
INTRODUCTION
Untreated, acute myeloid leukemia (AML) is a fatal hematological malignancy. Although remissions can be achieved with intensive chemotherapy, the disease relapses in a large number of cases and progression and death frequently occurs [1-5]. The current treatment strategies, involving combinations of cytarabine with an anthracycline, result in substantial toxicity and morbidity. This is a particularly serious problem in the case of older adults with the disease, who frequently have less favorable outcomes than younger patients. Undoubtedly, there is an urgent need for new treatments and therapeutic approaches. AML appears to result from mutations of key genes that ultimately lead to deregulation and constitutive activation of cellular cascades that promote cell growth and mediate anti-apoptotic and pro-survival responses. Such changes result in deregulation of normal hematopoiesis and promote malignant transformation leukemogenesis [6-10].
A major problem in efforts to treat and cure AML is the inability to efficiently target and eliminate leukemia initiating cells (LICs), which are the cells that initiate and maintain the leukemic phenotype [10, 11]. The majority of LICs are quiescent and therefore not sensitive to various chemotherapeutic drugs that target and kill rapidly dividing cells [12, 13]. This fact explains in part the difficulty in eliminating leukemia with chemotherapy and the relapses seen in the majority of patients, despite initially achieving complete responses with classical chemotherapy regimens. Aberrant activation of pro-survival signaling cascades in leukemia stem cells and early committed leukemic precursors may also act protectively and promote their survival, providing a potential therapeutic outlet and elements that can be targeted for the treatment of leukemias [12, 14].
The PI3’ kinase/mTOR pathway
The PI3’ kinase/AKT/mTOR pathway is a key regulatory network of signaling cascades in mammalian cells, whose coordinated operations regulate and promote important cellular activities and functions [15, 16]. Via controlling activation of the mTOR kinase and its downstream effectors, this cellular network ultimately regulates mRNA translation of genes that encode for pro-oncogenic proteins and, thus, promote malignant cell survival [15-18]. Interestingly, this network of signals is also engaged and activated by growth suppressive cytokines such as interferons (IFNs) [19-21], suggesting a competition between factors that suppress growth and mitogenic signals for the use and regulation of this pathway. PI 3’ kinase (PI3’K) is a lipid kinase that controls formation of distinct signaling complexes on the membrane of cells [22]. Activation of PI3’K leads to engagement of the kinase PDK1 which subsequently phosphorylates AKT on threonine 308 (Thr 308), ultimately leading to engagement and activation of AKT [23], which in turn phosphorylates many and activates multiple downstream substrates and effectors, leading to the generation of signals that promote survival and proliferation [24, 25].
As aberrant activation of the PI3’K/AKT/mTOR pathway promotes malignant cell proliferation and survival [15, 26, 27], several studies have sought to examine the implications of constitutive activation of this pathway in tumorigenesis. There is now extensive evidence that deregulation of this pathway contributes to the tumorigenic potential, a more aggressive phenotype and poorer prognosis in several malignancies [28-30]. In addition, activation of this pathway has been associated with chemotherapy resistance [31, 32], underscoring the importance of this signaling cascade as a therapeutic target for the treatment of various tumors. For all these reasons, there has been a major interest in the development of pharmacologic inhibitors of the PI3K/AKT/mTOR pathway for various solid tumors and hematological malignancies, which has further intensified after the detailed mapping and characterization of the pathway the last several years.
The mammalian target of rapamycin (mTOR) is a central element of the pathway and a key kinase activated downstream of PI3K/AKT. This kinase was originally identified in yeast [33], and subsequent work established that it is conserved in eukaryotic organisms. mTOR is present in two distinct and functionally diverse cellular complexes: TORC1 and TORC2 (Fig. 1). Each of these 2 complexes have common and distinct subunits and effectors and ultimately engage different downstream elements and activate distinct effector pathways. As shown in Fig. 1, the interaction of mTOR with Raptor (regulatory-associated protein of mTOR) defines the TORC1 complex [34-37]. TORC1 is generally perceived as rapamycin-sensitive and beyond mTOR and Raptor, it also contains mLST8 [35-37]. The two major substrates for TORC1 are the S6 kinase (S6K) and the translational repressor 4E-BP1, which binds to and negatively controls the function of the eukaryotic initiation factor 4E (eIF4E) [16, 19, 35-39]. After its phosphorylation/activation by mTOR, S6K regulates downstream engagement of two major substrates, the S6 ribosomal protein (rpS6) and the eukaryotic initiation factor 4B (eIF4B) [16, 35-39] (Fig.2). In addition, there is recent evidence that it phosphorylates and negatively regulates the expression of PDCD4 (Fig. 2), a tumor suppressor protein with inhibitory activities on cap-dependent translation via its ability to block the function of the translation initiation factor eIF4A and the integration of eIF4A into the eIF4F complex [40-44]. This protein undergoes phosphorylation by S6K, followed by degradation by the ubiquitin ligase βTRCP (45), suggesting a mechanism by which the mTOR pathway may be targeting and inhibiting tumor suppressor elements with regulatory effects on mRNA translation. In addition, to regulating activation of S6K, the mTORC1 complex is responsible for phosphorylation of the (eIF4E)–binding proteins (4E-BP) 1 and 2 on several sites, leading to their de-activation and detachment from eIF4E [19, 35-39]. Such dissociation allows eIF4E activation, which is a critical event for the initiation of mRNA translation by oncogenic proteins in eukaryotes.
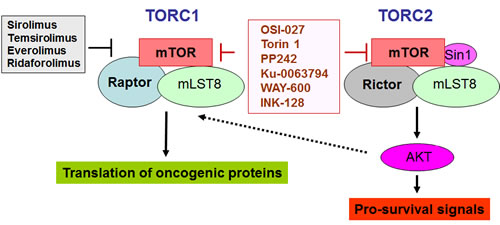
Figure 1: TORC1 and TORC2 complexes and inhibitory effects of different mTOR inhibitors. The rapalogs (shown in black), inhibit selectively TORC1, while the catalytic TOR inhibitors, (shown in red), inhibit both TORC1 and TORC2.
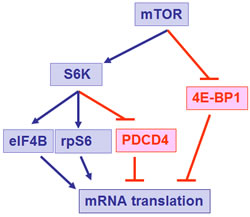
Figure 2: Effector elements downstream of mTOR that exhibit positive (in blue) or negative (in red) effects on mRNA translation.
In contrast to TORC1, whose major function is control of signals for the initiation of mRNA translation, the TORC2 complex plays a different role in normal and malignant cells. The TORC2 complex includes mTOR, Rictor (rapamycin-insensitive companion of mTOR), SIN1, and mLST8 [35-37, 46-48]. The primary function of TORC2 is the control of phosphorylation of AKT on Ser 473 [35-37], a site whose phosphorylation is essential for activation of AKT resulting in induction of its kinase domain. Beyond AKT, additional substrates for TORC2 activity have been identified, including PKCα, [49-51] and SGK1 [52, 53]. Notably it was recently shown that PKCα gene expression is inducible in AML cells resistant to chemotherapy [54].
Because of the high relevance of the mTOR pathway in malignancies, first generation mTOR inhibitors, including rapamycin and related drugs (rapalogs) have been tried extensively in various clinical contexts for the treatment of tumors of diverse cellular origin. Two rapalogs, temsirolimus (CCI-779) and everolimus (RAD001) have shown major activity and have been approved by the FDA for the treatment of renal cell carcinoma [55, 56]. Since then extensive clinical efforts have been ongoing in attempts to evaluate the clinical activity of the three major rapalogs (everolimus, temsirolimus and ridaforolimus) in the treatment of various solid tumors and hematological malignancies [57-60].
Targeting mTOR in AML
The simultaneous deregulation of pathways that control both transcription and mRNA translation of genes encoding for oncogenic proteins appears to play key roles in the pathogenesis and pathophysiology of AML. There has been extensive evidence that the PI3’K/AKT/mTOR pathway is aberrantly activated and deregulated in AML [61]. There is also some evidence that constitutive PI 3’K activation in AML is mainly due to the activity of the PI3K p110δ isoform [62, 63]. In one study, a large percentage of samples from patients with AML were found to have constitutive AKT activation [64]. In addition, the AKT pathway was among the signaling cascades whose simultaneous activation with other pathways, such as PKCα and ERK, was found to confer a poor prognosis in AML [65]. Other recent studies used proteomic analysis or single-cell network profiling (SCNP) with flow cytometry, to predict the likelihood of response to induction chemotherapy for patients with AML [66, 67]. Remarkably, lack of response to induction chemotherapy in patients older than 60 years or patients with secondary AML was associated with increased phosphorylation of AKT induced by FLT-3 ligand [67].
Recognition of aberrations in the AKT/mTOR pathway has led to clinical trials with rapalogs in AML. Recher et al showed that rapamycin resulted in blast clearance in some patients with AML. However, the length of response was limited and not all patients responded [68]. There has been also some evidence that rapamycin and etoposide exhibit synergistic/enhancing effects on AML cells in vitro and in AML mouse models in vivo [69]. However, when a clinical trial involving the addition rapamycin to salvage chemotherapy (mitoxantrone, etoposide, and cytarabine) for the treatment of relapsed and refractory AML was performed, the authors failed to observe synergistic activity by the combination [70].
New approaches to target TORC1 and TORC2 complexes in AML
Although approaches to optimize the administration of rapalogs with chemotherapy [71], in various settings are still being examined, the use of these agents has several limitations as discussed above. To overcome the limitations of the rapalogs, extensive efforts over recent years have been focused on the design and clinical development of agents that are catalytic inhibitors of mTOR and in addition to TORC1 suppress TORC2, or other agents that simultaneously target the PI3’K/AKT pathway. Several pan PI3K/AKT/mTOR inhibitors and dual TORC inhibitors have been developed and are being exploited [72-79]. Such efforts have also been extended to determine the effects of such compounds on leukemias. Recent studies demonstrated that the dual TORC1/TORC2 inhibitors PP242 [80] or OSI-027 [81] are potent suppressors of both TORC1 and TORC2 activities in BCR-ABL transformed cells. These catalytic inhibitors were shown to elicit potent antileukemic effects in vitro [80, 81] and in vivo [81] on CML or Ph+ ALL cells, including cells expressing the T315I BCR-ABL mutation, which is resistant to the kinase inhibitors currently approved for use in the treatment of CML and Ph+ ALL (imatinib mesylate, nilotinib, dasatinib).
The potent suppressive effects of dual TORC1/TORC2 inhibitors on BCR-ABL-transformed cells, have raised the possibility that such agents may have activity in other leukemias and prompted us to perform additional studies to examine the spectrum of the antileukemic properties of OSI-027 in AML. In recently published work [82], we examined the effects of dual TORC1/2 inhibition on various elements of the mTOR pathway in different AML cell lines and primary leukemia blasts from AML patients and compared them to the effects of the classic mTOR inhibitor rapamycin. As expected, only OSI-027 blocked TORC2-specific cellular events in AML cells, such as phosphorylation of AKT on Ser473 [82]. On the other hand, both OSI-027 and rapamycin were potent suppressors of the activation of the S6 kinase and the downstream phosphorylation of its target, S6 ribosomal protein [82] Importantly, phosphorylation of 4E-BP1 on Thr 37/46 was blocked by OSI-027, but not rapamycin, indicating that such phosphorylation is a rapamycin-insensitive cellular event in AML cells (79). This is consistent with the emerging evidence in other systems for rapamycin-insensitive TORC1-mediated signals [83, 84]. Our studies also established that OSI-027 is a potent suppressor of primitive leukemic precursors (CFU-L) from AML patients. Such effects were much more potent than the effects of rapamycin analyzed in parallel [82]. In addition, OSI-027 enhanced the inhibitory effects of low-dose cytarabine (Ara-C), suggesting that combinations of dual TORC1/2 inhibitors with chemotherapy may provide an approach to enhance antileukemic responses of chemotherapy [82].
Altogether, the results of such work raise the prospect of future clinical trials using dual TORC1/TORC2 inhibitors for the treatment of AML. Beyond OSI-027 there are additional TORC1/2 inhibitors in clinical or pre-clinical development [73-77, 85] that may be good candidates for such studies. Another potential approach to generate antileukemic responses by complete inhibition of the mTOR pathway would be to block the PI3’K/AKT axis [86]. In fact, approaches to simultaneously block PI3’K and mTOR have been developed [87]. NVPBEZ235 is a molecule that inhibits the PI3’K and also both TORC1 and TORC2 complexes [88]. Recent studies using this agent in AML have demonstrated potent inhibitory effects on PI3’K and TORC1/TORC2 complexes, including rapamycin-insensitive TORC1. It was also found to inhibit rapamycin-insensitive phosphorylation sites in 4E-BP1 [89]. Such potent effects were associated with decreased cell proliferation and survival of leukemia cells and suppressed leukemic progenitor clonogenicity [89], raising the prospect of using such pan P13’K/AKT/mTOR inhibitors as a potential future approach for the treatment of AML.
Summary
While inhibiting mTOR is a promising strategy for the treatment of malignancies, agents that selectively target TORC1 (rapalogs) have limited clinical activity and are unlikely to have major impact in the treatment of AML. The development of selective ATP-catalytic inhibitors, which have the capacity to block the functions of both TORC1 and TORC2 has resulted in new momentum in the research field of mTOR targeting in AML and is igniting important work with major therapeutic implications. Approaches to overcome the limitations of rapalogs for the treatment of leukemias are now possible, using either dual TORC1/2 inhibitors or pan–PI3K-TORC1/2 inhibitors. Our recent studies have established that beyond exhibiting potent antileukemic effects, dual TORC1/2 catalytic inhibition enhances the effects of cytarabine on primitive leukemic precursors from AML patients. These studies are very encouraging and suggest a potential role for these agents in the treatment of AML patients. They also raise the possibility that combinations of dual TORC1/2 inhibitors with chemotherapeutic agents may provide a novel approach to target leukemic initiating stem cells and increase the probability of cure for AML patients.
References
1. Tallman MS, Gilliland DG, Rowe JM. Drug therapy for acute myeloid leukemia. Blood. 2005; 106:1154-63.
2. Estey E. New drugs in acute myeloid leukemia. Semin Oncol. 2008; 35:439-48.
3. Shipley JL, Butera JN. Acute myelogenous leukemia. Exp Hematol. 2009; 37:649-58.
4. Godley LA, Larson RA. Therapy-related myeloid leukemia. Semin Oncol. 2008; 35:418-29.
5. Kohrt HE, Coutre SE. Optimizing therapy for acute myeloid leukemia. J Natl Compr Canc Netw 2008; 6:1003-16.
6. Gilliland DG, Tallman MS. Focus on acute leukemias. Cancer Cell. 2002; 1:417-20.
7. Scholl C, Gilliland DG, Fröhling S. Deregulation of signaling pathways in acute myeloid leukemia. Semin Oncol. 2008; 35:336-45.
8. Cammenga J. Gatekeeper pathways and cellular background in the pathogenesis and therapy of AML. Leukemia. 2005; 19:1719–28.
9. Fathi AT, Grant S, Karp JE. Exploiting cellular pathways to develop new treatment strategies for AML. Cancer Treat Rev. 2010; 36:142-50.
10. Britschgi C, Fey MF. Tumor suppressor genes in myeloid differentiation and leukemogenesis. Future Oncol. 2009; 5:245-57.
11. Roboz GJ, Guzman M. Acute myeloid leukemia stem cells: seek and destroy. Expert Rev Hematol. 2009; 2:663-72.
12. Valent P. Targeting of leukemia-initiating cells to develop curative drug therapies: straightforward but nontrivial concept. Curr Cancer Drug Targets. 2011; 11:56-71.
13. Misaghian N, Ligresti G, Steelman LS, Bertrand FE, Bäsecke J, Libra M, Nicoletti F, Stivala F, Milella M, Tafuri A, Cervello M, Martelli AM, McCubrey JA. Targeting the leukemic stem cell: the Holy Grail of leukemia therapy. Leukemia. 2009; 23:25-42.
14. Martelli AM, Evangelisti C, Chiarini F, McCubrey JA. The phosphatidylinositol 3-kinase/Akt/mTOR signaling network as a therapeutic target in acute myelogenous leukemia patients. Oncotarget. 2010; 1:89-103.
15. Martelli AM, Chiarini F, Evangelisti C, Grimaldi C, Ognibene A, Manzoli L, Billi AM, JA M. The phosphatidylinositol 3-kinase/AKT/mammalian target of rapamycin signaling network and the control of normal myelopoiesis. Histol Histopathol. 2010; 25:669-80.
16. Altman JK, Platanias LC. Exploiting the mammalian target of rapamycin pathway in hematologic malignancies. Curr Opin Hematol. 2008; 15:88-94.
17. Cantley LC. The phosphoinositide 3-kinase pathway. Science. 296 : 1655-57, 2002.
18. Deane JA, Fruman DA. Phosphoinositide 3-kinase: diverse roles in immune cell activation. Annu Rev Immunol. 2004; 22: 563-98.
19. Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signaling. Nat Rev Immunol. 2005; 5:375-86.
20. Kroczynska B, Kaur S, Platanias LC. Growth suppressive cytokines and the AKT/mTOR pathway. Cytokine. 2009; 48:138-43.
21. Kaur S, Uddin S, Platanias LC. The PI3’ kinase pathway in interferon signaling. J Interferon Cytokine Res. 2005; 25:780-87.
22. Carnero A, Blanco-Aparicio C, Renner O, Link W, Leal JF. The PTEN/PI3K/AKT signalling pathway in cancer, therapeutic implications. Curr Cancer Drug Targets. 2008; 8:187-98.
23. Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009; 8:627–44.
24. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007; 129:1261-74.
25. Naughton R, Quiney C, Turner SD, Cotter TG. Bcr-Abl-mediated redox regulation of the PI3K/AKT pathway. Leukemia. 2009; 23:1432-40.
26. Hay N. The Akt-mTOR tango and its relevance to cancer. Cancer Cell. 2005; 8:179-83.
27. Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006; 7:606-19.
28. Kim D, Dan HC, Park S, Yang L, Liu Q, Kaneko S, Ning J, He L, Yang H, Sun M, Nicosia SV, Cheng JQ. AKT/PKB signaling mechanisms in cancer and chemoresistance. Front Biosci. 2005; 10:975-87.
29. Samuels Y, Ericson K. Oncogenic PI3K and its role in cancer. Curr Opin Oncol. 2006; 18:77-82.
30. Horn S, Bergholz U, Jücker M, McCubrey JA, Trümper L, Stocking C, Bäsecke J. Mutations in the catalytic subunit of class IA PI3K confer leukemogenic potential to hematopoietic cells. Oncogene. 2008; 27:4096-06.
31. Brognard J, Clark AS, Ni Y, Dennis PA. Akt/protein kinase B is constitutively active in non-small cell lung cancer cells and promotes cellular survival and resistance to chemotherapy and radiation. Cancer Res. 2001; 61:3986-97.
32. Martelli AM, Tabellini G, Bortul R, Tazzari PL, Cappellini A, Billi AM, Cocco L. Involvement of the phosphoinositide 3-kinase/Akt signaling pathway in the resistance to therapeutic treatments of human leukemia. Histol Histopathol. 2005; 20:239-52.
33. Chiu MI, Katz H, Berlin V. RAPT1, a mammalian homolog of yeast Tor, interacts with the FKBP12/rapamycin complex. Proc Natl Acad Sci USA. 1994; 91:12574–78.
34. Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, Tokunaga C, Avruch J, Yonezawa K. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002; 110:177-89.
35. Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007; 12:9-22.
36. Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004; 18:1926-45.
37. Bhaskar PT, Hay N. The two TORCs and Akt. Dev Cell. 2007; 12:487-02.
38. Bjornsti MA, Houghton PJ. The TOR pathway: a target for cancer therapy. Nat Rev Cancer. 2004; 4: 335-48.
39. Jaccinto E, Hall MN. Tor signaling in bugs, brain and brawn. Nature Rev Mol Cell Biol. 2003; 4:117-26.
40. Cmarik JL, Min H, Hegamyer G, Zhan S, Kulesz-Martin M, Yoshinaga H, Matsuhashi S, and Colburn NH. Differentially expressed protein Pdcd4 inhibits tumor promoter-induced neoplastic transformation. Proc Natl Acad Sci U S A. 1999; 96:14037–42.
41. Yang HS, Knies JL, Stark C, Colburn N. Pdcd4 suppresses tumor phenotype in JB6 cells by inhibiting AP-1 transactivation. Oncogene. 2003; 22:3712–20.
42. Yang HS, Jansen AP, Komar AA, Zheng Xm Merrick WC, Costes S, Lockett SJ, Sonenberg N, Colburn NH. The transformation suppressor Pdcd4 is a novel eukaryotic translation initiation factor 4A binding protein that inhibits translation. Mol Cell Biol. 2003; 23:26–37.
43. Yang HS, Matthews CP, Clair T, Wang Q, Baker AR, Li CC, Tan TH, and Colburn NH. Tumorigenesis suppressor Pdcd4 down-regulates mitogen-activated protein kinase kinase kinase kinase 1 expression to suppress colon carcinoma cell invasion. Mol Cell Biol. 2006; 26:1297–06.
44. Yang HS, Cho MH, Zakowicz H, Hegamyer G, Sonenberg N, and Colburn NH. A novel function of the MA-3 domains in transformation and translation suppressor Pdcd4 is essential for its binding to eukaryotic translation initiation factor 4A. Mol Cell Biol. 2004; 24:3894–06.
45. Dorrello NV, Peschiaroli A, Guardavaccaro D, Colburn NH, Sherman NE, and Pagano M. S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science. 2006; 314:467-71.
46. Jacinto E, Loewith R, Schmidt A, Lin S, Rüegg MA, Hall A, Hall MN. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004; 6:1122-28.
47. Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004; 14:1296-02.
48. Guertin DA, Stevens DM, Saitoh M, Kinkel S, Crosby K, Sheen JH, Mullholland DJ, Magnuson MA, Wu H, Sabatini DM. mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell. 2009; 15:148-59.
49. Fan QW, Cheng C, Knight ZA, Haas-Kogan D, Stokoe D, James CD, McCormick F, Shokat KM, Weiss WA. EGFR signals to mTOR through PKC and independently of Akt in glioma. Sci Signal 2:ra4, 2009.
50. Facchinetti V, Ouyang W, Wei H, Soto N, Lazorchak A, Gould C, Lowry C, Newton AC, Mao Y, Miao RQ, Sessa WC, Qin J, Zhang P, Su B, Jacinto E. The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. EMBO J. 2008; 27:1932-43.
51. Ikenoue T, Inoki K, Yang Q, Zhou X, Guan KL. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J. 2008; 27:1919-31.
52. García-Martínez JM, Alessi DR. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem J. 2008; 416:375-85.
53. Yan L, Mieulet V, Lamb RF. mTORC2 is the hydrophobic motif kinase for SGK1. Biochem J. 2008; 416:e19-21.
54. Kornblau SM, Qiu YH, Bekele BN, Cade JS, Zhou X, Harris D, Jackson CE, Estrov Z, Andreeff M. Studying the right cell in acute myelogenous leukemia: dynamic changes of apoptosis and signal transduction pathway protein expression in chemotherapy resistant ex-vivo selected “survivor cells”. Cell Cycle. 2006; 5:2769-77.
55. Hudes G, Carducci M, Tomczak P, Dutcher J, Figlin R, Kapoor A, Staroslawska E, Sosman J, McDermott D, Bodrogi I, Kovacevic Z, Lesovoy V, Schmidt-Wolf IG, Barbarash O, Gokmen E, O’Toole T et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007; 356:2271-81.
56. Amato R. Everolimus for the treatment of advanced renal cell carcinoma. Expert Opin Pharmacother. 2011; 12:1143-55.
57. Azim H, Azim HA Jr, Escudier B. Targeting mTOR in cancer: renal cell is just a beginning. Target Oncol. 2010; 5:269-80.
58. Bhagwat SV, Crew AP. Novel inhibitors of mTORC1 and mTORC2. Curr Opin Investig Drugs. 2010;11:638-45.
59. Ciuffreda L, Di Sanza C, Incani UC, Milella M. The mTOR pathway: a new target in cancer therapy. Curr Cancer Drug Targets. 2010;10:484-95.
60. Dancey J. mTOR signaling and drug development in cancer. Nat Rev Clin Oncol. 2010;7:209-19.
61. Xu Q, Simpson S, Scialla TJ, Bagg A, Carroll M. Survival of acute myeloid leukemia requires PI3 kinase activation. Blood. 2003; 102:972-80.
62. Billottet C, Grandage VL, Gale RE, Quattropani A, Rommel C, Vanhaesebroeck B, Khwaja A. A selective inhibitor of the p110δ isoform of PI 3-kinase inhibits AML cell proliferation and survival and increases the cytotoxic effects of VP16. Oncogene. 2006; 25:6648–59.
63. Sujobert P, Bardet V, Cornillet-Lefebvre P, Hayflick JS, Prie N, Verdier F, Vanhaesebroeck B, Muller O, Pesce F, Ifrah N, Hunault-Berger M, Berthou C, Villemagne B, Jourdan E, Audhuy B, Solary E et al. Essential role for the p110delta isoform in phosphoinositide 3-kinase activation and cell proliferation in acute myeloid leukemia. Blood. 2005; 106:1063-66.
64. Min YH, Eom JI, Cheong JW, Maeng HO, Kim JY, Jeung HK, Lee ST, Lee MH, Hahn JS, Ko YW. Constitutive phosphorylation of Akt/PKB protein in acute myeloid leukemia: its significance as a prognostic variable. Leukemia. 2003; 17:995-97.
65. Kornblau SM, Womble M, Qiu YH, Jackson CE, Chen W, Konopleva M, Estey EH, Andreeff M. Simultaneous activation of multiple signal transduction pathways confers poor prognosis in acute myelogenous leukemia. Blood. 2006; 108:2358-65.
66. Kornblau SM, Tibes R, Qiu YH, Chen W, Kantarjian HM, Andreeff M, Coombes KR, Mills GB. Functional proteomic profiling of AML predicts response and survival. Blood. 2009; 113:154-64.
67. Kornblau SM, Minden MD, Rosen DB, Putta S, Cohen A, Covey T, Spellmeyer DC, Fantl WJ, Gayko U, Cesano A. Dynamic single-cell network profiles in acute myelogenous leukemia are associated with patient response to standard induction therapy. Clin Cancer Res. 2010; 16:3721-33.
68. Récher C, Beyne-Rauzy O, Demur C, Chicanne G, Dos Santos C, Mas VM, Benzaquen D, Laurent G, Huguet F, Payrastre B. Antileukemic activity of rapamycin in acute myeloid leukemia. Blood. 2005; 105:2527-34.
69. Xu Q, Thompson JE, Carroll M. mTOR regulates cell survival after etoposide treatment in primary AML cells. Blood. 2005;106:4261-68.
70. Perl AE, Kasner MT, Tsai DE, Vogl DT, Loren AW, Schuster SJ, Porter DL, Stadtmauer EA, Goldstein SC, Frey NV, Nasta SD, Hexner EO, Dierov JK, Swider CR, Bagg A, Gewirtz AM et al. A phase I study of the mammalian target of rapamycin inhibitor sirolimus and MEC chemotherapy in relapsed and refractory acute myelogenous leukemia. Clin Cancer Res. 2009; 15:6732-39.
71. Apontes P, Leontieva OV, Demidenko ZN, Li F, Blagosklonny MV. Exploring long-term protection of normal human fibroblasts and epithelial cells from chemotherapy in cell culture. Oncotarget. 2011; 2:222-33.
72. Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, Shokat KM. tive-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009; 7:e38.
73. García-Martínez JM, Moran J, Clarke RG, Gray A, Cosulich SC, Chresta CM, Alessi DR. Ku-0063794 is a specific inhibitor of the mammalian target of rapamycin (mTOR). Biochem J. 2009; 421:29-42.
74. Yu K, Toral-Barza L, Shi C, Zhang WG, Lucas J, Shor B, Kim J, Verheijen J, Curran K, Malwitz DJ, Cole DC, Ellingboe J, Ayral-Kaloustian S, Mansour TS, Gibbons JJ, Abraham RT et al. Biochemical, cellular, and in vivo activity of novel ATP-competitive and selective inhibitors of the mammalian target of rapamycin. Cancer Res. 2009; 69:6232-40.
75. Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, Gray NS. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009; 284:8023-32.
76. Falcon BL, Barr S, Gokhale PC, Chou J, Fogarty J, Depeille P, Miglarese M, Epstein DM, McDonald DM. Reduced VEGF production, angiogenesis, and vascular regrowth contribute to the antitumor properties of dual mTORC1/mTORC2 inhibitors. Cancer Res. 2011;71: 1573-83.
77. Markman B, Dienstmann R, Tabernero J. Targeting the PI3K/Akt/mTOR pathway-beyond rapalogs. Oncotarget. 2010; 1:530-43.
78. Sacco A, Roccaro A, Ghobrial IM. Role of dual PI3/Akt and mTOR inhibition in Waldenstrom’s Macroglobulinemia. Oncotarget. 2010;1: 578-82.
79. Feldman ME, Shokat KM. New inhibitors of the PI3K-Akt-mTOR pathway: insights into mTOR signaling from a new generation of Tor Kinase Domain Inhibitors (TORKinibs). Curr Top Microbiol Immunol. 2010; 347:241-62.
80. Janes MR, Limon JJ, So L, Chen J, Lim RJ, Chavez MA, Vu C, Lilly MB, Mallya S, Ong ST, Konopleva M, Martin MB, Ren P, Liu Y, Rommel C, Fruman DA. Effective and selective targeting of leukemia cells using a TORC1/2 kinase inhibitor. Nat Med. 2010; 16:205-13.
81. Carayol N, Vakana E, Sassano A, Kaur S, Goussetis DJ, Glaser H, Druker BJ, Donato NJ, Altman JK, Barr S, Platanias LC. Critical roles for mTORC2- and rapamycin-insensitive mTORC1-complexes in growth and survival of BCR-ABL-expressing leukemic cells. Proc Natl Acad Sci U S A. 2010; 107:12469-74.
82. Altman JK, Sassano A, Kaur S, Glaser H, Kroczynska B, Redig AJ, Russo S, Barr S, Platanias LC. Dual mTORC2/mTORC1 targeting results in potent suppressive effects on acute myeloid leukemia (AML) progenitors. Clin Cancer Res. 2011; Epub Mar 17.
83. Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, Gray NS. An ATP-competitive mTOR inhibitor reveals rapamycin-insensitive functions of mTORC1. J Biol. Chem. 2009; 284:8023-32.
84. Thoreen CC, Sabatini DM. Rapamycin inhibits mTORC1, but not completely. Autophagy. 2009; 5:725-26.
85. Dose Escalation Study of INK128 in Relapsed or Refractory Multiple Myeloma or Waldenstrom Macroglobulinemia. http://www.clinicaltrials.gov./ct2/show/NCT01118689?term=ink-128&rank=3
86. Tamburini J, Chapuis N, Bardet V, Park S, Sujobert P, Willems L, Ifrah N, Dreyfus F, Mayeux P, Lacombe C, Bouscary D. Mammalian target of rapamycin (mTOR) inhibition activates phosphatidylinositol 3-kinase/Akt by up-regulating insulin-like growth factor-1 receptor signaling in acute myeloid leukemia: rationale for therapeutic inhibition of both pathways. Blood. 2008; 111:379-82.
87. Mazzoletti M, Bortolin F, Brunelli L, Pastorelli R, Di Giandomenico S, Erba E, Ubezio P, Broggini M. Combination of PI3K/mTOR inhibitors: antitumor activity and molecular correlates. Cancer Res. 2011; May 20:Epub ahead of print.
88. Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, Brachmann S, Chène P, De Pover A, Schoemaker K, Fabbro D, Gabriel D, Simonen M, Murphy L, Finan P, Sellers W et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008; 7:1851-63.
89. Chapuis N, Tamburini J, Green AS, Vignon C, Bardet V, Neyret A, Pannetier M, Willems L, Park S, Macone A, Maira SM, Ifrah N, Dreyfus F, Herault O, Lacombe C, Mayeux P et al. Dual inhibition of PI3K and mTORC1/2 signaling by NVP-BEZ235 as a new therapeutic strategy for acute myeloid leukemia. Clin Cancer Res. 2010; 16:5424-35.