
Introduction
Initial broad-based clinical studies found that only a minority of individuals with non-small cell lung cancer (NSCLC) responded to either gefitinib or erlotinib, which are tyrosine kinase inhibitors (TKIs) blocking activation of the epidermal growth factor (EGFR) [1-4]. Asian, non-smoking females were noted to be particularly sensitive to this class of drugs [2, 5]. Subsequently, investigators discovered that sensitivity to these TKIs was correlated with the presence of somatic mutations that affect the kinase domain of EGFR, especially either deletions within exon 19 or a L858R mutation in exon 21 [6-8]. Individuals with these tumors often respond to TKI therapy, but usually have progressive disease after 6-12 months of therapy. These resistant tumors frequently acquired either an additional mutation (T790M) in exon 20 of EGFR or a second mutation in the downstream pathway of EGFR, both resulting in lack of response to EGFR-TKI [9-11]. Other mechanisms of resistance include either amplification of c-met [12, 13] or deletion of PTEN [14, 15]. A major therapeutic priority is to identify drugs that can reverse the TKI resistance in exon 20 mutant EGFR. In order to discover additional drugs and pathways that might inhibit growth of NSCLC with an exon 20 mutant EGFR, we screened with a short-hairpin library for synergism with gefitinib [16-20] and identified several molecular biological pathways and pharmaceutical compounds that inhibited these TKI-resistant NSCLC cells.
Results
The focus of our study was to identify target genes and/or agents that can overcome TKI-resistance in NSCLC. Initial growth curves (MTT assays) showed that H1975 NSCLC cells [TKI-resistant, harbors EGFR exon 20 (T790M) and exon 21 (L858R) mutations] was more resistant to gefitinib than PC14 cells [TKI-sensitive, EGFR exon 19 (delE746-A750)] (Fig. 1A). A screen was conducted for genes that, when silenced, enhanced TKI sensitivity in H1975 cells. The cells were transduced with a small hairpin RNAs (shRNAs) pooled library and a synthetic lethality screen in the presence of gefitinib was performed. Hairpins targeting 35 genes (from 16,000 genes) reproducibly conferred gefitinib sensitivity in H1975 cells (threshold relative fold depletion in gefitinib versus vehicle as log2 ratio and p-value < 0.05; Table 1).
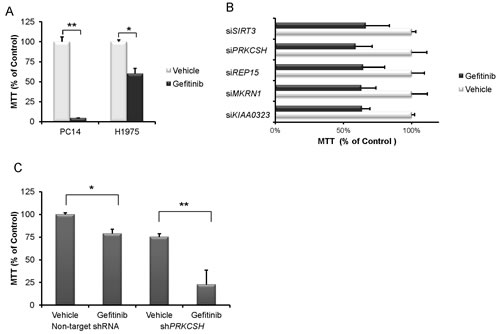
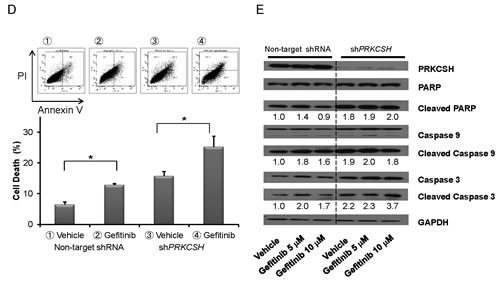
Fig.1: PRKCSH knockdown cells have increased sensitivity to gefitinib. (A) MTT assay was performed using H1975 NSCLC cells [TKI-resistant, EGFR exon 20 (T790M) and exon 21 mutation] and PC14 NSCLC cells [TKI-sensitive, EGFR exon 19 (delE746-A750)]. Cells were treated with either vehicle or 10 μM gefitinib for 3 days. Data represent the mean and SD of experiments done in triplicate. (B) H1975 cells were cultured with either vehicle or 10 μM gefitinib after transduction with either a non-target siRNA or gene-specific siRNA. Relative cell viability (vehicle – gefitinib) / non-target siRNA vehicle x 100 % was measured. (C) H1975 NSCLC with stable silencing of PRKCSH (see panel E) were cultured for 3 days either with or without gefitinib (10 μM) and cell proliferation was measured (MTT assay). (D) H1975 NSCLC cells either with or without stable silencing of PRKCSH (see panel E) were cultured for 6 days either with or without gefitinib (10 μM) and total cell death [apoptosis and necrosis (Annexin V+)] (%) was measured. (E) H1975 NSCLC cells transduced with either a non-target shRNA or a stable PRKCSH shRNA were cultured with either 5 or 10 μM gefitinib for 24 hours. Lysates were western blotted and probed with antibodies against either PRKCSH, PARP, cleaved PARP, caspase 9, cleaved caspase 9, caspase 3, cleaved caspase 3 and GAPDH (loading control). Densitometry of bands of cleaved PARP, cleaved caspase 9, cleaved caspase 3 and GAPDH was performed with image J software. Band intensities were normalized to GAPDH band intensity. * p < 0.05, ** p value < 0.01.
Table 1: List of shRNA primary screen hairpin hits in gefitinib-treated NSCLC cells.
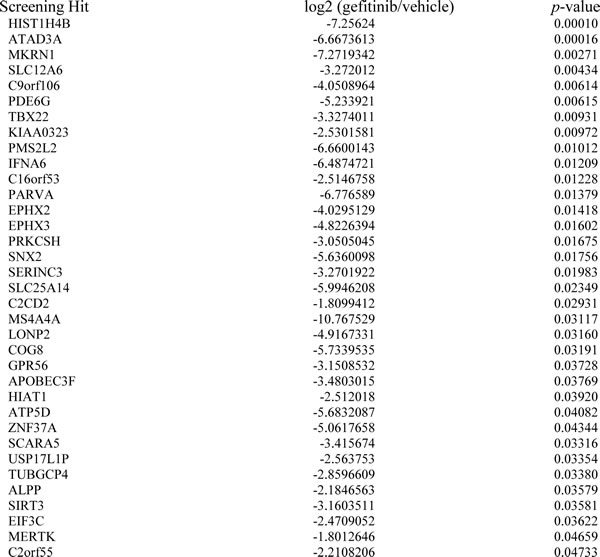
H1975 cells treated with either gefitinib or vehicle were infected with a pooled shRNA library consisting of 80,000 hairpins. Primary screen hairpin hits displayed as relative fold depletion in gefitinib versus vehicle expressed as log2 ratio and p-value <0.05.
Twenty genes from the initial 35 target genes identified in the primary screen were examined for their growth inhibitory effects in gefitinib treated H1975 cells using independent siRNAs. Five genes (SIRT3, PRKCSH, REP15, MKRN1, KIAA0323) were confirmed to enhance the gefitinib-induced inhibition of cell proliferation (Fig. 1B). Silencing the PRKCSH gene produced the most profound growth inhibition. Therefore, expression of the PRKCSH gene was stably silenced in H1975 cells using lentiviral shRNA vector which markedly enhanced the gefitinib-related growth inhibition (50% decrease compared to gefitinib alone) and apoptosis (27% versus 17% with gefitinib alone) (Figs. 1C, D). Silencing of PRKCSH enhanced the expression of cleaved PARP, caspase-9 and -3 which was only slightly enhanced by addition of gefitinib (5, 10 μM) (Fig. 1E).
We next investigated which pathways were linked to gefitinib-induced cell death based on shRNA library data using NCI / Nature Pathway Interaction Database (PID) (http://pid.nci.nih.gav) (Table 2) [21]. The CD27 signaling pathway (including BCL2L1, MAP3K7, MAP3K10, MAPK10, TRAF5, IKBKB) was identified as the most significantly affected (p-value: 9.3325 E-05; Fig. 2, Table 2). This pathway relates to activation of NF-κB and pro-survival signaling. To explore the role of NF-κB pathway in gefitinib-induced cell death, cells were treated with dasatinib. Dasatinib is a known inhibitor of multiple tyrosine kinases (BCR-ABL, SRC family, c-Kit etc) which inhibits the NF-κB pathway upstream of Src pathway [22, 23].
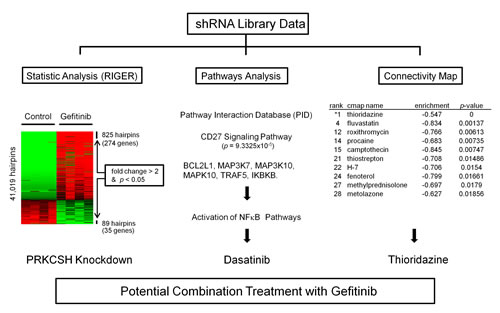
Fig.2: Three different approaches to identify a pathway or drug that can overcome gefitinib resistance of NSCLC. H1975 cells were transduced with shRNA pooled library (MISSION® shRNA) and a synthetic lethality screen with gefitinib was performed. shRNA library data were statistically analyzed by RIGER software (left panel). “Heat-map”: red and green represent either accumulation or depletion of the shRNA, respectively, when comparing the gefitinib treated and untreated cohort of H1975 NSCLC cells. Arrows point to those shRNA vectors whose abundance was significantly changed compared to control. Pathway analysis of the shRNA screen was done using NCI / Nature Pathway Interaction Database (PID) (middle panel). Connectivity Map software (right panel) was used to identify small-molecule combination therapy with gefitinib based on shRNA library data. All three approaches found effective combination treatment with gefitinib.
Table 2: Analysis of gefitinib-treatment related signaling pathway in NSCLC cells.
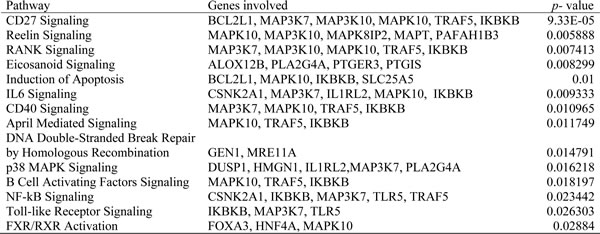
Gefitinib-treatment related signaling pathway was analyzed to identify new candidate inhibitors. shRNA primary hairpin screen” hits” were analyzed by using NCI / Nature Pathway Interaction Database (PID).
Treatment of cells with dasatinib promoted gefitinib-induced cell inhibition of proliferation of NSCLC (Fig. 3A). Dasatinib treatment alone did not have a potent ability to induce cell death of H1975 cells; but when combined with gefitinib, they significantly increased cell death (Fig. 3B). Our studies using multiple concentration of gefitinib and dasatinib showed that co-treatment with gefitinib (5 or 10 μM) and dasatinib (100, 200 or 400 nM) synergistically inhibited growth of H1975 cells (CI<1 1-6 columns, Fig. 3C). Also, in contrast to either drug alone, this drug combination induced apoptosis, as shown by an increase in caspase 3 cleavage (Fig. 3D). Exposure to dasatinib attenuated phospho-Src, as well as phospho-Rel A (Fig. 3D).
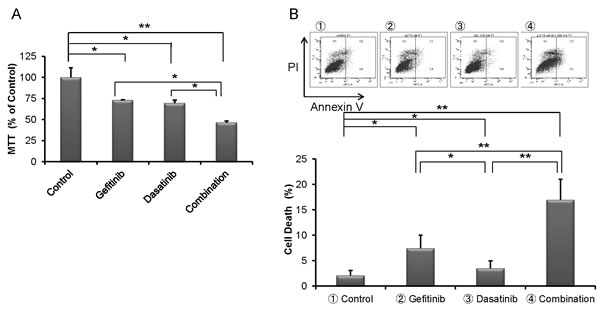
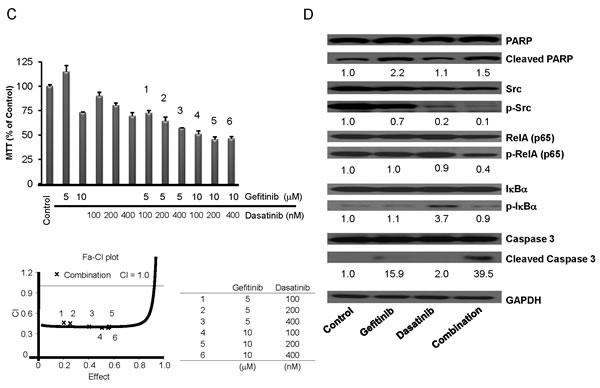
Fig.3: Combination of dasatinib and gefitinib synergistically inhibited cell proliferation of TKI-resistant H1975 NSCLC cells. (A) Co-treatment with gefitinib (10 μM) and dasatinib (400 nM) for 3 days inhibited growth of H1975 cells greater than either inhibitor alone (MTT assays, mean ± SD of 3 experiments done in quadruplicate). (B) H1975 cells were cultured with gefitinib (10 μM), dasatinib (400 nM) and combination (gefitinib 10 μM, dasatinib 400 nM) for 6 days; cells were harvested and stained with Annexin-V and PI, and analyzed by flow cytometry. Cells in lower right quadrant (Annexin-V+/PI-) was defined as apoptotic cells and upper right quadrant (Annexin-V+/PI+) was defined as necrotic cells. Total cell death (%) [apoptotic cells and necrotic cells] was measured. Representative results are shown. (C) H1975 cells were exposed to gefitinib alone (5 μM or 10 μM), dasatinib alone (100, 200 or 400 nM) and combination treatment gefitinib (5 μM or 10 μM) and dasatinib (100, 200 or 400 nM) for 3 days. Combination treatment inhibited growth of H1975 cells greater than either inhibitor alone. Combination effect was measured by Calcuysin software. CI < 1, CI = 1 and CI > 1 represent synergism, additive, and antagonism of the two compounds, respectively. (D) H1975 cells were exposed to gefitinib (10 μM), dasatinib (400 nM) and combination (gefitinib 10 μM and dasatinib 400 nM) for 24 hours. Lysates were western blotted and probed with antibody against PARP, cleaved PARP, Src, phospho-Src, caspase 3, cleaved caspase 3 and GAPDH (loading control). Densitometry of bands of cleaved PARP, phospho-Src, cleaved caspase 3 and GAPDH was done with image J software, and band intensities were normalized to GAPDH band intensity. * p < 0.05, ** p value < 0.01.
In-silico screening using the Connectivity Map of our shRNA library data identified drugs that could enhance cell kill by gefitinib (Overview, Fig. 2). In brief, this correlation-based pattern-matching software utilizes the input gene signatures from our shRNA library analysis by RIGER. Based on the degree of difference between gefitinib and the pharmaceutical perturbagen, a connectivity score is assigned, and the negative enrichment score is used to identify a perturbagen inducing a synergistic effect with gefitinib (Table 3). The top pharmaceutical compounds were examined. Surprisingly, the first compound on the list was “thioridazine”, which has been reported to have antiproliferative activity against tumor cells [24]. Exposure of H1975 cells to either thioridazine (10 μM) or gefitinib (10 μM) treatment produced an approximate 25% growth inhibition; when the two drugs were combined, an appropriate 65% decreased growth occurred (Fig. 4A). Similarly, the combination of the two drugs caused cell death (annexin V/propidium iodine positivity) of about 40% of the lung cancer cells (H1975) compared to either gefitinib (10 μM) or thioridazine (15 μM) alone (~ 12% and 14%, respectively) (Fig. 4B). Furthermore, co-treatment with thioridazine and gefitinib for 3 days synergistically inhibited the growth of the lung cancer cells (CI<1, columns labeled 1-4; (Fig. 4C,). Thioridazine (10 μM) and gefitinib (10 μM) alone (24 hours) did not affect expression of either p-Akt or p-p70S6K in lung cancer cells; but together, they reduced levels of these activated signaling pathways (Fig 4D).
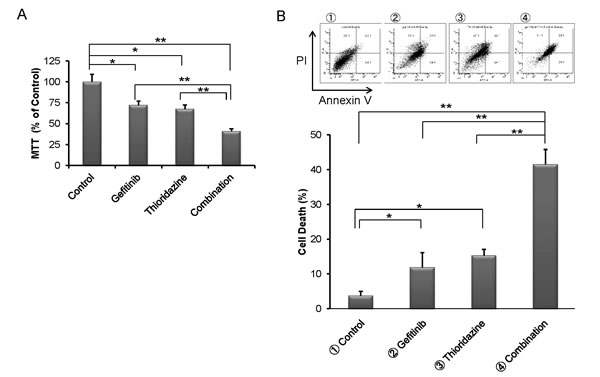
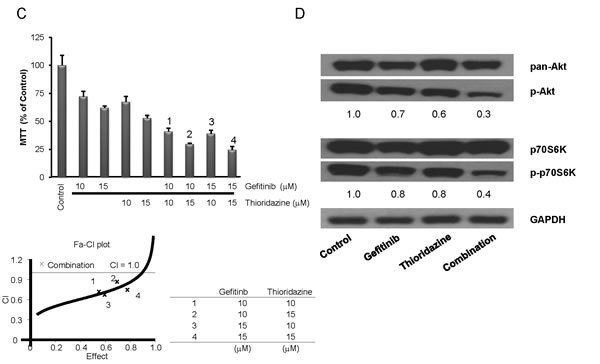
Fig.4: Effect of the combination of thioridazine and gefitinib on TKI-resistant NSCLC cells. (A) Incubation with gefitinib (10 μM) and thioridazine (10 μM) for 3 days inhibited growth (MTT assay) of H1975 NSCLC cells greater than either inhibitor alone (MTT assays, mean ± SD of 3 experiments done in quadruplicate). (B) H1975 NSCLC cells were exposed to either gefitinib (10 μM) or thioridazine (10 μM), or their combination (gefitinib 10 μM, thioridazine 10 μM) for 6 days; cells were harvested and stained with Annexin-V and PI, and analyzed by flow cytometry. Lower right quadrant (Annexin-V+/PI-) was defined as apoptotic cells and upper right quadrant (Annexin-V+/PI+) was defined as necrotic cells. Total cell death (%) [apoptotic cells and necrotic cells] is displayed graphically. Representative results are shown. (C) Incubation with gefitinib (10 μM or 15 μM) and/or thioridazine (10 μM or 15 μM) for 3 days inhibited growth of H1975 cells measured in MTT assay (top of panel C). Combination effect was measured by Calcuysin software. CI < 1, CI = 1 and CI > 1 represent synergism, additive, and antagonism of the two compounds, respectively. (D) H1975 cells were cultured with either gefitinib (10 μM) and/or thioridazine (10 μM) for 24 hours. Lysates were western blotted and probed with antibody against, pan-Akt, phospho-Akt, p70S6K, phospho-p70S6K and GAPDH (loading control). Densitometry of bands of phospho-Akt,, phospho-p70S6K, and GAPDH was done with image J software. Band intensities were normalized to GAPDH band intensity. * p < 0.05, ** p value < 0.01.
Table 3: Connectivity Map: Potential therapeutic agents enhancing gefitinib activity.
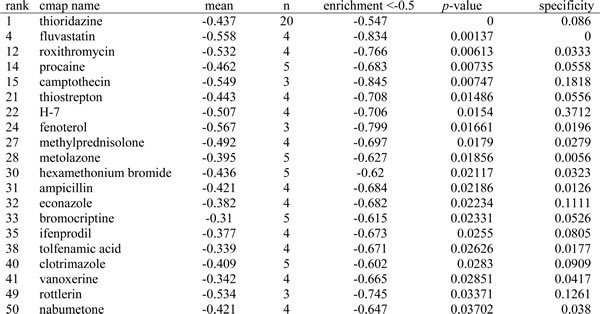
The Connectivity Map is correlation-based pattern-matching software. Data from shRNA primary hairpin screen hits were analyzed by the Connectivity Map. This ranking (left column) reflects the ability of the pharmaceutical perturbagens to induce a synergistic effect with gefitinib.
Discussion
Almost all patients with a newly diagnosed NSCLC with either an exon 19 or 21 mutation of EGFR have an initial response to TKI therapy. However, invariably after 6-12 months, the tumor becomes resistant to therapy, associated with additional genomic changes especially a T790M EGFR mutation. Similar to the lessons learned with HIV therapy, treatment of these NSCLC with more than one drug may prevent emergence of a drug-resistant clone. Therefore, we performed a synthetic lethality screen with gefitinib using a pooled short-hairpin RNA library in NSCLC cells to help identify pathways druggable in TKI resistant NSCLC. Furthermore, the shRNA library data was used for gefitinib-related pathway analysis and in-silico gene signature based examination.
From the shRNA library screen and siRNA validation experiments in NSCLC cells, we identified and validated PRKCSH as a candidate gene that synergizes with gefitinib (Fig. 1). PRKCSH encodes the non-catalytic β-subunit of glucosidase II (GIIβ). Glucosidase II facilitates protein translocation and “quality control” pathways of the endoplasmic reticulum (ER). Mutation of this gene is associated with autosomal dominant polycystic liver disease (PCLD) [25-27]. Silencing of PRKCSH in NSCLC cells caused decreased cell proliferation and increase in the proportion of apoptotic cells, and when combined with gefitinib both these effects were significantly enhanced (Figs. 1C and D). Identification of PRKCSH led us to examine PKD2 [28-30]. Both these molecules are involved in autosomal dominant polycystic kidney and liver diseases, and PRKCSH acts as a chaperone-like molecule to regulate PKD2 expression. PKD2 controls the endoplasmic reticulum (ER) regulated Ca2+ mediated apoptosis by decreasing the Ca2+ concentration in the ER [26]. Silencing of PKD2 in NSCLC cells augmented apoptosis when combined with gefitinib (Supplement Fig. 1B, C). In addition, PKD2 knockdown inhibited activation of the ERK pathway, which is associated with cell survival (Supplement Fig. 1C). To the best of our knowledge, neither PRKCSH nor PKD2 has been reported as therapeutic targets for NSCLC. The use of either PRKCSH or PKD2 inhibitors in combination with gefitinib to treat NSCLC requires further study.
Our shRNA library screen also identified the CD27 signaling pathway which relates to activation of NF-κB and pro-survival signals (Fig. 2 and Table 2). Dasatinib can modulate the NF-κB pathway; but alone, the drug had little effect on either apoptosis or proliferation of NSCLC. However, the combination of dasatinib and gefitinib synergistically induced cell death and inhibited cell growth (Fig. 3). Taken together, these experiments suggest that the combination of NF-κB inhibitors and gefitinib may be therapeutically effective for EGFR-mutant NSCLC.
The Connectivity Map is a genomic screening tool for linking genes associated with a selected phenotype with potential therapeutic agents [31, 32]. To discover additional therapeutic compounds that may synergistically interact with gefitinib, the shRNA library data were analyzed using the Connectivity Map software. Thioridazine, a phenothiazine derivative, was one of the prominent “hits”. Several reports have noted that the drug can have anticancer activity [33-35]. The mechanism by which this is mediated is unclear, although it may occur by inhibiting the PI3K/Akt pathway. The combined treatment of thioridazine and gefitinib synergistically inhibited proliferation of NSCLC cells (Fig. 4C). Neither thioridazine nor gefitinib suppressed levels of activated Akt and p70S6K in NSCLC, but their combination reduced phospho-Akt and phospho-p70S6K levels in these cells (Fig. 4D). Phenothiazines can cause severe extrapyramidal side-effects such as tardive dyskinesia. Thus, further modification of this molecule should be explored to incorporate the anti-NSCLC effects without neurological toxicity. Alternatively, phenothiazines activate PP2A; we (unpublished data) and others [36] have shown that drugs that activate PP2A have anti-cancer activity. Novel activators of this phosphatase may synergize with gefitinib to inhibit growth of EGFR mutant NSCLC.
In summary, shRNA library screen identified therapeutic partners of gefitinib for treatment of EGFR mutant NSCLC including EGFR T790M. Furthermore, the shRNA library data were utilized for gene signature based in-silico query which identified PRKCSH as an interesting partner with gefitinib. Finally, the shRNA results were used to interrogate the Connectivity Map. This pathway analysis identified thioridazine as a synergistic partner of gefitinib. In summary, this study using several experimental approaches has identified exciting drugs and pathways for further study in EGFR mutant NSCLC.
Materials and Methods
Reagents and NSCLC cell lines
The following compounds were used in this study: gefitinib; dasatinib [LC Laboratories (MA, USA)]; thioridazine (10-[21-methyl-2-piperidyl) ethyl]-2-methylthio-phenothiazine) [Sigma Aldrich (Singapore)]. All reagents were dissolved in dimethyl sulfoxide (DMSO) and the final concentration of DMSO never exceeded 0.1% in culture. Antibodies: Akt [36], phospho-Akt (Ser473), caspase-3, 8, 9, cleaved caspase-3, 8, 9, ERK, phospho-ERK, IκBα, phospho-IκBα, p70S6K, phospho-p70S6K (Thr421 / Ser424), PKD2, PRKCSH, PARP, cleaved-PARP, RelA, phospho-RelA, Src, phospho-Src, GAPDH were purchased from Cell Signaling (MA, USA).
The human NSCLC cell lines H1975 and PC14 were purchased from the American Type Culture Collection (Manassas,VA). These cells were cultured in RPMI 1640 supplemented with 10% fetal calf serum, 100 U/ml of penicillin, and 100 μg/ml of streptomycin. The H1975 cell line has an EGFR T790M mutations in exon 20, associated with gefitinib and erlotinib resistance. The PC14 cell line has an EGFR delE746-A750 mutation in exon 19, associated with TKI sensitivity.
Pooled short-hairpin RNA library screen
H1975 cells were transduced in quadruplicates with a pooled shRNA library (MISSION® shRNA), consisting of 80,000 hairpins targeting 16,000 genes at an MOI of 0.3 hairpins/cell. After 48 hours, puromycin was added to the media (5 μg/ml) to select for transduced cells.
Puromycin was continuously in the media until cells were harvested. Following selection, each replicate was treated with either vehicle or gefitinib (8 μM) for 1 week. This concentration of gefitinb was identified as the IC50 to H1975 cells [37]. Cells were harvested and genomic DNA was isolated both prior to drug treatment and at day 10. shRNA sequences were amplified by PCR using specific primers. The purified PCR products were subjected to high-throughput sequencing using Illumina Genome Analyzer IIx. The quality of the data was checked, normalized and statistically analyzed by RNAi gene enrichment ranking (RIGER) software [38]. Significant hits were defined as those altered by 2 fold, p<0.05
PRKCSH and PKD2 knockdown cells
Stably knockdown of the expression of the PRKCSH and PKD2 genes in H1975 cells used lentiviral shRNA vectors. PRKCSH shRNA plasmid was kindly provided by Prof. Gard Walz (Renal Division, University Hospital Freiburg). The PRKCSH shRNA sequence (5’-CCGGAAGTTCAGTGCCATGAAGTATCT CGAGATACTTCATGGCACTGAACTTTT TTTG-3’) was cloned into pLKO.1 vector. PKD2 shRNA plasmid was constructed according to selected sequences within the gene and cloned into pLKO.1 vector. (5’-CCGGCACGACCAACAGATACTATCTCGAGATAGTATCTGTTGGTCGTGTTTTTG-3’). The negative control shRNA [SHC002 (MISSION® shRNA)] was purchased from Sigma. Cells were transduced and selected with puromycin (2 μg / ml, 10 days). Silencing of the genes were confirmed by western blotting.
Cell proliferation assays
NSCLC cells were seeded at a density of 10,000 per well in 96-well plates. After 24 hours, these cells were cultured with multiple concentrations of drugs for 72 hours. Cell proliferation was determined using CellTiter 96® Non-Radioactive Cell Proliferation Assay (Promega) according to the manufacturer’s instructions; and the plates were read by a fluorescence spectrometer. Half-maximal inhibitory concentrations (“IC50-values”) were determined from the images under the growth inhibition curves using Prism 4.0 software. Calcusyn software (Biosoft) was used to analyze combination drug treatment data. CI<1, CI=1 and CI>1 represent synergism, additive, and antagonism of the two compounds, respectively (Fig. 3C, Fig. 4C)
Cell death assays
Cells were harvested and stained with Annexin-V and PI (BD Biosciences), and analyzed by a FACSCalibur flow cytometer (BD Sciences). Lower right quadrant (Annexin-V+/PI-) and upper right quadrant (Annexin-V+/PI+) were defined as apoptotic cells and necrotic cells, respectively.
Western blot analysis
Cells were harvested, washed with PBS, and lysed in ProteoJET Mammalian Cell Lysis Regent (Fermentas) supplemented with ProteoBlock Protease Inhibitor Cocktail (Fermentas). Protein concentrations were measured by Bradford assay (Bio-Rad, Richmond, CA). Equal amounts of protein were dissolved in SDS-polyacrylamide gel electrophoresis sample loading buffer and electrophoresed in a polyacrylamide gel (7.5% or 10%). After electrophoresis, the proteins were electrotransferred to a polyvinylidene difluoride membrane (Immobilon, Millipore, Bedford, MA). Immunoblotting was performed using antibodies mentioned above, and detected by ECL-Plus reagent (Amersham, Boston, MA).
Statistical analysis
Data were collected using a minimum of three experiments and used to calculate the mean ± S.D. Statistical significance was calculated using either ANOVA or student-t test and was considered significant at p values < 0.05.
Acknowledgements
We would like to thank the members of the Koeffler lab at both Cancer Science Institute of Singapore and Cedars-Sinai Medical Center for advice and helpful support. This work was funded by the Singapore Ministry of Health’s National Medical Research Council (NMRC) under its Singapore Translational Research (STaR) Investigator Award to H. Phillip Koeffler and the NMRC Centre Grant awarded to National University Cancer Institute of Singapore, the National Research Foundation Singapore and the Singapore Ministry of Education under its Research Centres of Excellence initiatives, and NIH grant 2R01CA026038-35.
References
1. Sordella R, Bell DW, Haber DA and Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science 2004; 305(5687):1163-1167.
2. Fukuoka M, Yano S, Giaccone G, Tamura T, Nakagawa K, Douillard JY, Nishiwaki Y, Vansteenkiste J, Kudoh S, Rischin D, Eek R, Horai T, Noda K, Takata I, Smit E, Averbuch S, et al. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (The IDEAL 1 Trial) [corrected]. Journal of Clinical Oncology. 2003; 21(12):2237-2246.
3. Blackhall F, Ranson M and Thatcher N. Where next for gefitinib in patients with lung cancer? The Lancet Oncology. 2006; 7(6):499-507.
4. Krause DS and Van Etten RA. Tyrosine kinases as targets for cancer therapy. The New England Journal of Medicine. 2005; 353(2):172-187.
5. Kris MG, Natale RB, Herbst RS, Lynch TJ, Jr., Prager D, Belani CP, Schiller JH, Kelly K, Spiridonidis H, Sandler A, Albain KS, Cella D, Wolf MK, Averbuch SD, Ochs JJ and Kay AC. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. The Journal of the American Medical Association. 2003; 290(16):2149-2158.
6. Greulich H, Chen TH, Feng W, Janne PA, Alvarez JV, Zappaterra M, Bulmer SE, Frank DA, Hahn WC, Sellers WR and Meyerson M. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Medicine. 2005; 2(11):e313.
7. Ji H, Li D, Chen L, Shimamura T, Kobayashi S, McNamara K, Mahmood U, Mitchell A, Sun Y, Al-Hashem R, Chirieac LR, Padera R, Bronson RT, Kim W, Janne PA, Shapiro GI, et al. The impact of human EGFR kinase domain mutations on lung tumorigenesis and in vivo sensitivity to EGFR-targeted therapies. Cancer Cell. 2006; 9(6):485-495.
8. Mitsudomi T, Kosaka T, Endoh H, Horio Y, Hida T, Mori S, Hatooka S, Shinoda M, Takahashi T and Yatabe Y. Mutations of the epidermal growth factor receptor gene predict prolonged survival after gefitinib treatment in patients with non-small-cell lung cancer with postoperative recurrence. Journal of Clinical Oncology. 2005; 23(11):2513-2520.
9. Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG and Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. The New England Journal of Medicine. 2005; 352(8):786-792.
10. Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG and Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Medicine. 2005; 2(3):e73.
11. Kwak EL, Sordella R, Bell DW, Godin-Heymann N, Okimoto RA, Brannigan BW, Harris PL, Driscoll DR, Fidias P, Lynch TJ, Rabindran SK, McGinnis JP, Wissner A, Sharma SV, Isselbacher KJ, Settleman J, et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102(21):7665-7670.
12. Ma PC, Maulik G, Christensen J and Salgia R. c-Met: structure, functions and potential for therapeutic inhibition. Cancer Metastasis Reviews. 2003; 22(4):309-325.
13. Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, Chitale D, Motoi N, Szoke J, Broderick S, Balak M, Chang WC, Yu CJ, Gazdar A, Pass H, Rusch V, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proceedings of the National Academy of Sciences of the United States of America. 2007; 104(52):20932-20937.
14. Yamasaki F, Johansen MJ, Zhang D, Krishnamurthy S, Felix E, Bartholomeusz C, Aguilar RJ, Kurisu K, Mills GB, Hortobagyi GN and Ueno NT. Acquired resistance to erlotinib in A-431 epidermoid cancer cells requires down-regulation of MMAC1/PTEN and up-regulation of phosphorylated Akt. Cancer Research. 2007; 67(12):5779-5788.
15. Kokubo Y, Gemma A, Noro R, Seike M, Kataoka K, Matsuda K, Okano T, Minegishi Y, Yoshimura A, Shibuya M and Kudoh S. Reduction of PTEN protein and loss of epidermal growth factor receptor gene mutation in lung cancer with natural resistance to gefitinib (IRESSA). British Journal of Cancer. 2005; 92(9):1711-1719.
16. Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK, Carpenter AE, Foo SY, Stewart SA, Stockwell BR, Hacohen N, Hahn WC, et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 2006; 124(6):1283-1298.
17. Ngo VN, Davis RE, Lamy L, Yu X, Zhao H, Lenz G, Lam LT, Dave S, Yang L, Powell J and Staudt LM. A loss-of-function RNA interference screen for molecular targets in cancer. Nature. 2006; 441(7089):106-110.
18. Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, Frohling S, Chan EM, Sos ML, Michel K, Mermel C, Silver SJ, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009; 462(7269):108-112.
19. Bivona TG, Hieronymus H, Parker J, Chang K, Taron M, Rosell R, Moonsamy P, Dahlman K, Miller VA, Costa C, Hannon G and Sawyers CL. FAS and NF-kappaB signalling modulate dependence of lung cancers on mutant EGFR. Nature. 2011; 471(7339):523-526.
20. Meylan E, Dooley AL, Feldser DM, Shen L, Turk E, Ouyang C and Jacks T. Requirement for NF-kappaB signalling in a mouse model of lung adenocarcinoma. Nature. 2009; 462(7269):104-107.
21. Schaefer CF, Anthony K, Krupa S, Buchoff J, Day M, Hannay T and Buetow KH. PID: the Pathway Interaction Database. Nucleic Acids Research. 2009; 37(Database issue):D674-679.
22. Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA and Connelly PA. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. The Journal of Biological Chemistry. 1996; 271(2):695-701.
23. Lluis JM, Buricchi F, Chiarugi P, Morales A and Fernandez-Checa JC. Dual role of mitochondrial reactive oxygen species in hypoxia signaling: activation of nuclear factor-{kappa}B via c-SRC and oxidant-dependent cell death. Cancer Research. 2007; 67(15):7368-7377.
24. Rho SB, Kim BR and Kang S. A gene signature-based approach identifies thioridazine as an inhibitor of phosphatidylinositol-3’-kinase (PI3K)/AKT pathway in ovarian cancer cells. Gynecologic Oncology. 2011; 120(1):121-127.
25. Gao H, Wang Y, Wegierski T, Skouloudaki K, Putz M, Fu X, Engel C, Boehlke C, Peng H, Kuehn EW, Kim E, Kramer-Zucker A and Walz G. PRKCSH/80K-H, the protein mutated in polycystic liver disease, protects polycystin-2/TRPP2 against HERP-mediated degradation. Human Molecular Genetics. 2010; 19(1):16-24.
26. Wegierski T, Steffl D, Kopp C, Tauber R, Buchholz B, Nitschke R, Kuehn EW, Walz G and Kottgen M. TRPP2 channels regulate apoptosis through the Ca2+ concentration in the endoplasmic reticulum. The EMBO journal. 2009; 28(5):490-499.
27. Fedeles SV, Tian X, Gallagher AR, Mitobe M, Nishio S, Lee SH, Cai Y, Geng L, Crews CM and Somlo S. A genetic interaction network of five genes for human polycystic kidney and liver diseases defines polycystin-1 as the central determinant of cyst formation. Nature Genetics. 2011; 43(7):639-647.
28. Chen J, Lu L, Feng Y, Wang H, Dai L, Li Y and Zhang P. PKD2 mediates multi-drug resistance in breast cancer cells through modulation of P-glycoprotein expression. Cancer Letters. 2011; 300(1):48-56.
29. Tsiokas L. Function and regulation of TRPP2 at the plasma membrane. American Journal of Physiology. 2009; 297(1):F1-9.
30. Streets AJ, Needham AJ, Gill SK and Ong AC. Protein kinase D-mediated phosphorylation of polycystin-2 (TRPP2) is essential for its effects on cell growth and calcium channel activity. Molecular Biology of the Cell. 2010; 21(22):3853-3865.
31. Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, Reich M, Hieronymus H, Wei G, Armstrong SA, Haggarty SJ, Clemons PA, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science 2006; 313(5795):1929-1935.
32. Lamb J. The Connectivity Map: a new tool for biomedical research. Nature Reviews. 2007; 7(1):54-60.
33. Nagel D, Spranger S, Vincendeau M, Grau M, Raffegerst S, Kloo B, Hlahla D, Neuenschwander M, Peter von Kries J, Hadian K, Dorken B, Lenz P, Lenz G, Schendel DJ and Krappmann D. Pharmacologic inhibition of MALT1 protease by phenothiazines as a therapeutic approach for the treatment of aggressive ABC-DLBCL. Cancer Cell. 2012; 22(6):825-837.
34. Sachlos E, Risueno RM, Laronde S, Shapovalova Z, Lee JH, Russell J, Malig M, McNicol JD, Fiebig-Comyn A, Graham M, Levadoux-Martin M, Lee JB, Giacomelli AO, Hassell JA, Fischer-Russell D, Trus MR, et al. Identification of drugs including a dopamine receptor antagonist that selectively target cancer stem cells. Cell. 2012; 149(6):1284-1297.
35. Kang S, Dong SM, Kim BR, Park MS, Trink B, Byun HJ and Rho SB. Thioridazine induces apoptosis by targeting the PI3K/Akt/mTOR pathway in cervical and endometrial cancer cells. Apoptosis. 2012; 17(9):989-997.
36. Gutierrez A, Pan L, Groen RW, Baleydier F, Kentsis A, Marineau J, Grebliunaite R, Kozakewich E, Reed C, Pflumio F, Poglio S, Uzan B, Clemons P, VerPlank L, An F, Burbank J, et al. Phenothiazines induce PP2A-mediated apoptosis in T cell acute lymphoblastic leukemia. Journal of Clinical Investigation. 2014; 124(2):644-655.
37. Sudo M, Chin TM, Mori S, Doan NB, Said JW, Akashi M and Koeffler HP. Inhibiting proliferation of gefitinib-resistant, non-small cell lung cancer. Cancer Chemotherapy and Pharmacology. 2013; 71(5):1325-1334.
38. Luo B, Cheung HW, Subramanian A, Sharifnia T, Okamoto M, Yang X, Hinkle G, Boehm JS, Beroukhim R, Weir BA, Mermel C, Barbie DA, Awad T, Zhou X, Nguyen T, Piqani B, et al. Highly parallel identification of essential genes in cancer cells. Proceedings of the National Academy of Sciences of the United States of America. 2008; 105(51):20380-20385.