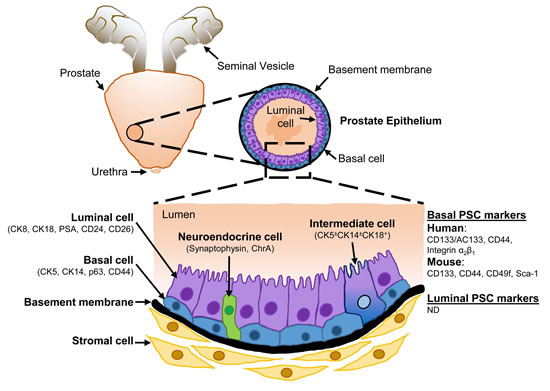
Figure 1: Schematic representation of the cellular architecture of the prostate epithelium. The prostate epithelium consists of an inner layer of secretory luminal cells. Basal cells form a continuous layer of cells around the luminal cells and in contact with the basement membrane, which serves as a barrier between the epithelium and stromal compartment. Intermediate cells display both basal and luminal markers, while rare neuroendocrine cells are scattered throughout the epithelium. Markers used to distinguish each cell type are indicated in brackets, along with the surface markers used to identify human and murine prostate stem cells (PSCs). ChrA Chromogranin A; ND not determined.
Prostate stem cells in murine tissues
Tissue-specific stem cells are defined by their capacity for long-term self-renewal and to produce mature progeny, which include non-renewing progenitors and terminally-differentiated cells that constitute distinct cell types within the tissue of interest [20]. Self-renewal is the ability of stem cells to maintain an undifferentiated state through cell division without losing their identity or functional potential, thus ensuring maintenance of the stem cell population during clonal growth [21-23]. The concept of a stem cell compartment in the prostate epithelium was first realized upon evaluating the regenerative capacity of the prostate following castration-induced atrophy in adult rats [24, 25]. Castration results in prostate regression in response to androgen deprivation, with a stable number of cells remaining in a regressed state. Upon re-administration of androgen, the prostate epithelium regenerated over a two-week period [24, 25]. The ability for the prostate to undergo several rounds of regression and regeneration following androgen ablation and restoration, respectively [26], indicates that the prostate contains a long-term surviving population of PSCs that are resistant to castration.
In the mouse prostate, there is evidence for distinct PSCs with either a basal or luminal phenotype. Prostate cells expressing stem cell antigen-1 (Sca-1) reconstitute secretory-producing prostatic ducts lined with basal and luminal cells, which form upon combining Sca-1+ cells with embryonic urogenital sinus mesenchyme (UGSM) cells under the renal capsule of mice [27]. Using specific cell surface markers to further discriminate prostate basal (CD49f+) Sca-1+ cells from luminal (CD24+CD49f-), stromal (CD34+), haematopoietic (CD45+, Ter119+), and endothelial (CD31+) cell lineages (Lin), purified Sca-1+CD49f+Lin- cells demonstrated self-renewal ability in vitro and formed in vivo prostatic ducts containing basal and luminal cells [28]. Furthermore, a single murine prostate cell, defined by the Sca-1+CD133+CD44+CD117+Lin- marker profile, generated a secretion-producing prostate when transplanted with UGSM cells under the kidney capsule [29].
Although the functional prostate regeneration assay has demonstrated that murine prostate basal cells are capable of being bipotent, generating both basal and luminal cell lineages, such tissue reconstitution assays involve co-culturing basal cells with UGSM cells [27-29] which provides a strong inductive influence on prostate cells during engraftment [30]. To avoid any unexpected plasticity that may manifest upon removing prostate cells from their endogenous tissue microenvironment, genetic lineage-tracing experiments have explored the nature of prostate basal or luminal cells towards forming the prostate epithelium in situ following castration-driven prostate regression and androgen-mediated prostate regeneration studies. Expression of a tamoxifen (TAM)-inducible Cre-recombinase (Cre) driven by the CK5 promoter labelled rare basal cells within the prostate epithelium that produced both basal and luminal cell progeny in situ following androgen-mediated regeneration [26]. Similarly, basal cells in the developing and adult mouse prostate were observed to be multipotent, giving rise to basal, luminal and neuroendocrine cells following cell lineage analysis [17, 31], while prostate luminal progenitors contribute to luminal cell expansion during postnatal development [17]. These findings contrast with the results of recent reports indicating that prostate basal and luminal cell lineages are self-sustaining (unipotent) in the adult mouse prostate and do not typically undergo lineage conversion in situ [18, 32], with prostate basal cells requiring inflammatory cues to demonstrate plasticity and generate luminal cells [18].
Additional evidence supports the existence of PSCs that are of luminal cell origin. The p63-null prostate lacks basal cells, however, prostatic ducts containing luminal cells formed upon grafting p63-null prostate epithelial cells along with UGSM cells and supplementing with testosterone [33]. Furthermore, expression of a TAM-inducible Cre driven by the PSA promoter labelled prostate luminal cells that were capable of surviving castration and reconstituting the luminal cell compartment in situ following androgen treatment [34]. A population of castration-resistant Nkx3.1-expressing (CARN) cells, which display a luminal phenotype in the regressed prostate, generated prostate basal and luminal cells following androgen-mediated regeneration, indicating that CARN cells are bipotent in nature [35]. Therefore, regenerated prostate luminal cells appear to be derived from pre-existing luminal cells that survive castration [32, 34, 35]. The reason for these discrepancies is unclear at present and suggests that the prostate cell lineage hierarchy has not been clearly characterized, with distinct PSCs with different plasticities existing in the mouse prostate.
Prostate stem cells in human tissues
In the human prostate, initial evidence supported PSCs confined to the basal cell compartment. Human prostate cells with a basal phenotype undergo self-renewal in vitro [36], with the capacity to reconstitute the prostate epithelium containing basal and luminal cells in a prostate regeneration assay [36, 37]. The recent establishment of organoid cultures using human prostate epithelial cells isolated from primary tissues has demonstrated that both basal (CD49f+) and luminal (CD24+, CD26+) cell populations contain bipotent cells which retain the ability to differentiate towards prostate basal and luminal cell lineages in vitro, as well as in tissue reconstitution assays involving UGSM cells [38]. However, prostate organoids derived from human and mouse prostate epithelial cells do not generate prostate neuroendocrine cells [38, 39], suggesting that organoid culture conditions need to be optimized to promote neuroendocrine differentiation. Despite this incomplete reconstitution of the prostate epithelium by prostate epithelial cells in culture, organoids derived from prostate luminal cells more closely resemble prostate glands than those derived from prostate basal cells [38, 39], and therefore, they may be more physiologically relevant in studies which attempt to recapitulate in situ histology in an in vitro setting.
A number of cell surface markers have been used to identify prospective human PSCs. CD44, which binds the extracellular matrix component hyaluronate [40], enriched for human PSCs in the basal cell layer possessing prostate regenerative activity [36]. Rapid adhesion to type I collagen isolated a population of prostate epithelial cells expressing high cell surface levels of integrin α2β1 (α2β1hi), another prospective PSC marker. Primary prostate α2β1hi-expressing cells formed slow-proliferating colonies in culture displaying a basal phenotype (PSA-CK5/14+) and produced epithelia consisting of basal and secretory luminal (PSA+AR+) cells when combined with stromal cells and implanted in recipient mice [41], which was considered as evidence of PSC activity. CD133, a five-transmembrane domain cell surface glycoprotein [42], is another putative PSC marker. Monoclonal antibodies have been developed against distinct epitopes of CD133, the most commonly used being AC133 (CD133/1) and AC141 (CD133/2), with CD133/AC133 enriching for human haematopoietic and neural stem cells [43, 44]. CD133/AC133+ human prostate epithelial cells, which are restricted to the integrin α2β1-expressing basal cell subpopulation and account for less than 1% of prostate basal cells, generated prostate acini in vivo that resemble prostate epithelia consisting of basal and luminal cells [45] (Figure 1). Whether these cell surface markers highlight basal-derived PSCs in human prostate organoid cultures will likely be addressed in future studies, while the discovery of novel cell surface markers corresponding to PSCs of luminal cell origin, which at present are unknown, are enthusiastically anticipated.
CELL-OF-ORIGIN IN PROSTATE CANCER
The PCa cell-of-origin refers to the cell(s) within the prostate epithelium, whether it be a PSC or its lineage-restricted progeny, which acquires tumor-promoting mutations and subsequently initiates tumorigenesis [8]. At present, there is insufficient evidence supporting prostate cells which display prospective PSC markers as serving as the cell-of-origin in human PCa. Taylor and colleagues reported that the CD133/AC133+ PSC population may not serve as the cellular target responsible for prostate tumor initiation [46]. CD133/AC133+ cells from SV40-immortalized prostate cells were observed to be incapable of generating tumors when recombined with UGSM cells or cancer-associated fibroblasts in immunocompromised mice, whereas CD133/AC133- cells formed xenograft tumors [46]. While the CD133/AC133+ PSC population may acquire oncogenic mutations, its differentiated (CD133/AC133-) progeny may subsequently undergo oncogenic transformation and correspond to the PCa cell-of-origin. Similarly, genetic lineage-tracing in a glioma mouse model has shown that neural stem cells acquire oncogenic mutations, however, it is their descendent oligodendrocyte progenitor cells that act as tumor-initiating cells during gliomagenesis [47]. Therefore, an alternative approach towards investigating the cell-of-origin for PCa has involved studying the vulnerability of prostate basal and luminal epithelial cells towards tumor initiation using transgenic mouse models of PCa, with cell surface marker expression being utilized to characterize the tumor-propagating cell population which sustains prostate tumorigenesis.
Basal versus luminal cell origin for PCa
Elucidation of the cell lineage relationships within the prostate epithelium is relevant for understanding the origins of PCa. Given that prostate adenocarcinoma has a luminal phenotype, the cell-of-origin for PCa should correspond to a luminal cell that has undergone oncogenic transformation, or a basal cell that differentiates and proliferates as luminal progeny following tumorigenesis [5]. A basal cell origin for PCa has been suggested as human prostate basal cells expressing AR, AKT and ERG oncogenes formed carcinoma in tissue reconstitution assays that is histologically similar to clinical human PCa [37]. Human prostate luminal cells appear to be resistant to AKT and MYC-induced transformation, suggesting that additional oncogenes like eIF4E, a target of PI3K/AKT signaling, may be required to promote neoplastic transformation of prostate luminal cells [48].
Genetically-engineered animal models have been utilized to address the cell-of-origin in PCa. Murine prostate basal cells have been reported to undergo neoplastic transformation [26, 32, 49]. In contrast, deletion of the Pten (Phosphatase and tensin homolog deleted on chromosome 10) tumor suppressor gene in luminal CARN cells resulted in carcinoma formation following androgen treatment [35]. While prostate carcinomas generated following Pten deletion in prostate basal or luminal cells are histologically similar [26, 32], basal cells are relatively resistant towards oncogene-induced transformation in situ [32] with prostate basal cell-derived carcinoma arising slowly [26]. This long latency may be due to the loss of Pten activity inducing murine prostate basal cells to differentiate into transformation-competent luminal cells [32], which requires prostatic inflammation to accelerate this differentiation process [18].
Animal models also support a luminal cell origin for PCa. Loss of the Nkx3.1 tumor suppressor gene, whose expression is restricted to luminal cells in human prostate glands [50], induced the formation of pre-neoplastic PIN-like lesions in murine prostates [50-52], with concurrent loss of Pten [53, 54] or overexpression of MYC oncoprotein [55] promoting the development of invasive carcinoma. PTEN ablation in prostate luminal cells is capable of driving PCa formation itself, as PTEN loss following PSA promoter-driven TAM-inducible Cre expression (PSACreERT2Ptenlox/lox) resulted in PIN formation (2-3 months) followed by invasive adenocarcinoma development (8-10 months) [56]. More recently, Shen and colleagues demonstrated through genetic lineage-tracing that prostate luminal cells, but not prostate basal cells, were consistently observed as the PCa cell-of-origin in a diverse range of mouse models (Pten+/-, Nkx3.1+/-Pten+/-, TRAMP, and transgenic ARR2/probasin-driven Myc) in situ [57]. Furthermore, luminal cell-derived murine PCa displays a molecular gene signature that is highly correlated with poor patient prognosis (reduced biochemical-free survival) and upregulated in high-risk patients [26].
In addition to influencing the tumor cell’s malignant potential, recent evidence suggests that the nature of the cellular target within the prostate tumor may also influence tumor histopathology. Basal tumor cells can generate tumor lesions exhibiting a mixture of squamous and adenocarcinoma regions in tissue reconstitution assays, while luminal-like tumor cells generate tumors with solely an adenocarcinoma phenotype [48]. Therefore, cell-of-origin models propose that distinct cell types within the prostate cell lineage hierarchy are vulnerable towards tumorigenesis and may give rise to PCa subtypes with different histopathologies, prognoses and/or treatment responses. At least in a mouse model, luminal cell-derived prostate tumors seem to recapitulate the more aggressive types of human PCa [26], while NKX3.1 haploinsufficiency, PTEN loss and/or allelic gain of the c-MYC gene in human PCa are associated with increased biochemical relapse (recurrence of PSA serum levels) following radiotherapy [58, 59]. This implies a scenario whereby human prostate tumors, which are associated with different prognoses and phenotypes, may originate from prostate basal and/or luminal cells, respectively.
EVIDENCE OF PCSCs IN HUMAN PROSTATE CANCERS
PCa is characterized by the excessive and uncontrolled growth of abnormal cells that invade the basement membrane, metastasizing primarily to the lymph nodes or bone. Like many other carcinomas, PCa exhibits a functional hierarchy of cells with tumor-propagating cells positioned at the apex and having the ability to differentiate into a spectrum of less tumorigenic progeny [60, 61]. The cancer stem cell (CSC) hypothesis postulates that only a subset of cells within the tumor are capable of sustaining tumorigenesis and driving disease progression, while establishing the cellular heterogeneity that constitutes the primary tumor. Stem-like cancer cells, often referred to as CSCs or tumor-propagating cells, are defined by their ability to initiate tumors upon implanting in immunocompromised mice [62]. CSCs generate non-tumorigenic progeny, resulting in intrinsically different populations of tumorigenic and non-tumorigenic cells within the tumor [63]. Despite the limited experimental means to identify and evaluate CSCs in situ, the ability of an isolated, phenotypically distinct population of cancer cells to initiate tumor formation in immunocompromised mice is currently the standard assay available to define human CSCs in vivo [62].
Accumulating evidence supports the existence of human PCSCs. A small population of primary PCa cells expressing the cell surface antigenic profile CD133/AC133+CD44+α2β1hi were suggested to be prospective candidates of PCSCs [64], as CSCs are known to share similar cell surface antigens with their normal tissue stem cell counterparts [65]. Consistent with this concept, human prostate epithelial cells and malignant cells express PSC markers CD44, CD133 and/or integrin α2β1 [66-68], and these prospective PCSCs generated xenograft tumors that resembled the original tumor [66]. These observations are supported by reports that CD44+ cells isolated from PCa cell lines display increased tumorigenicity in vivo compared to CD44- cells [60], with the CD44+α2β1+ cell population being enriched in PCSCs [61]. Furthermore, PCa cells expressing CD133/AC141 and CD44 displayed an increased ability to form tumors in vivo compared to an equal number of CD133/AC141-CD44- cells [69]. Despite these observations supporting the candidacy of CD133+CD44+α2β1hi cells as prospective PCSCs, it still remains to be demonstrated whether CD133+CD44+α2β1hi cells isolated from primary PCa tissues are capable of tumor initiation in vivo.
Due to the constraints in obtaining a sufficient amount of human PCa material to isolate and characterize prospective PCSCs, established PCa cell lines and/or xenograft models have become useful surrogate sources for investigating human PCSCs. Immortalized human prostate cells expressing integrin α2β1, but not CD133/AC133, formed xenografts with malignant characteristics [46], implicating proliferative CD133/AC133- cells [45] as being more susceptible to tumorigenesis compared to CD133/AC133+ stem-like cells [64]. Human PCa cells derived from androgen-insensitive DU145 cells and propagated under serum-free conditions as non-adherent spheres express surface markers CD44, integrin α2β1 and CD24, both basal and luminal cytokeratins, and display increased tumorigenicity in vivo [70]. Using human primary PCa xenografts, CD133+ or CD24+ PCa cells, isolated from tumor growths and serially diluted, initiate secondary tumor xenograft formation at comparable frequencies [71]. Moreover, a small subset of cells isolated from the CWR22 orthotopic tumor xenograft model display TRA-1-60, CD166 and CD151 markers, with TRA-1-60+CD166+CD151+ cells initiating tumor formation in vivo at increased efficiency following serial dilution of cells compared to TRA-1-60+ cells [72]. Tumor xenografts derived from primary human PCa tissue also recapitulate histological features present in the original tumor, with castration resulting in tumor regression leaving residual stem-like cells which display tumor-regenerating potential following testosterone re-administration [73]. Therefore, PCa xenografts serve as an in vivo source of human PCa cells, which can be propagated following serial transplantation in a number of grafting sites and subsequently used to isolate prospective human PCSCs. However, the recent generation of patient-derived PCa organoid cultures that recapitulate the diverse mutational landscape and histology observed in their original PCa tissue samples [74] will likely further aid in the characterization of PCSCs, as well as improve the understanding of the molecular determinants of therapeutic and castration resistance.
EVALUATION OF PCSC ACTIVITY
In vitro propagation and evaluation of self-renewal activity of PCSCs
Different culture methods have been utilized to propagate PCSCs derived from human PCa cell lines including suspension or low adherence culture [67, 68], and media cultures containing either fetal bovine serum [60, 61, 66] or serum-free media supplemented with epidermal growth factor [75, 76] and basic fibroblast growth factor [67, 69, 70, 72]. However, the formation of free-floating spheres under serum-free conditions is routinely used as an in vitro approach towards propagating and evaluating the stem-like properties of putative CSCs derived from solid tumors [62], including those derived from PCa cells [69, 70, 72]. Despite its limitations [77], the sphere formation assay allows for the clonality and self-renewal capacity of stem-like cells to be evaluated upon culturing individual cells, reducing the impact of progenitor-like cells which display limited self-renewal potential [78]. The self-renewal capacity of sphere-derived human PCSCs has been evaluated [70, 76], which display enhanced tumor formation in vivo [69, 70, 72, 76].
Functional identification of prostate cancer cells with stem-like properties
While purification of human PCa cells based on the expression of surface markers isolates stem-like cells with tumor-initiating ability (Table 1), flow cytometry can also enrich for prospective PCSCs based on their ability to efflux the Hoechst 33342 dye. These cells, also known as side population (SP) cells [79], isolated from androgen-sensitive LAPC9 cells are capable of in vivo tumor formation unlike the non-SP cell population [80]. The SP phenotype is associated with the expression of the ATP-binding cassette membrane transporter family of proteins, particularly ABCG2 [81], which are involved in drug efflux and confer multi-drug resistance [82]. However, ABCG2+ and ABCG2- PCa cells displayed similar tumorigenic potential in vivo [80], implicating the expression of additional ABC transporter proteins and/or other means of Hoechst 33342 dye efflux by these PCa cells.
An alternative strategy towards purifying PCSCs involves measuring their aldehyde dehydrogenase (ALDH) activity. Elevated ALDH activity has been associated with CSCs in various solid tumors [83] and is required to maintain the drug-tolerant cancer cell subpopulation [84]. ALDH1A1, which is expressed by a minor basal cell population in human prostates [85], was observed to be heterogeneously expressed in prostate adenocarcinomas, with elevated ALDH1A1 expression being positively correlated with the severity (Gleason score and pathologic stage) of the disease and inversely correlated with patient survival [85, 86]. Primary and metastatic PCa also express ALDH7A1 protein [87], with knockdown of ALDH7A1 reducing the tumor-propagating and metastatic abilities of androgen-independent PCa cells [88]. Using a functional (Aldefluor™) assay validated to assess ALDH1 isoform activities [89, 90], PCa cells demonstrating high ALDH activity (ALDHhi) were isolated and displayed increased tumorigenicity in vivo [87]. In addition, as few as 101 ALDHhiCD44+α2β1+ cells isolated from LAPC9 (low PSA-expressing; PSAlow) tumor xenografts generated secondary tumors in castrated mice whereas 104 ALDHlowCD44-α2β1- cells were required for tumor formation, indicating that the ALDHhiCD44+α2β1+ phenotype in a PSAlow cell population is enriched for castration-resistant PCSCs [91]. While 19 ALDH isoforms have been identified in humans [83], other ALDH isoforms have yet to be shown to be functionally-active in PCSCs or required for tumor formation in vivo. As no individual method isolates bona-fide human PCSCs exclusively, a combination of methods (sphere culture, and isolation based on cell surface marker expression and/or functional activity) can be used to enrich for and characterize human PCSCs, with subsequent implantation in immunocompromised mice to evaluate their tumor-propagating activity and serial transplantation to assess their long-term self-renewal capability in vivo [62].
Table 1: Marker profiles of prospective human prostate cancer stem-like cells (PCSCs).
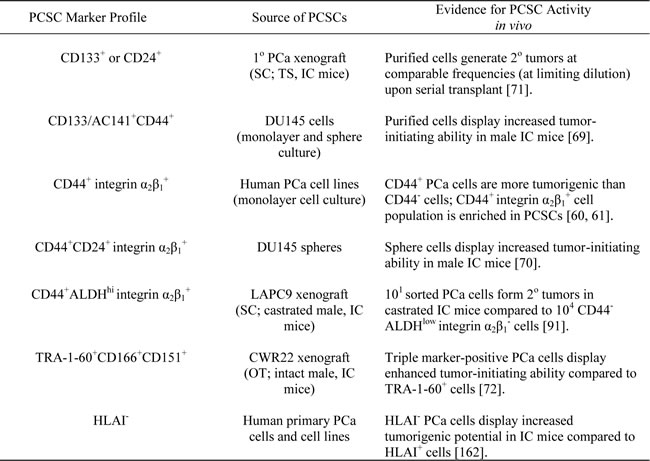
Abbreviations: 1o primary; 2o secondary; hi high activity; HLAI human leukocyte antigen class I; IC immunocompromised; OT orthotopic; PCa prostate cancer; TS testosterone-supplemented; SC subcutaneous.
CELL SIGNALING PATHWAYS UTILIZED BY PROSTATE CANCER AND THEIR TUMOR-PROPAGATING CELLS
A number of cell signaling pathways have been implicated in PCa progression towards an androgen-resistant state including receptor tyrosine kinases, like epidermal growth factor receptor (EGFR) [92-94], and developmental pathways including Wnt, Notch and Hedgehog signaling [95-97]. These developmental pathways have also been implicated in PCSC maintenance [98-100]. Human PSCs and PCSCs display low levels or lack AR expression [45, 64, 66, 72], with evidence to suggest that murine PSCs and PCSCs are capable of surviving in an androgen-independent state [26, 32, 101]. While not to undermine the importance of these developmental pathways and AR signaling in PCa, we will focus on the involvement of intracellular PI3K/AKT, RAS/MAPK and STAT3 pathways in maintaining PCSCs. Given that details of these signaling pathways and their ability to regulate various hallmarks of PCa have been reviewed thoroughly elsewhere [102-104], we highlight key proteins within these pathways that regulate PCSC activity (Figure 2), and the animal models that support their role in PCa development (Figure 3).
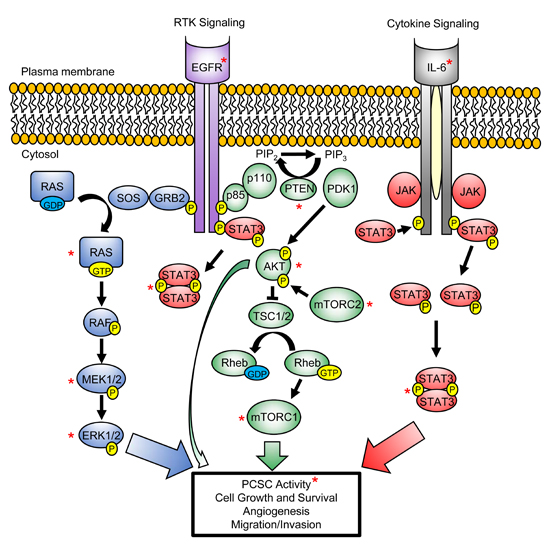
Figure 2: PI3K/AKT, RAS/MAPK and STAT3 signaling pathways converge to regulate PCSC maintenance and promote tumorigenesis. Activation of PI3K/AKT (green), RAS/MAPK (blue) and STAT3 (red) signaling pathways, mediated by the activation of growth factor-driven receptor tyrosine kinase (RTK) (e.g. Epidermal growth factor receptor (EGFR)) or cytokine (e.g. IL-6) signaling, promote PCSC self-renewal activity and the various hallmarks of PCa development. These signaling pathways act directly, or through cross-talk activation, to mediate prostate tumorigenesis. P denotes phosphorylation of protein at specific residue(s), which is required for its activation (yellow). Red asterisk (*) marks key proteins within these signaling pathways that have been implicated in PCSC activity.
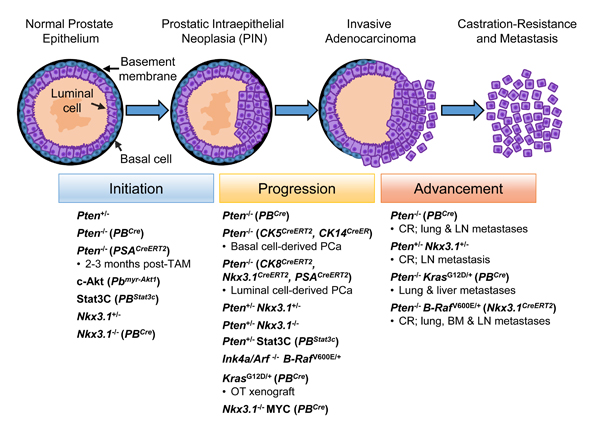
Figure 3: Prostate cancer initiation, progression and advancement are associated with proto-oncogene activation and inhibition of tumor suppressor genes involved in PI3K/AKT, RAS/MAPK and STAT3 signaling. Abnormalities in the prostate epithelium result in pre-neoplastic lesions called prostatic intraepithelial neoplasia (PIN), which feature luminal epithelial hyperplasia and a reduction in the number of basal cells. PIN lesions progress to invasive adenocarcinoma (luminal phenotype) with loss of the basal cell layer and basement membrane resulting in various tumor grades, beginning with indolent to more aggressive forms of PCa, and subsequent development of metastasis and castration resistance. Studies involving murine PCa models provide support for the role of proteins involved in PI3K/AKT, RAS/MAPK and STAT3 signaling at various stages of PCa development. Murine PCa models discussed in the text are illustrated, with specific promoter-driven gene knockout or transgene overexpression indicated in brackets. Figure is adapted from ref. [5]. BM bone marrow; c-Akt constitutive-active Akt; CR castration-resistant; Cre Cre-recombinase; CreER/CreERT2 TAM-inducible Cre; LN lymph node; OT orthotopic; PB probasin promoter; TAM tamoxifen.
PI3K/AKT signaling
Dysregulation of PI3K/AKT signaling has been implicated in PCa given the discovery that its negative regulator, PTEN, is mutated and frequently lost in PCa. Loss of PTEN function, as a result of mutation, deletion or reduced expression, occurs at a frequency of ~40% in PCa [105, 106]. Integrative analysis of mRNA expression, copy number and exon sequencing for somatic mutations conducted on a cohort of PCa patients revealed that the PI3K/AKT pathway was altered in 42% of primary cancers and 100% of metastases, while the RAS/MAPK pathway was altered in 43% of primary specimens and 90% of metastases [106]. Both PTEN loss and AKT activation, as measured by Ser 473 phosphorylation, are associated with biochemical relapse following radical prostatectomy [107, 108], while PTEN deletion (along with allelic c-MYC gain) is associated with biochemical relapse following local therapy [59]. Intense staining patterns for activated AKT are correlated with higher Gleason grade PCa [109] and tumor progression [110], while PTEN loss is associated with PCa progression and predicts a shorter time for metastasis-free survival in patients [111].
The role of PI3K/AKT signaling in PCa has also been examined in animal models. Pten+/- mice were first reported to display hyperplastic lesions in the prostate, with benign PIN lesions being observed in some mice at a young age (≤14 weeks) [112, 113]. Pten deletion in prostate basal or luminal cells has also been carried out to promote PI3K/AKT signal activation in order to determine the prostate cell lineage responsible for subsequent tumor initiation [26, 32, 35], as previously discussed. Prior to these studies, probasin (PB) promoter-driven Pten deletion was conducted, resulting in PI3K/AKT signal activation specifically in both prostate basal and luminal cells [114]. By 6 weeks of age, these mice formed PIN lesions, developed adenocarcinoma that invades stromal regions (9 weeks) and displays androgen-independent survival following castration, and generated metastases in the lymph nodes and lungs (12 weeks) [114]. Moreover, prostate epithelial cells lacking Pten require mammalian target of rapamycin (mTOR) complex 2 (mTORC2) for neoplastic transformation [115], thereby implicating downstream mTOR signaling in murine PCa development. Expression of a constitutively-active form of Akt1 (c-Akt) resulted in PIN formation when expression is driven by the PB promoter [116], or after reconstituting c-Akt-expressing prostate basal cells with UGSM cells and grafting under the kidney capsule of mice [49]. Therefore, the murine prostate epithelium has been shown to be vulnerable to Pten loss, with the propensity to undergo AKT-driven tumorigenesis.
The role of PI3K/AKT signaling in the proliferation and maintenance of PCSCs has been examined in human PCa. PTEN knockdown in DU145 PCa cells resulted in an increased ability to enrich for CD133/AC141+CD44+ stem-like cells [69]. Treatment with PI3K inhibitor LY294002 reduced sphere formation [69] while the dual PI3K/mTOR inhibitor NVP-BEZ235 reduced the CD133/AC141+CD44+ cell population in vivo and subsequently delayed tumor formation [117]. However, DU145 spheres display low levels of AKT activation while PTEN knockdown marginally increased AKT activation and did not affect sphere maintenance, suggesting that PTEN function may not be critical for PCSC self-renewal in vitro [70]. While PTEN may not be required to negatively-regulate PI3K/AKT-dependent PCSC maintenance, whether PTEN loss/inactivity and subsequent PI3K/AKT signal activation promotes PCSC differentiation into non-tumorigenic bulk tumor cells has not been thoroughly evaluated, despite PTEN knockdown increasing tumor formation in vivo [69].
RAS/MAPK signaling
Mitogen-activated protein kinases (MAPKs) are cytoplasmic Ser/threonine kinases that transduce signals by activating extracellular signal-regulated kinases (ERKs) through the RAS-RAF-MEK-ERK (RAS/MAPK) signal transduction cascade [118]. Similar to PI3K/AKT signaling, MAPK signal activation correlates with disease progression, with elevated staining for phosphorylated (active) forms of ERK1 and ERK2 (ERK1/2) being detected with increasing tumor stage of human PCa specimens [119]. Increased ERK1/2 activation in patients receiving neoadjuvant hormone therapy, and in recurrent CRPC patients, has been observed [120]. Mutations of all three RAS isoforms (KRAS, HRAS and NRAS) have been detected in human PCa specimens (3 – 30%), particularly among Japanese men [121-124], while the activating mutant B-RAF(V600E) has been observed in ~10% of Korean PCa patients [122]. These correlative studies reinforce that activated RAS/MAPK signaling contributes towards prostate tumorigenesis.
Further evidence in support of a relationship between RAS/MAPK signaling and PCa progression towards an androgen-independent state has been demonstrated in the preclinical setting. Ectopic expression of HRAS(T35S) in androgen-sensitive LNCaP cells maintained MAPK signal activation under serum-free conditions and increased their in vivo tumorigenicity compared to parental cells. More than two-thirds of these HRAS-expressing PCa xenografts were resistant to castration and displayed robust activation of MAPK signaling post-castration [125]. Conversely, inducible expression of dominant-negative HRAS(S17N) in the androgen-insensitive, highly tumorigenic derivative LNCaP cell line, C4-2, inhibited MAPK signaling and restored androgen sensitivity to these cells, causing tumor regression in surgically-castrated mice [126]. Furthermore, ERK1/2 activation is necessary and sufficient to mediate RAF-induced AR downregulation in PCa cells [127], supporting MAPK signaling in PCa advancement.
As studies suggest that RAS-driven MAPK activation plays a substantial role in PCa progression towards CRPC development and metastasis, the role of MAPK signaling in prostate tumor formation is less defined. DU145 PCSCs require MAPK signaling for their propagation in vitro, as treatment with MEK inhibitor U0126, ectopic expression of dominant-negative MEK1(K97M), or knockdown of either ERK1 or ERK2 reduced their sphere-forming ability [75]. Murine PCSCs generated as a result of expressing constitutively-active Kras(G12D) (PBCreKrasG12D/+) or deleting Pten (PBCrePtenlox/lox) in prostate epithelial cells initiated orthotopic tumor formation, while neither PCSC population was capable of generating detectable macrometastases by 10 weeks post-transplantation. However, PCSCs demonstrating concurrent activation of both PI3K/AKT and RAS/MAPK signaling (PBCrePtenlox/loxKrasG12D/+) promoted metastasis to the lymph nodes, lung and liver, with MEK inhibitor (PD325901) treatment reducing orthotopically-engrafted tumors and inhibiting their ability to metastasize [120]. Similarly, PTEN loss and B-Raf(V600E) expression following Nkx3.1-driven TAM-inducible Cre expression (Nkx3.1CreERT2Ptenlox/loxB-RafV600E/+) resulted in murine prostate tumors that were inherently castration-resistant and which metastasized to the lung, lymph nodes and bone marrow [128]. While these studies implicate concomitant activation of PI3K/AKT and RAS/MAPK signaling in PCa advancement, activation of MAPK signaling itself has been demonstrated to promote basal (p63+) cell proliferation and the development of invasive prostate adenocarcinoma independently of PI3K/AKT signal activation [129], supporting its involvement in prostate tumorigenesis.
STAT3 signaling
Signal transducer and activator of transcription (STAT) proteins have been implicated in prostate tumorigenesis, particularly STAT3 protein [130, 131]. Activated STAT3 (phosphorylation at tyrosine 705) is associated with higher Gleason score and pathological stage of the disease [132, 133]. STAT3 activation is also associated with decreased survival in PCa patients [133], particularly in CRPC patients [134], and a shorter time to death from biochemical relapse [133]. In patients having undergone radical prostatectomy or hormonal therapy, recurrence-free survival rates were lower in patients displaying increased levels of JAK1 and STAT3 activation [133]. Elevated serum levels of IL-6, a known activator of STAT3 signaling, were observed in CRPC and metastatic PCa patients compared to patients bearing benign or non-malignant forms [135-137], suggesting that activated STAT3 signaling correlates with the clinicopathologic features of PCa.
In preclinical studies, STAT3 signaling promotes PCa development. Treating mice bearing subcutaneously-engrafted TRAMP-C1 PCa cells (TC1) with flutamide, an AR antagonist, immediately following engraftment resulted in elevated STAT3 activation compared to vehicle-treated mice. Moreover, knockdown of STAT3 in TC1 cells reduced tumor growth [138]. Substitution of alanine 662 and asparagine 664 for cysteine residues in Stat3 promotes its dimerization and generates a constitutively-active transcription factor (Stat3C) [139], with PB promoter-driven Stat3C expression promoting the development of PIN lesions while invasive adenocarcinoma developed when combined with Pten heterozygosity [140]. In human PCa cells, androgen-insensitive DU145 and PC3 cells display elevated STAT3 activation compared to androgen-sensitive LNCaP cells [141], with inhibition of STAT3 expression or activity inducing apoptosis of DU145 cells [141, 142]. Conversely, expression of constitutively-active Stat3C in LNCaP cells promoted androgen-independent tumor growth in castrated mice [143].
While STAT3 activation has been implicated in PCa development, its role in PCSCs has only recently been investigated. Pre-treating human PCSCs (CD133/AC133+α2β1hi) with increasing concentrations of STAT3 inhibitor LLL12 abrogated their tumor-propagating ability in vivo [144]. Spheres derived from human PCa cells display elevated STAT3 activation [75, 145] with STAT3 knockdown reducing sphere formation, while treatment with a soluble IL-6 receptor fusion protein inhibited STAT3 activation and prevented tumor growth in vivo [138]. Whether activated by cytokines such as IL-6 [144], stressors like reactive oxygen species [145] or driven by the progression towards castration resistance [138], STAT3 activation plays a vital role in the self-renewal and tumor-propagating capacity of PCSCs.
Cell signaling pathways: maintenance of the PCSC pool versus drivers of PCa development
Prostate tumorigenesis is a complex process which involves many hallmarks that are important beyond simply maintaining PCSCs within the tumor. Many factors are necessary for PCSC generation in vivo, and it can be envisaged that secondary events govern the growth and sustenance of the subsequent tumor mass. While it is intriguing that PCSCs utilize PI3K/AKT, RAS/MAPK and STAT3 signaling to self-renew and maintain their presence within the tumor, these same pathways are also required for PCa cell proliferation and survival, tumor angiogenesis and metastasis (Figure 2). Whether these signaling pathways play a critical role in forming and/or sustaining the prostate tumor, drive PCSC maintenance or simply are required for generating the bulk of the less tumorigenic cells (non-PCSCs) within the tumor will need to be addressed further in future studies. Recently, STAT3 signal blockade has been shown to inhibit the prostate tumor-propagating and bulk tumor cell populations in patient-derived PCa xenograft models, as well as block tumor angiogenesis [146]. Moreover, IL-6 driven STAT3 signaling induced the formation of a cancer stem-like population from non-stem PCa cells [146, 147], which could be inhibited by treatment with the small molecule STAT3 inhibitor, Stattic [146]. As CSC-like cells can arise de novo from non-tumorigenic cells [148] or following induction as a result of IL-6 secretion by CSCs [147], a dynamic equilibrium between CSCs and non-CSCs is suggested to exist in order to maintain a constant proportion of tumor-propagating cells over successive generations [147, 148]. The extent to which PI3K/AKT and RAS/MAPK signaling contribute towards this dynamic equilibrium along with STAT3 signaling in PCa, driven by IL-6 or perhaps other molecules, remains to be elucidated. Taken together, activated signaling appears to be required by both PCSC and non-PCSCs in order to drive and sustain prostate tumorigenesis through its various hallmarks, and is likely to be relevant in heterogeneous prostate cancers that maintain a balanced, yet small, number of PCSCs within a larger population of non-tumorigenic PCa cells.
LESSONS FOR PROSTATE CANCER THERAPY: INHIBITION OF SIGNALING PATHWAYS AS A STRATEGY TO TARGET PCSCs
Active surveillance with serial PSA monitoring and prostate biopsies is a reasonable management option for patients with indolent, low-grade (≤Gleason 6) PCa [149]. However, 30% of patients diagnosed with low-grade PCa will transition to non-indolent cancers suggesting the presence of non-detected, aggressive tumor cell variants at the time of diagnosis [149] that are not reflected solely by Gleason score. This leads to the hypothesis that a higher proportion and/or activity of PCSCs may be present within these transitioning tumors. Higher grade PCa present as multifocal tumors [150] with genetic heterogeneity among foci [7], which suggests of multiple neoplastic transformation events occurring within the prostate and an increased number of PCSCs that may (or not) be genetically-distinct.
The standard treatment for non-metastatic, localized (intermediate and high risk) PCa remains to be external beam radiotherapy or radical prostatectomy, with strong evidence indicating that the efficacy of these therapies is related to the extent of killing local, proliferating tumor clones with PCSC properties [151-153]. Increased radiotherapy dose leads to increased clonogenic killing and the use of molecularly-targeted agents may lead to a better outcome by sterilizing radioresistant PCSCs [154, 155], providing improved local control and preventing the dissemination of resistant, proliferating stem-like PCa cells [156, 157]. A number of patients will also undergo biochemical relapse following localized therapy leading to the use of ADT, with progression-free survival lasting only several months following ADT compared to control treatment [158, 159] due to CRPC development. Similarly, docetaxel, an anti-mitotic chemotherapeutic agent, which is routinely used in treating patients whose disease progresses towards metastatic CRPC, provides a modest improvement in patient survival [160, 161]. Docetaxel-resistant PCa cells possess an increased number of cells with an undifferentiated phenotype due to the lack of low molecular weight cytokeratins (CK18 and CK19) and human leukocyte antigen (HLA) class I proteins, display elevated levels of AKT activation and pro-survival Bcl-2 protein expression, and demonstrate a higher tumor-propagating capacity in vivo compared to their parental, docetaxel-sensitive cells [162]. Therefore, along with enhanced radioresistance and chemoresistance [154, 162], development of castration-resistant disease serves as an adaptive or selective pressure for increasing the number and/or activity of PCSCs.
The progression towards an androgen-resistant state is associated with elevated PI3K/AKT, RAS/MAPK and STAT3 signaling, signaling pathways that promote PCSC self-renewal and tumorigenesis, as previously described. Indeed, a reciprocal feedback loop exists between PI3K/AKT and AR signaling which regulates castration-resistant PCa growth [163, 164]. Neoplastic transformation of the prostate by cell autonomous PI3K/AKT signal activation occurs in the absence of epithelial AR expression or following androgen withdrawal [164], suggesting that androgen-targeted therapy may not be able to ablate PCSCs that display activated PI3K/AKT signaling and survive in an androgen-independent state. AKT also promotes phosphorylation of B-RAF (Ser 445) and its subsequent activation, with ERK1/2 activation requiring androgen deprivation in androgen-sensitive PCa cells [165]. Phosphorylation of Ser 727 residue on STAT3, which enhances its transcriptional activity [166], occurs in an ERK-dependent fashion [167], while ERK1/2 can activate mTOR complex 1 (mTORC1) directly [168] or through functional inactivation of tuberous sclerosis factor 2 (TSC2) [169] in order to promote RAS-dependent mTOR signaling. Furthermore, downregulation of AR signaling itself has been shown to promote a stem-like phenotype and increase the tumorigenicity of PCa cells through a STAT3 signal-dependent mechanism [138]. Taken together, a model can be proposed whereby reduced dependence on AR signaling promotes activation and subsequent cross-talk between PI3K/AKT, RAS/MAPK and STAT3 pathways to regulate PCSC maintenance and tumorigenesis, with sustained activation of these pathways in CRPC (Figure 4).
A combinatorial approach towards blocking multiple signaling pathways that are coordinately dysregulated in PCa has shown to be an effective strategy towards inhibiting PCa growth in the preclinical setting. Targeting PI3K/AKT/mTOR and MAPK pathways by combined PD325901 (MEK inhibitor) and rapamycin (mTOR inhibitor) treatment reduced prostate tumor burden of Nkx3.1+/-Pten+/- mice, particularly following castration [110]. Furthermore, the combination of PD325901 and rapamycin treatment of mice bearing orthotopically-transplanted PCSCs displaying conditional Pten loss and oncogenic Kras(G12D) expression showed reduced primary tumor burden compared to PD325901 treatment alone, and ablated their ability for macrometastatic colonization [120]. This combined treatment was also effective in reducing the number of metastatic cases by one-third in mice displaying conditional Pten loss combined with oncogenic B-Raf(V600E) expression [128]. Finally, NVP-BEZ235 (PI3K/mTOR inhibitor) treatment in combination with docetaxel was more effective than chemotherapy alone at decreasing the human PCSC population and preventing tumor formation in vivo [117]. Taken together, these preclinical studies provide lessons and promise for treating advanced stage PCa (including metastatic CRPC) with the hope that a combinatorial strategy for PI3K/AKT/mTOR, RAS/MAPK and/or STAT3 signal inhibition will impose a series of blockades that will impact the tumor-propagating ability and maintenance of PCSCs, thereby lowering PCSC burden by altering the dynamic balance, and subsequent conversion, between non-PCSC and PCSC pools. Whether a pathway-targeted treatment regime, alone or in combination with current local, ADT, immunological or chemotherapeutic regimens, would lead to cure for individual patients at risk of systemic spread remains an area for future investigation. Therefore, pre-screening and subsequent monitoring of patient tumors for activated signaling and response to specific therapy will be required to determine the efficacy of such combinatorial therapy as a first-line or secondary treatment within the context of precision cancer medicine.
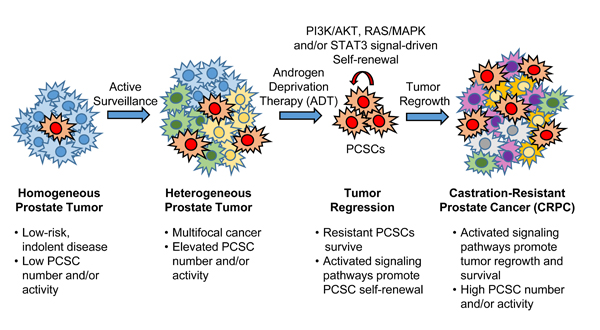
Figure 4: Reduced dependence on AR signaling promotes activation and subsequent cross-talk between PI3K/AKT, RAS/MAPK and STAT3 pathways to regulate PCSC maintenance and tumorigenesis. Low-risk (indolent) PCa progresses during active surveillance to a heterogeneous tumor that is multifocal in nature. Conventional androgen-deprivation therapy (ADT) causes tumor regression by targeting androgen-responsive PCa cells. Inhibition of androgen receptor (AR) signaling promotes activation of cell survival pathways like PI3K/AKT, RAS/MAPK and STAT3, either directly or through cross-talk, in surviving androgen-independent PCSCs. Surviving PCSCs drive tumor regrowth and the development of castration-resistant prostate cancer (CRPC), with cell signal activation being sustained in castration-resistant disease.
CONCLUDING REMARKS
Despite PCa being a prevalent malignancy in men, our understanding of prostate tumorigenesis remains limited, including knowledge of its origin, as well as the factors and signaling pathways that regulate its initiation. Based on the reported tumor-propagating abilities demonstrated for CSCs in various solid tumors, PCSCs are believed to sustain prostate tumorigenesis and efforts have been made using cell-based and animal models to address their existence. Evidence in support of the existence of PSCs has been demonstrated in prostate regeneration experiments following castration and re-administration of androgen, with candidate PSCs displaying either basal or luminal cell characteristics in prostate glands that survive androgen depletion. Although aberrant cytokine-driven and receptor tyrosine kinase-driven signaling in prostate cells promotes PCa development, there is insufficient evidence demonstrating that PSCs acquire tumor-promoting mutations and serve as the PCa cell-of-origin, or whether it is their differentiated progeny that initiate prostate tumorigenesis. While PCa is generally accepted to be heterogeneous in nature, distinct subtypes formed due to the activation of different oncogenic pathways remain undefined, and whether human prostate basal and luminal epithelial cells are equally vulnerable to oncogenic transformation and subsequently account for the multifocality of neoplastic lesions within a given section of PCa tissue remains unclear. In addition, PCSCs are believed to be dynamic entities that co-exist in equilibrium with non-PCSC populations, which are likely to display variable phenotypes depending on their cell-of-origin and produce different PCa subtypes. A deeper understanding of PCa heterogeneity and multifocality, patient tumor profiling to determine the most effective combinatorial treatment, and subsequent monitoring of their tumors during treatment will be critical in achieving an efficacious response in personalized therapy.
ACKNOWLEDGEMENTS
We would like to thank Dr. Jean-Claude Cutz (McMaster University) and Dr. Gaetano Zafarana (University Health Network, Toronto, ON, Canada) for their constructive scientific input during the preparation of this paper. This work was supported by research funding from the McMaster Institute of Urology and St. Joseph’s Hospital.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
1. Siegel R, Ma J, Zou Z and Jemal A. Cancer statistics, 2014. CA: a cancer journal for clinicians. 2014; 64:9-29.
2. Fraser M, Berlin A, Bristow RG and van der Kwast T. Genomic, pathological, and clinical heterogeneity as drivers of personalized medicine in prostate cancer. Urologic oncology. 2014.
3. Scher HI and Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2005; 23(32):8253-8261.
4. McNeal JE and Bostwick DG. Intraductal dysplasia: a premalignant lesion of the prostate. Hum Pathol. 1986; 17:64-71.
5. Shen MM and Abate-Shen C. Molecular genetics of prostate cancer: new prospects for old challenges. Genes Dev. 2010; 24:1967-2000.
6. Humphrey PA. Histological variants of prostatic carcinoma and their significance. Histopathology. 2012; 60(1):59-74.
7. Macintosh CA, Stower M, Reid N and Maitland NJ. Precise microdissection of human prostate cancers reveals genotypic heterogeneity. Cancer Res. 1998; 58:23-28.
8. Visvader JE. Cells of origin in cancer. Nature. 2011; 469(7330):314-322.
9. Bonkhoff H, Stein U and Remberger K. Multidirectional differentiation in the normal, hyperplastic, and neoplastic human prostate: simultaneous demonstration of cell-specific epithelial markers. Hum Pathol. 1994; 25:42-46.
10. Armbruster DA. Prostate-specific antigen: biochemistry, analytical methods, and clinical application. Clin Chem. 1993; 39:181-195.
11. De Marzo AM, Nelson WG, Meeker AK and Coffey DS. Stem cell features of benign and malignant prostate epithelial cells. J Urol. 1998; 160:2381-2392.
12. Liu AY, Roudier MP and True LD. Heterogeneity in primary and metastatic prostate cancer as defined by cell surface CD profile. Am J Pathol. 2004; 165(5):1543-1556.
13. Sherwood ER, Berg LA, Mitchell NJ, McNeal JE, Kozlowski JM and Lee C. Differential cytokeratin expression in normal, hyperplastic and malignant epithelial cells from human prostate. J Urol. 1990; 143:167-171.
14. Liu AY and True LD. Characterization of prostate cell types by CD cell surface molecules. Am J Pathol. 2002; 160:37-43.
15. Yang A, Kaghad M, Wang Y, Gillett E, Fleming MD, Dotsch V, Andrews NC, Caput D and McKeon F. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell. 1998; 2:305-316.
16. Xue Y, Smedts F, Debruyne FM, de la Rosette JJ and Schalken JA. Identification of intermediate cell types by keratin expression in the developing human prostate. Prostate. 1998; 34:292-301.
17. Ousset M, Van Keymeulen A, Bouvencourt G, Sharma N, Achouri Y, Simons BD and Blanpain C. Multipotent and unipotent progenitors contribute to prostate postnatal development. Nat Cell Biol. 2012; 14(11):1131-1138.
18. Kwon OJ, Zhang L, Ittmann MM and Xin L. Prostatic inflammation enhances basal-to-luminal differentiation and accelerates initiation of prostate cancer with a basal cell origin. Proc Natl Acad Sci U S A. 2014; 111(5):E592-600.
19. Noordzij MA, van Steenbrugge GJ, van der Kwast TH and Schröder FH. Neuroendocrine cells in the normal, hyperplastic and neoplastic prostate. Urological research. 1995; 22:333-341.
20. Wagers AJ and Weissman IL. Plasticity of adult stem cells. Cell. 2004; 116:639-648.
21. Kreso A and Dick JE. Evolution of the cancer stem cell model. Cell stem cell. 2014; 14:275-291.
22. Sneddon JB, Borowiak M and Melton DA. Self-renewal of embryonic-stem-cell-derived progenitors by organ-matched mesenchyme. Nature. 2012; 491:765-768.
23. Chambers I and Smith A. Self-renewal of teratocarcinoma and embryonic stem cells. Oncogene. 2004; 23:7150-7160.
24. Sugimura Y, Cunha GR and Donjacour AA. Morphological and histological study of castration-induced degeneration and androgen-induced regeneration in the mouse prostate. Biol Reprod. 1986; 34:973-983.
25. English HF, Santen RJ and Isaacs JT. Response of glandular versus basal rat ventral prostatic epithelial cells to androgen withdrawal and replacement. Prostate. 1987; 11:229-242.
26. Wang ZA, Mitrofanova A, Bergren SK, Abate-Shen C, Cardiff RD, Califano A and Shen MM. Lineage analysis of basal epithelial cells reveals their unexpected plasticity and supports a cell-of-origin model for prostate cancer heterogeneity. Nat Cell Biol. 2013; 15:274-283.
27. Burger PE, Xiong X, Coetzee S, Salm SN, Moscatelli D, Goto K and Wilson EL. Sca-1 expression identifies stem cells in the proximal region of prostatic ducts with high capacity to reconstitute prostatic tissue. Proc Natl Acad Sci U S A. 2005; 102:7180-7185.
28. Lawson DA, Xin L, Lukacs RU, Cheng D and Witte ON. Isolation and functional characterization of murine prostate stem cells. Proc Natl Acad Sci U S A. 2007; 104:181-186.
29. Leong KG, Wang BE, Johnson L and Gao WQ. Generation of a prostate from a single adult stem cell. Nature. 2008; 456:804-808.
30. Xin L, Ide H, Kim Y, Dubey P and Witte ON. In vivo regeneration of murine prostate from dissociated cell populations of postnatal epithelia and urogenital sinus mesenchyme. Proc Natl Acad Sci U S A. 2003; 100 Suppl 11896-11903.
31. Pignon J-C, Grisanzio C, Geng Y, Song J, Shivdasani RA and Signoretti S. p63-expressing cells are the stem cells of developing prostate, bladder, and colorectal epithelia. Proceedings of the National Academy of Sciences of the United States of America. 2013; 110:8105-8110.
32. Choi N, Zhang B, Zhang L, Ittmann M and Xin L. Adult murine prostate basal and luminal cells are self-sustained lineages that can both serve as targets for prostate cancer initiation. Cancer Cell. 2012; 21:253-265.
33. Kurita T, Medina RT, Mills AA and Cunha GR. Role of p63 and basal cells in the prostate. Development. 2004; 131:4955-4964.
34. Liu J, Pascal LE, Isharwal S, Metzger D, Ramos Garcia R, Pilch J, Kasper S, Williams K, Basse PH, Nelson JB, Chambon P and Wang Z. Regenerated luminal epithelial cells are derived from preexisting luminal epithelial cells in adult mouse prostate. Mol Endocrinol. 2011; 25:1849-1857.
35. Wang X, Kruithof-de Julio M, Economides KD, Walker D, Yu H, Halili MV, Hu YP, Price SM, Abate-Shen C and Shen MM. A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature. 2009; 461:495-500.
36. Garraway IP, Sun W, Tran CP, Perner S, Zhang B, Goldstein AS, Hahm SA, Haider M, Head CS, Reiter RE, Rubin MA and Witte ON. Human prostate sphere-forming cells represent a subset of basal epithelial cells capable of glandular regeneration in vivo. Prostate. 2010; 70:491-501.
37. Goldstein AS, Huang J, Guo C, Garraway IP and Witte ON. Identification of a cell of origin for human prostate cancer. Science. 2010; 329:568-571.
38. Karthaus WR, Iaquinta PJ, Drost J, Gracanin A, van Boxtel R, Wongvipat J, Dowling CM, Gao D, Begthel H, Sachs N, Vries RG, Cuppen E, Chen Y, Sawyers CL and Clevers HC. Identification of Multipotent Luminal Progenitor Cells in Human Prostate Organoid Cultures. Cell. 2014.
39. Chua CW, Shibata M, Lei M, Toivanen R, Barlow LJ, Bergren SK, Badani KK, McKiernan JM, Benson MC, Hibshoosh H and Shen MM. Single luminal epithelial progenitors can generate prostate organoids in culture. Nat Cell Biol. 2014; 16(10):951-961, 951-954.
40. Aruffo A, Stamenkovic I, Melnick M, Underhill CB and Seed B. CD44 is the principal cell surface receptor for hyaluronate. Cell. 1990; 61:1303-1313.
41. Collins AT, Habib FK, Maitland NJ and Neal DE. Identification and isolation of human prostate epithelial stem cells based on alpha(2)beta(1)-integrin expression. J Cell Sci. 2001; 114:3865-3872.
42. Miraglia S, Godfrey W, Yin AH, Atkins K, Warnke R, Holden JT, Bray RA, Waller EK and Buck DW. A novel five-transmembrane hematopoietic stem cell antigen: isolation, characterization, and molecular cloning. Blood. 1997; 90:5013-5021.
43. Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J and Dirks PB. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003; 63:5821-5828.
44. Yin AH, Miraglia S, Zanjani ED, Almeida-Porada G, Ogawa M, Leary AG, Olweus J, Kearney J and Buck DW. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood. 1997; 90:5002-5012.
45. Richardson GD, Robson CN, Lang SH, Neal DE, Maitland NJ and Collins AT. CD133, a novel marker for human prostatic epithelial stem cells. J Cell Sci. 2004; 117:3539-3545.
46. Taylor RA, Toivanen R, Frydenberg M, Pedersen J, Harewood L, Australian Prostate Cancer B, Collins AT, Maitland NJ and Risbridger GP. Human epithelial basal cells are cells of origin of prostate cancer, independent of CD133 status. Stem Cells. 2012; 30:1087-1096.
47. Liu C, Sage JC, Miller MR, Verhaak RG, Hippenmeyer S, Vogel H, Foreman O, Bronson RT, Nishiyama A, Luo L and Zong H. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell. 2011; 146(2):209-221.
48. Stoyanova T, Cooper AR, Drake JM, Liu X, Armstrong AJ, Pienta KJ, Zhang H, Kohn DB, Huang J, Witte ON and Goldstein AS. Prostate cancer originating in basal cells progresses to adenocarcinoma propagated by luminal-like cells. Proc Natl Acad Sci U S A. 2013; 110(50):20111-20116.
49. Lawson DA, Zong Y, Memarzadeh S, Xin L, Huang J and Witte ON. Basal epithelial stem cells are efficient targets for prostate cancer initiation. Proc Natl Acad Sci U S A. 2010; 107:2610-2615.
50. Abdulkadir SA, Magee JA, Peters TJ, Kaleem Z, Naughton CK, Humphrey PA and Milbrandt J. Conditional loss of Nkx3.1 in adult mice induces prostatic intraepithelial neoplasia. Molecular and cellular biology. 2002; 22:1495-1503.
51. Bhatia-Gaur R, Donjacour AA, Sciavolino PJ, Kim M, Desai N, Young P, Norton CR, Gridley T, Cardiff RD, Cunha GR, Abate-Shen C and Shen MM. Roles for Nkx3.1 in prostate development and cancer. Genes & development. 1999; 13:966-977.
52. Kim MJ, Bhatia-Gaur R, Banach-Petrosky WA, Desai N, Wang Y, Hayward SW, Cunha GR, Cardiff RD, Shen MM and Abate-Shen C. Nkx3.1 mutant mice recapitulate early stages of prostate carcinogenesis. Cancer research. 2002; 62:2999-3004.
53. Kim MJ, Cardiff RD, Desai N, Banach-Petrosky WA, Parsons R, Shen MM and Abate-Shen C. Cooperativity of Nkx3.1 and Pten loss of function in a mouse model of prostate carcinogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2002; 99:2884-2889.
54. Abate-Shen C, Banach-Petrosky WA, Sun X, Economides KD, Desai N, Gregg JP, Borowsky AD, Cardiff RD and Shen MM. Nkx3.1; Pten mutant mice develop invasive prostate adenocarcinoma and lymph node metastases. Cancer research. 2003; 63:3886-3890.
55. Anderson PD, McKissic SA, Logan M, Roh M, Franco OE, Wang J, Doubinskaia I, van der Meer R, Hayward SW, Eischen CM, Eltoum I-E and Abdulkadir SA. Nkx3.1 and Myc crossregulate shared target genes in mouse and human prostate tumorigenesis. The Journal of clinical investigation. 2012; 122:1907-1919.
56. Ratnacaram CK, Teletin M, Jiang M, Meng X, Chambon P and Metzger D. Temporally controlled ablation of PTEN in adult mouse prostate epithelium generates a model of invasive prostatic adenocarcinoma. Proceedings of the National Academy of Sciences of the United States of America. 2008; 105:2521-2526.
57. Wang ZA, Toivanen R, Bergren SK, Chambon P and Shen MM. Luminal cells are favored as the cell of origin for prostate cancer. Cell reports. 2014; 8(5):1339-1346.
58. Locke JA, Zafarana G, Ishkanian AS, Milosevic M, Thoms J, Have CL, Malloff CA, Lam WL, Squire JA, Pintilie M, Sykes J, Ramnarine VR, Meng A, Ahmed O, Jurisica I, van der Kwast T, et al. NKX3.1 haploinsufficiency is prognostic for prostate cancer relapse following surgery or image-guided radiotherapy. Clinical cancer research. 2012; 18:308-316.
59. Zafarana G, Ishkanian AS, Malloff CA, Locke JA, Sykes J, Thoms J, Lam WL, Squire JA, Yoshimoto M, Ramnarine VR, Meng A, Ahmed O, Jurisca I, Milosevic M, Pintilie M, van der Kwast T, et al. Copy number alterations of c-MYC and PTEN are prognostic factors for relapse after prostate cancer radiotherapy. Cancer. 2012; 118:4053-4062.
60. Patrawala L, Calhoun T, Schneider-Broussard R, Li H, Bhatia B, Tang S, Reilly JG, Chandra D, Zhou J, Claypool K, Coghlan L and Tang DG. Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene. 2006; 25:1696-1708.
61. Patrawala L, Calhoun-Davis T, Schneider-Broussard R and Tang DG. Hierarchical organization of prostate cancer cells in xenograft tumors: the CD44+alpha2beta1+ cell population is enriched in tumor-initiating cells. Cancer Res. 2007; 67:6796-6805.
62. Visvader JE and Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008; 8:755-768.
63. Magee JA, Piskounova E and Morrison SJ. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell. 2012; 21:283-296.
64. Collins AT, Berry PA, Hyde C, Stower MJ and Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005; 65:10946-10951.
65. Zhao RC, Zhu YS and Shi Y. New hope for cancer treatment: exploring the distinction between normal adult stem cells and cancer stem cells. Pharmacol Ther. 2008; 119:74-82.
66. Gu G, Yuan J, Wills M and Kasper S. Prostate cancer cells with stem cell characteristics reconstitute the original human tumor in vivo. Cancer Res. 2007; 67:4807-4815.
67. Miki J, Furusato B, Li H, Gu Y, Takahashi H, Egawa S, Sesterhenn IA, McLeod DG, Srivastava S and Rhim JS. Identification of putative stem cell markers, CD133 and CXCR4, in hTERT-immortalized primary nonmalignant and malignant tumor-derived human prostate epithelial cell lines and in prostate cancer specimens. Cancer Res. 2007; 67:3153-3161.
68. Li H, Zhou J, Miki J, Furusato B, Gu Y, Srivastava S, McLeod DG, Vogel JC and Rhim JS. Telomerase-immortalized non-malignant human prostate epithelial cells retain the properties of multipotent stem cells. Exp Cell Res. 2008; 314:92-102.
69. Dubrovska A, Kim S, Salamone RJ, Walker JR, Maira SM, Garcia-Echeverria C, Schultz PG and Reddy VA. The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations. Proc Natl Acad Sci U S A. 2009; 106:268-273.
70. Rybak AP, He L, Kapoor A, Cutz J-C and Tang D. Characterization of sphere-propagating cells with stem-like properties from DU145 prostate cancer cells. Biochimica et biophysica acta. 2011; 1813:683-694.
71. Maitland NJ, Frame FM, Polson ES, Lewis JL and Collins AT. Prostate cancer stem cells: do they have a basal or luminal phenotype? Hormones & cancer. 2011; 2:47-61.
72. Rajasekhar VK, Studer L, Gerald W, Socci ND and Scher HI. Tumour-initiating stem-like cells in human prostate cancer exhibit increased NF-kappaB signalling. Nat Commun. 2011; 2:162.
73. Toivanen R, Frydenberg M, Murphy D, Pedersen J, Ryan A, Pook D, Berman DM, Australian Prostate Cancer B, Taylor RA and Risbridger GP. A preclinical xenograft model identifies castration-tolerant cancer-repopulating cells in localized prostate tumors. Science translational medicine. 2013; 5(187):187ra171.
74. Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, Gopalan A, Dowling C, Wanjala JN, Undvall EA, Arora VK, Wongvipat J, Kossai M, Ramazanoglu S, Barboza LP, Di W, Cao Z, et al. Organoid Cultures Derived from Patients with Advanced Prostate Cancer. Cell. 2014.
75. Rybak AP, Ingram AJ and Tang D. Propagation of human prostate cancer stem-like cells occurs through EGFR-mediated ERK activation. PloS one. 2013; 8:e61716.
76. Rybak AP and Tang D. SOX2 plays a critical role in EGFR-mediated self-renewal of human prostate cancer stem-like cells. Cell Signal. 2013; 25:2734-2742.
77. Pastrana E, Silva-Vargas V and Doetsch F. Eyes wide open: a critical review of sphere-formation as an assay for stem cells. Cell Stem Cell. 2011; 8:486-498.
78. Reynolds BA and Rietze RL. Neural stem cells and neurospheres--re-evaluating the relationship. Nat Methods. 2005; 2:333-336.
79. Goodell MA, Rosenzweig M, Kim H, Marks DF, DeMaria M, Paradis G, Grupp SA, Sieff CA, Mulligan RC and Johnson RP. Dye efflux studies suggest that hematopoietic stem cells expressing low or undetectable levels of CD34 antigen exist in multiple species. Nature medicine. 1997; 3:1337-1345.
80. Patrawala L, Calhoun T, Schneider-Broussard R, Zhou J, Claypool K and Tang DG. Side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2+ and ABCG2- cancer cells are similarly tumorigenic. Cancer Res. 2005; 65:6207-6219.
81. Zhou S, Schuetz JD, Bunting KD, Colapietro AM, Sampath J, Morris JJ, Lagutina I, Grosveld GC, Osawa M, Nakauchi H and Sorrentino BP. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nature medicine. 2001; 7:1028-1034.
82. Holohan C, Van Schaeybroeck S, Longley DB and Johnston PG. Cancer drug resistance: an evolving paradigm. Nature reviews Cancer. 2013; 13:714-726.
83. Marcato P, Dean CA, Giacomantonio CA and Lee PWK. Aldehyde dehydrogenase: its role as a cancer stem cell marker comes down to the specific isoform. Cell cycle (Georgetown, Tex). 2011; 10:1378-1384.
84. Raha D, Wilson TR, Peng J, Peterson D, Yue P, Evangelista M, Wilson C, Merchant M and Settleman J. The cancer stem cell marker aldehyde dehydrogenase is required to maintain a drug-tolerant tumor cell subpopulation. Cancer research. 2014.
85. Li T, Su Y, Mei Y, Leng Q, Leng B, Liu Z, Stass SA and Jiang F. ALDH1A1 is a marker for malignant prostate stem cells and predictor of prostate cancer patients’ outcome. Laboratory investigation. 2010; 90:234-244.
86. Le Magnen C, Bubendorf L, Rentsch CA, Mengus C, Gsponer J, Zellweger T, Rieken M, Thalmann GN, Cecchini MG, Germann M, Bachmann A, Wyler S, Heberer M and Spagnoli GC. Characterization and clinical relevance of ALDHbright populations in prostate cancer. Clinical cancer research. 2013; 19:5361-5371.
87. van den Hoogen C, van der Horst G, Cheung H, Buijs JT, Lippitt JM, Guzmán-Ramírez N, Hamdy FC, Eaton CL, Thalmann GN, Cecchini MG, Pelger RCM and van der Pluijm G. High aldehyde dehydrogenase activity identifies tumor-initiating and metastasis-initiating cells in human prostate cancer. Cancer research. 2010; 70:5163-5173.
88. van den Hoogen C, van der Horst G, Cheung H, Buijs JT, Pelger RCM and van der Pluijm G. The aldehyde dehydrogenase enzyme 7A1 is functionally involved in prostate cancer bone metastasis. Clinical & experimental metastasis. 2011; 28:615-625.
89. Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, Schott A, Hayes D, Birnbaum D, Wicha MS and Dontu G. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell stem cell. 2007; 1:555-567.
90. Marcato P, Dean CA, Pan D, Araslanova R, Gillis M, Joshi M, Helyer L, Pan L, Leidal A, Gujar S, Giacomantonio CA and Lee PWK. Aldehyde dehydrogenase activity of breast cancer stem cells is primarily due to isoform ALDH1A3 and its expression is predictive of metastasis. Stem cells (Dayton, Ohio). 2011; 29:32-45.
91. Qin J, Liu X, Laffin B, Chen X, Choy G, Jeter CR, Calhoun-Davis T, Li H, Palapattu GS, Pang S, Lin K, Huang J, Ivanov I, Li W, Suraneni MV and Tang DG. The PSA(-/lo) prostate cancer cell population harbors self-renewing long-term tumor-propagating cells that resist castration. Cell stem cell. 2012; 10:556-569.
92. Hernes E, Fossa SD, Berner A, Otnes B and Nesland JM. Expression of the epidermal growth factor receptor family in prostate carcinoma before and during androgen-independence. Br J Cancer. 2004; 90:449-454.
93. Di Lorenzo G, Tortora G, D’Armiento FP, De Rosa G, Staibano S, Autorino R, D’Armiento M, De Laurentiis M, De Placido S, Catalano G, Bianco AR and Ciardiello F. Expression of epidermal growth factor receptor correlates with disease relapse and progression to androgen-independence in human prostate cancer. Clin Cancer Res. 2002; 8:3438-3444.
94. Shah RB, Ghosh D and Elder JT. Epidermal growth factor receptor (ErbB1) expression in prostate cancer progression: correlation with androgen independence. Prostate. 2006; 66:1437-1444.
95. Karhadkar SS, Bova GS, Abdallah N, Dhara S, Gardner D, Maitra A, Isaacs JT, Berman DM and Beachy PA. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature. 2004; 431:707-712.
96. Kypta RM and Waxman J. Wnt/β-catenin signalling in prostate cancer. Nature reviews Urology. 2012.
97. Leong KG and Gao W-Q. The Notch pathway in prostate development and cancer. Differentiation; research in biological diversity. 2008; 76:699-716.
98. Bisson I and Prowse DM. WNT signaling regulates self-renewal and differentiation of prostate cancer cells with stem cell characteristics. Cell Res. 2009; 19:683-697.
99. Luo W, Rodriguez M, Valdez JM, Zhu X, Tan K, Li D, Siwko S, Xin L and Liu M. Lgr4 is a key regulator of prostate development and prostate stem cell differentiation. Stem cells (Dayton, Ohio). 2013; 31:2492-2505.
100. Nanta R, Kumar D, Meeker D, Rodova M, Van Veldhuizen PJ, Shankar S and Srivastava RK. NVP-LDE-225 (Erismodegib) inhibits epithelial-mesenchymal transition and human prostate cancer stem cell growth in NOD/SCID IL2Rγ null mice by regulating Bmi-1 and microRNA-128. Oncogenesis. 2013; 2:e42.
101. Kwon OJ, Valdez JM, Zhang L, Zhang B, Wei X, Su Q, Ittmann MM, Creighton CJ and Xin L. Increased Notch signalling inhibits anoikis and stimulates proliferation of prostate luminal epithelial cells. Nat Commun. 2014; 5:4416.
102. Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009; 9:550-562.
103. Miklossy G, Hilliard TS and Turkson J. Therapeutic modulators of STAT signalling for human diseases. Nature reviews Drug discovery. 2013; 12:611-629.
104. Lee JT, Lehmann BD, Terrian DM, Chappell WH, Stivala F, Libra M, Martelli AM, Steelman LS and McCubrey JA. Targeting prostate cancer based on signal transduction and cell cycle pathways. Cell Cycle. 2008; 7:1745-1762.
105. Teng DH, Hu R, Lin H, Davis T, Iliev D, Frye C, Swedlund B, Hansen KL, Vinson VL, Gumpper KL, Ellis L, El-Naggar A, Frazier M, Jasser S, Langford LA, Lee J, et al. MMAC1/PTEN mutations in primary tumor specimens and tumor cell lines. Cancer research. 1997; 57:5221-5225.
106. Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, Antipin Y, Mitsiades N, Landers T, Dolgalev I, Major JE, Wilson M, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010; 18:11-22.
107. Ayala G, Thompson T, Yang G, Frolov A, Li R, Scardino P, Ohori M, Wheeler T and Harper W. High levels of phosphorylated form of Akt-1 in prostate cancer and non-neoplastic prostate tissues are strong predictors of biochemical recurrence. Clin Cancer Res. 2004; 10:6572-6578.
108. Bedolla R, Prihoda TJ, Kreisberg JI, Malik SN, Krishnegowda NK, Troyer DA and Ghosh PM. Determining risk of biochemical recurrence in prostate cancer by immunohistochemical detection of PTEN expression and Akt activation. Clin Cancer Res. 2007; 13:3860-3867.
109. Malik SN, Brattain M, Ghosh PM, Troyer DA, Prihoda T, Bedolla R and Kreisberg JI. Immunohistochemical demonstration of phospho-Akt in high Gleason grade prostate cancer. Clinical cancer research. 2002; 8:1168-1171.
110. Kinkade CW, Castillo-Martin M, Puzio-Kuter A, Yan J, Foster TH, Gao H, Sun Y, Ouyang X, Gerald WL, Cordon-Cardo C and Abate-Shen C. Targeting AKT/mTOR and ERK MAPK signaling inhibits hormone-refractory prostate cancer in a preclinical mouse model. J Clin Invest. 2008; 118:3051-3064.
111. Lotan TL, Gurel B, Sutcliffe S, Esopi D, Liu W, Xu J, Hicks JL, Park BH, Humphreys E, Partin AW, Han M, Netto GJ, Isaacs WB and De Marzo AM. PTEN protein loss by immunostaining: analytic validation and prognostic indicator for a high risk surgical cohort of prostate cancer patients. Clin Cancer Res. 2011; 17:6563-6573.
112. Di Cristofano A, Pesce B, Cordon-Cardo C and Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet. 1998; 19:348-355.
113. Podsypanina K, Ellenson LH, Nemes A, Gu J, Tamura M, Yamada KM, Cordon-Cardo C, Catoretti G, Fisher PE and Parsons R. Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc Natl Acad Sci U S A. 1999; 96:1563-1568.
114. Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, Thomas GV, Li G, Roy-Burman P, Nelson PS, Liu X and Wu H. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell. 2003; 4:209-221.
115. Guertin DA, Stevens DM, Saitoh M, Kinkel S, Crosby K, Sheen J-H, Mullholland DJ, Magnuson MA, Wu H and Sabatini DM. mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer cell. 2009; 15:148-159.
116. Majumder PK, Yeh JJ, George DJ, Febbo PG, Kum J, Xue Q, Bikoff R, Ma H, Kantoff PW, Golub TR, Loda M and Sellers WR. Prostate intraepithelial neoplasia induced by prostate restricted Akt activation: the MPAKT model. Proceedings of the National Academy of Sciences of the United States of America. 2003; 100:7841-7846.
117. Dubrovska A, Elliott J, Salamone RJ, Kim S, Aimone LJ, Walker JR, Watson J, Sauveur-Michel M, Garcia-Echeverria C, Cho CY, Reddy VA and Schultz PG. Combination therapy targeting both tumor-initiating and differentiated cell populations in prostate carcinoma. Clin Cancer Res. 2010; 16:5692-5702.
118. Roskoski Jr. R. ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol Res. 2012; 66:105-143.
119. Gioeli D, Mandell JW, Petroni GR, Frierson Jr. HF and Weber MJ. Activation of mitogen-activated protein kinase associated with prostate cancer progression. Cancer Res. 1999; 59:279-284.
120. Mulholland DJ, Kobayashi N, Ruscetti M, Zhi A, Tran LM, Huang J, Gleave M and Wu H. Pten loss and RAS/MAPK activation cooperate to promote EMT and metastasis initiated from prostate cancer stem/progenitor cells. Cancer Res. 2012; 72:1878-1889.
121. Anwar K, Nakakuki K, Shiraishi T, Naiki H, Yatani R and Inuzuka M. Presence of ras oncogene mutations and human papillomavirus DNA in human prostate carcinomas. Cancer research. 1992; 52:5991-5996.
122. Cho N-Y, Choi M, Kim B-H, Cho Y-M, Moon KC and Kang GH. BRAF and KRAS mutations in prostatic adenocarcinoma. International journal of cancer. 2006; 119:1858-1862.
123. Shiraishi T, Muneyuki T, Fukutome K, Ito H, Kotake T, Watanabe M and Yatani R. Mutations of ras genes are relatively frequent in Japanese prostate cancers: pointing to genetic differences between populations. Anticancer research. 1998; 18:2789-2792.
124. Suzuki H, Aida S, Akimoto S, Igarashi T, Yatani R and Shimazaki J. State of adenomatous polyposis coli gene and ras oncogenes in Japanese prostate cancer. Japanese journal of cancer research : Gann. 1994; 85:847-852.
125. Bakin RE, Gioeli D, Sikes RA, Bissonette EA and Weber MJ. Constitutive activation of the Ras/mitogen-activated protein kinase signaling pathway promotes androgen hypersensitivity in LNCaP prostate cancer cells. Cancer Res. 2003; 63:1981-1989.
126. Bakin RE, Gioeli D, Bissonette EA and Weber MJ. Attenuation of Ras signaling restores androgen sensitivity to hormone-refractory C4-2 prostate cancer cells. Cancer Res. 2003; 63:1975-1980.
127. Hong SK, Kim JH, Lin MF and Park JI. The Raf/MEK/extracellular signal-regulated kinase 1/2 pathway can mediate growth inhibitory and differentiation signaling via androgen receptor downregulation in prostate cancer cells. Exp Cell Res. 2011; 317:2671-2682.
128. Wang J, Kobayashi T, Floc’h N, Kinkade CW, Aytes A, Dankort D, Lefebvre C, Mitrofanova A, Cardiff RD, McMahon M, Califano A, Shen MM and Abate-Shen C. B-Raf activation cooperates with PTEN loss to drive c-Myc expression in advanced prostate cancer. Cancer research. 2012; 72:4765-4776.
129. Jeong JH, Wang Z, Guimaraes AS, Ouyang X, Figueiredo JL, Ding Z, Jiang S, Guney I, Kang GH, Shin E, Hahn WC, Loda MF, Abate-Shen C, Weissleder R and Chin L. BRAF activation initiates but does not maintain invasive prostate adenocarcinoma. PloS one. 2008; 3:e3949.
130. Dhir R, Ni Z, Lou W, DeMiguel F, Grandis JR and Gao AC. Stat3 activation in prostatic carcinomas. Prostate. 2002; 51:241-246.
131. Ni Z, Lou W, Leman ES and Gao AC. Inhibition of constitutively activated Stat3 signaling pathway suppresses growth of prostate cancer cells. Cancer Res. 2000; 60:1225-1228.
132. Horinaga M, Okita H, Nakashima J, Kanao K, Sakamoto M and Murai M. Clinical and pathologic significance of activation of signal transducer and activator of transcription 3 in prostate cancer. Urology. 2005; 66:671-675.
133. Liu X, He Z, Li C-H, Huang G, Ding C and Liu H. Correlation analysis of JAK-STAT pathway components on prognosis of patients with prostate cancer. Pathology oncology research : POR. 2012; 18:17-23.
134. Tam L, McGlynn LM, Traynor P, Mukherjee R, Bartlett JMS and Edwards J. Expression levels of the JAK/STAT pathway in the transition from hormone-sensitive to hormone-refractory prostate cancer. British journal of cancer. 2007; 97:378-383.
135. Akimoto S, Okumura A and Fuse H. Relationship between serum levels of interleukin-6, tumor necrosis factor-alpha and bone turnover markers in prostate cancer patients. Endocrine journal. 1998; 45:183-189.
136. Adler HL, McCurdy MA, Kattan MW, Timme TL, Scardino PT and Thompson TC. Elevated levels of circulating interleukin-6 and transforming growth factor-beta1 in patients with metastatic prostatic carcinoma. J Urol. 1999; 161:182-187.
137. Nakashima J, Tachibana M, Horiguchi Y, Oya M, Ohigashi T, Asakura H and Murai M. Serum interleukin 6 as a prognostic factor in patients with prostate cancer. Clinical cancer research. 2000; 6:2702-2706.
138. Schroeder A, Herrmann A, Cherryholmes G, Kowolik C, Buettner R, Pal S, Yu H, Mueller-Newen G and Jove R. Loss of androgen receptor expression promotes a stem-like cell phenotype in prostate cancer through STAT3 signaling. Cancer Res. 2014; 74(4):1227-1237.
139. Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C and Darnell JE. Stat3 as an oncogene. Cell. 1999; 98:295-303.
140. Blando JM, Carbajal S, Abel E, Beltran L, Conti C, Fischer S and DiGiovanni J. Cooperation between Stat3 and Akt signaling leads to prostate tumor development in transgenic mice. Neoplasia (New York, NY). 2011; 13:254-265.
141. Mora LB, Buettner R, Seigne J, Diaz J, Ahmad N, Garcia R, Bowman T, Falcone R, Fairclough R, Cantor A, Muro-Cacho C, Livingston S, Karras J, Pow-Sang J and Jove R. Constitutive activation of Stat3 in human prostate tumors and cell lines: direct inhibition of Stat3 signaling induces apoptosis of prostate cancer cells. Cancer research. 2002; 62:6659-6666.
142. Barton BE, Karras JG, Murphy TF, Barton A and Huang HF-S. Signal transducer and activator of transcription 3 (STAT3) activation in prostate cancer: Direct STAT3 inhibition induces apoptosis in prostate cancer lines. Molecular cancer therapeutics. 2004; 3:11-20.
143. DeMiguel F, Lee SO, Lou W, Xiao X, Pflug BR, Nelson JB and Gao AC. Stat3 enhances the growth of LNCaP human prostate cancer cells in intact and castrated male nude mice. The Prostate. 2002; 52:123-129.
144. Kroon P, Berry PA, Stower MJ, Rodrigues G, Mann VM, Simms M, Bhasin D, Chettiar S, Li C, Li P-K, Maitland NJ and Collins AT. JAK-STAT blockade inhibits tumor initiation and clonogenic recovery of prostate cancer stem-like cells. Cancer research. 2013; 73:5288-5298.
145. Qu Y, Oyan AM, Liu R, Hua Y, Zhang J, Hovland R, Popa M, Liu X, Brokstad KA, Simon R, Molven A, Lin B, Zhang W-D, McCormack E, Kalland K-H and Ke X-S. Generation of Prostate Tumor-Initiating Cells Is Associated with Elevation of Reactive Oxygen Species and IL-6/STAT3 Signaling. Cancer research. 2013; 73:7090-7100.
146. Han Z, Wang X, Ma L, Chen L, Xiao M, Huang L, Cao Y, Bai J, Ma D, Zhou J and Hong Z. Inhibition of STAT3 signaling targets both tumor-initiating and differentiated cell populations in prostate cancer. Oncotarget. 2014; 5(18):8416-8428.
147. Iliopoulos D, Hirsch HA, Wang G and Struhl K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc Natl Acad Sci U S A. 2011; 108(4):1397-1402.
148. Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, Brooks M, Reinhardt F, Su Y, Polyak K, Arendt LM, Kuperwasser C, Bierie B and Weinberg RA. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci U S A. 2011; 108:7950-7955.
149. Klotz L and Emberton M. Management of low risk prostate cancer: active surveillance and focal therapy. Current opinion in urology. 2014; 24:270-279.
150. Djavan B, Susani M, Bursa B, Basharkhah A, Simak R and Marberger M. Predictability and significance of multifocal prostate cancer in the radical prostatectomy specimen. Techniques in urology. 1999; 5:139-142.
151. Baumann M, Krause M and Hill R. Exploring the role of cancer stem cells in radioresistance. Nat Rev Cancer. 2008; 8(7):545-554.
152. Peitzsch C, Kurth I, Kunz-Schughart L, Baumann M and Dubrovska A. Discovery of the cancer stem cell related determinants of radioresistance. Radiotherapy and oncology : journal of the European Society for Therapeutic Radiology and Oncology. 2013; 108(3):378-387.
153. Cho YM, Kim YS, Kang MJ, Farrar WL and Hurt EM. Long-term recovery of irradiated prostate cancer increases cancer stem cells. Prostate. 2012; 72(16):1746-1756.
154. Frame FM, Pellacani D, Collins AT, Simms MS, Mann VM, Jones GDD, Meuth M, Bristow RG and Maitland NJ. HDAC inhibitor confers radiosensitivity to prostate stem-like cells. British journal of cancer. 2013; 109:3023-3033.
155. Cojoc M, Mabert K, Muders MH and Dubrovska A. A role for cancer stem cells in therapy resistance: Cellular and molecular mechanisms. Seminars in cancer biology. 2014:In press.
156. Blagosklonny MV. Target for cancer therapy: proliferating cells or stem cells. Leukemia. 2006; 20(3):385-391.
157. Trumpp A and Wiestler OD. Mechanisms of Disease: cancer stem cells--targeting the evil twin. Nature clinical practice Oncology. 2008; 5(6):337-347.
158. Crawford ED, Eisenberger MA, McLeod DG, Spaulding JT, Benson R, Dorr FA, Blumenstein BA, Davis MA and Goodman PJ. A controlled trial of leuprolide with and without flutamide in prostatic carcinoma. The New England journal of medicine. 1989; 321:419-424.
159. Eisenberger MA, Blumenstein BA, Crawford ED, Miller G, McLeod DG, Loehrer PJ, Wilding G, Sears K, Culkin DJ, Thompson IM, Bueschen AJ and Lowe BA. Bilateral orchiectomy with or without flutamide for metastatic prostate cancer. The New England journal of medicine. 1998; 339:1036-1042.
160. Petrylak DP, Tangen CM, Hussain MHA, Lara PN, Jones JA, Taplin ME, Burch PA, Berry D, Moinpour C, Kohli M, Benson MC, Small EJ, Raghavan D and Crawford ED. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. The New England journal of medicine. 2004; 351:1513-1520.
161. Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, Oudard S, Théodore C, James ND, Turesson I, Rosenthal MA and Eisenberger MA. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. The New England journal of medicine. 2004; 351:1502-1512.
162. Domingo-Domenech J, Vidal SJ, Rodriguez-Bravo V, Castillo-Martin M, Quinn SA, Rodriguez-Barrueco R, Bonal DM, Charytonowicz E, Gladoun N, de la Iglesia-Vicente J, Petrylak DP, Benson MC, Silva JM and Cordon-Cardo C. Suppression of acquired docetaxel resistance in prostate cancer through depletion of notch- and hedgehog-dependent tumor-initiating cells. Cancer Cell. 2012; 22:373-388.
163. Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, Arora VK, Le C, Koutcher J, Scher H, Scardino PT, Rosen N and Sawyers CL. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer cell. 2011; 19:575-586.
164. Mulholland DJ, Tran LM, Li Y, Cai H, Morim A, Wang S, Plaisier S, Garraway IP, Huang J, Graeber TG and Wu H. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer cell. 2011; 19:792-804.
165. Hong S-K, Jeong JH, Chan AM and Park J-I. AKT upregulates B-Raf Ser445 phosphorylation and ERK1/2 activation in prostate cancer cells in response to androgen depletion. Experimental cell research. 2013; 319:1732-1743.
166. Decker T and Kovarik P. Serine phosphorylation of STATs. Oncogene. 2000; 19:2628-2637.
167. Chung J, Uchida E, Grammer TC and Blenis J. STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Molecular and cellular biology. 1997; 17:6508-6516.
168. Carriere A, Romeo Y, Acosta-Jaquez HA, Moreau J, Bonneil E, Thibault P, Fingar DC and Roux PP. ERK1/2 phosphorylate Raptor to promote Ras-dependent activation of mTOR complex 1 (mTORC1). The Journal of biological chemistry. 2011; 286:567-577.
169. Ma L, Chen Z, Erdjument-Bromage H, Tempst P and Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005; 121:179-193.