
Introduction
Colorectal cancer (CRC) is the third leading cause of cancer-related deaths in both men and women, with a projected 154,270 new cases, accounting for approximately 8% of all new cancer diagnoses in the United States in 2025 alone. Notably, 1 in 24 Americans are estimated to develop invasive CRC in their lifetime, and the prevalence of CRC is rising among adults under 55 years of age [1]. This shift suggests a changing CRC landscape and highlights the need to further understand the mechanisms underlying CRC and the development of better therapies to improve patient outcomes.
Collagen triple helix repeat containing 1 (CTHRC1) is an inducible, 28 kDa glycosylated secreted protein that is detectable in human circulation and is basally expressed by osteoblasts, osteocytes, neurons in the paraventricular and supraoptic nucleus of the hypothalamus, and episodically by fibroblasts following tissue injury or in cancer-associated stroma [2–4]. CTHRC1 contains twelve G-X-Y repeats that facilitate trimer formation, and a propeptide-encoding N terminus that, when cleaved, activates the protein [2, 4]. Most circulating CTHRC1 exists in its cleaved form, which can increase glycolysis and decrease the respiratory exchange ratio in mice [2], enhance cell migration, and regulate ECM synthesis and organization [4, 5]. Since CTHRC1 was first found to be expressed by fibroblasts in balloon-injured arteries in 2005 [4], its roles in a variety of physiological processes have been established, including metabolism [2, 6, 7], bone and joint homeostasis [8, 9], tissue remodeling and ECM synthesis and organization [4, 5, 10], and oncogenesis [3, 11, 12].
Many studies have identified CTHRC1 as a biomarker for metastasis and poor prognosis in cancers including breast [12, 13], head and neck, kidney, liver, lung, stomach, endometrial [14], and CRC [15]. However, many of these studies used bulk RNA-sequencing to identify CTHRC1 expression, leaving the identity of the cell type expressing CTHRC1 in the tumor unknown. CTHRC1-positive cancer-associated fibroblasts (CAFs) have been shown to modulate macrophage polarization to the M2 lineage in the tumor microenvironment (TME), which is characterized by anti-inflammatory, angiogenic, and immunosuppressive properties [12, 16]. Using commercially available antibodies that have not been rigorously validated some groups have claimed tumor cells express CTHRC1. However, in our own studies we detected CTHRC1 expression only in CAFs [3], and others agree that CTHRC1 is expressed by CAFs and is a key regulator of CRC progression [15]. In the present study, we investigated the role of host-derived CTHRC1 in CRC progression using a rigorously created and validated CTHRC1 global knockout (KO) mouse and a monoclonal antibody that has been thoroughly validated for specificity to CTHRC1 (clone Vli55) [3]. This antibody has been used to demonstrate that CTHRC1 is specifically and highly expressed by activated stromal cells in the TME of many cancers including gastric, breast, endometrial, pancreatic, kidney, lung, skin, and colon, but is not expressed by the malignant cells [3]. Currently, the precise function of stromal-derived CTHRC1 remains unknown and the consequences of inhibiting CTHRC1 in the TME on tumor outcomes have never been assessed. Additionally, enhanced understanding of how CTHRC1 may promote an immunosuppressive environment could influence future CRC treatments. Thus, we have herein tested the hypothesis that removal of CTHRC1 from the TME will reduce CRC tumor growth and increase anti-tumor immune responses.
Results
Human colorectal tumor cells and MC38 cells do not express CTHRC1
In the literature, there is debate whether the tumor cells themselves express CTHRC1, or only the activated cancer-associated fibroblasts (CAFs), which can make up a large portion of solid tumors. We have performed CTHRC1 immunohistochemistry (IHC) on multiple human tumor samples from our MaineHealth Institute for Research (MHIR) Biobank and consistently observed that tumor cells are negative for CTHRC1, while CAFs are strongly positive (Figure 1A, 1B, Supplementary Figure 1A–1I). We have also observed positive CTHRC1 expression in dermal fibroblasts during skin wound healing in a representative wild type (WT) mouse at days 5 and 8 post-injury, while dermal fibroblasts from a representative Cthrc1 global knockout (KO) mouse at day 5 post-injury lack detectable CTHRC1 expression (Supplementary Figure 1J–1L).
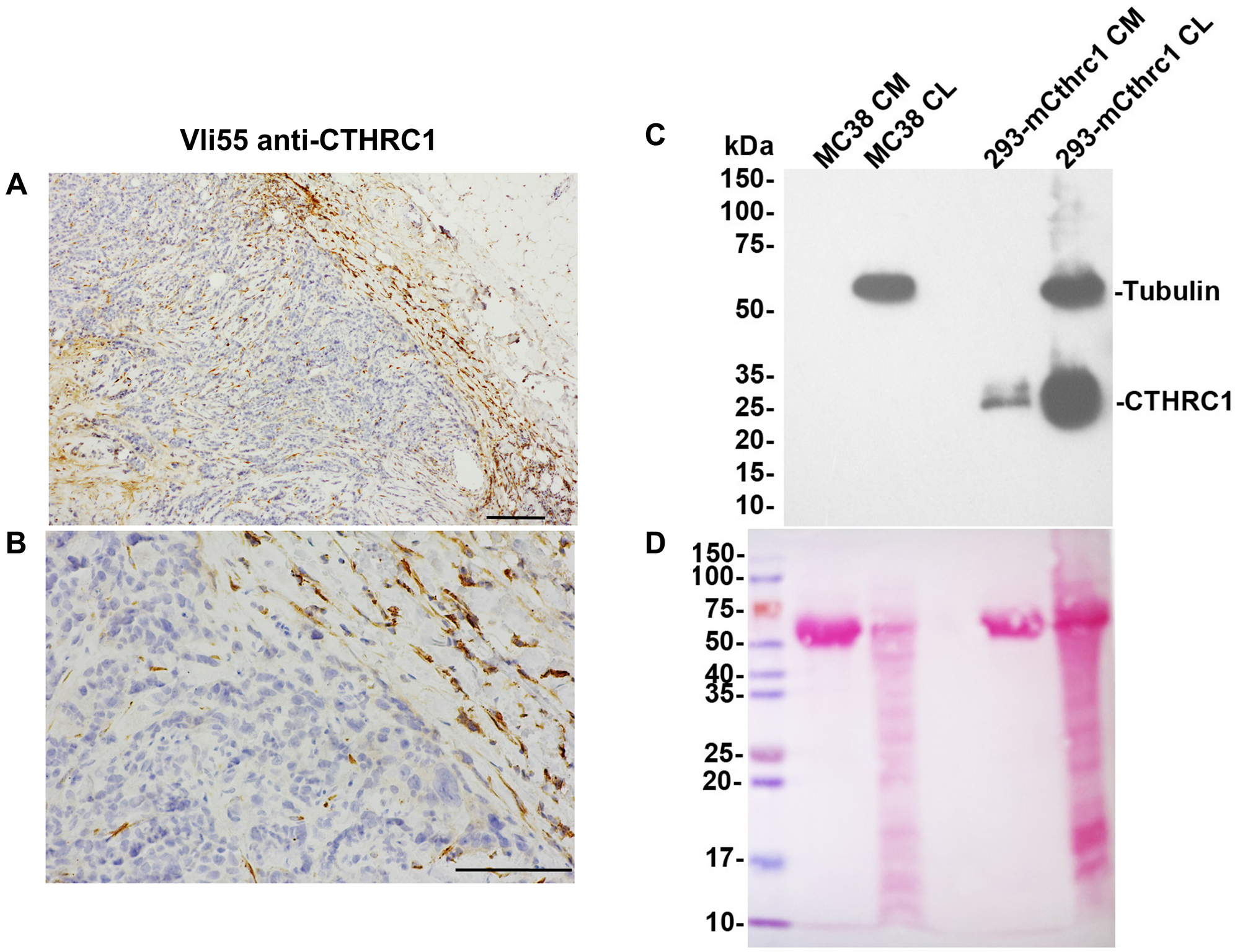
Figure 1: CTHRC1 is not detected in human colorectal tumor cells or MC38 cells.
CTHRC1 immunostaining of human colorectal tumor sections with VLi55 at 50X (A) and 200X (B) magnification shows no CTHRC1 protein present in tumor cells (stained blue). Positive dark brown staining is present in fibroblasts in the stroma surrounding and infiltrating the tumor. Scale bar (A) 200 μm and (B) 100 μm. (C) CTHRC1 protein was not detected in MC38 cells in either conditioned medium (CM) or cell lysates (CL) by Western blot analysis using VLi55 anti-CTHRC1 antibody recognizing the completely conserved C terminus of CTHRC1. CTHRC1 is detected in CM and CL from HEK293T cells transfected with an expression construct encoding mouse Cthrc1. Molecular weights are shown in kDa on the left. No bands corresponding to the expected 28 kDa CTHRC1 protein were observed in MC38 samples in the Western blot, even though proteins were present in the lanes. Tubulin staining is used as a loading control for CL samples. (D) Ponceau S staining of the same membrane confirms protein presence in each lane. A strong band at ~67 kDa is observed in samples of conditioned medium, which is consistent with albumin as part of the fetal bovine serum present in the culture medium.
We also performed Western blot analysis of the MC38 cells used in this study and found that they do not produce CTHRC1, either intracellularly (cell lysate; CL) or secreted into the medium (conditioned media; CM). MC38 cells showed no CTHRC1 protein expression, whereas positive control samples of Cthrc1-transfected cells showed CTHRC1 at the expected molecular weight (Figure 1C). Ponceau S staining of the same blot confirmed that protein was present in each lane (Figure 1D).
Knocking out Cthrc1 reduces subcutaneous tumor burden and increases mouse survival
To determine if host-derived CTHRC1 contributes to CRC tumor growth and mouse survival, we conducted an in vivo study tracking tumor growth with BLI and survival using Kaplan-Meier analysis (Figure 2 and Supplementary Figure 2). Cthrc1 KO mice had reduced tumor burden at Day 19 post-inoculation by longitudinal BLI compared to WT mice (Figure 2A and Supplementary Figure 2A). This was a result of Cthrc1 KO mice having either greatly delayed/slowed/absent tumor growth, or tumor growth and then regression, which can be clearly visualized in some mice when comparing days 6 to 13 (Figure 2B). This difference in tumor burden was confirmed by volumetric tumor measurements (Figure 2C and Supplementary Figure 2B). A correlation between BLI-quantified tumor burden and caliper-measured tumor volume is shown in Supplementary Figure 2C. When mice were individually monitored and euthanized upon meeting survival endpoint criteria, Cthcr1 KO mice survived significantly longer than WT mice, with 60% of Cthrc1 KO mice never reaching survival endpoint criteria by end of study (Day 69) (Figure 2D). Despite these differences in time of euthanasia, we still compared post-mortem tumor and spleen masses. Surprisingly, these were also reduced in Cthrc1 KO mice (Figure 2E, 2F). No significant weight differences were seen between WT and Cthrc1 KO mice, within sexes, (Supplementary Figure 2D), and no adverse effects of Cthrc1 KO were observed.
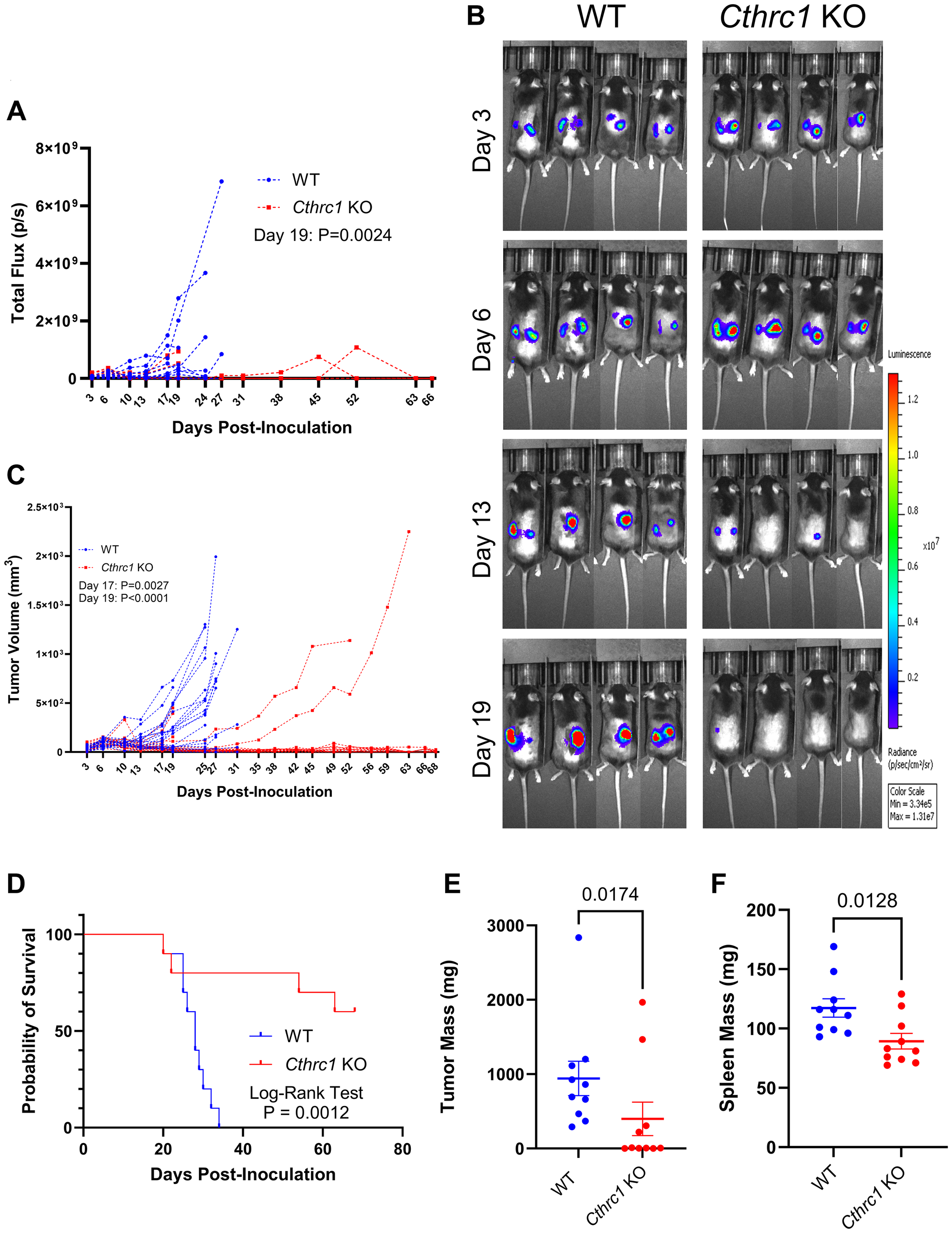
Figure 2: Knocking out Cthrc1 leads to decreased CRC tumor burden and increases survival.
Graph (A) shows longitudinal BLI following MC38Luc+/RFP+ cell inoculation with significance determined by two-way ANOVA through Day 19, the final day all mice were alive. Panel (B) shows representative BLI images of WT and Cthrc1 KO mice at Days 3, 6, 13, and 19 post-inoculation, with a luminescence scale of 3.34 × 106–1.31 × 107. The representative mice exhibited the smallest deviation from cohort averages. Graph (C) shows individual measurements of tumor volume over time. Statistical significance was determined by two-way ANOVA through Day 19, the last day all mice were alive. Panel (D) shows a Kaplan-Meier survival curve determined by endpoint criteria as described in Methods, with statistics determined by Log-Rank Test. Graphs of post-mortem (E) tumors and (F) spleen weights show significant differences between WT and Cthrc1 KO mice, as determined by Mann-Whitney and unpaired t-tests, respectively. (n = 10 per group).
Knocking out Cthrc1 consistently reduces subcutaneous tumor burden and induces a more active anti-tumor immune microenvironment
In an effort to directly compare time-matched tumor and spleen tissues between groups, we then conducted an endpoint study (Figure 3). By Day 13 post-inoculation, Cthrc1 KO mice exhibited significantly less BLI-quantified tumor burden compared to WT mice (Figure 3A and Supplementary Figure 3A). Cthrc1 KO mice in this cohort exhibited a tumor regression trend similar to that observed in the survival study, which occurred between Days 9–13, while tumors in WT mice kept growing. This phenomenon was clearly visible on Day 14 in representative BLI images (Figure 3B). Tumor regression in Cthrc1 KO mice was further captured by tumor volumes measured with electronic calipers (Figure 3C and Supplementary Figure 3B), which also confirmed significantly less tumor burden in Cthrc1 KO mice by Day 13. Overall reduced tumor volume in the Cthrc1 KO cohort was further validated by post-mortem tumor weights, which confirmed that tumors from Cthrc1 KO mice weighed significantly less than those from WT counterparts (Figure 3D). No significant differences were detected in spleen weights or sex-matched body weights between groups (Figure 3E and Supplementary Figure 3C). Further analysis of tumors and spleens collected at sacrifice investigated immune cell differences between groups (Figure 3F). Compared to WT mice, Cthrc1 KO mice exhibited an increased percentage of CD3+ T cells in both tumors and spleens, and decreased Gr-1+ myeloid cells in the spleen, indicating a potential inhibitory role for CTHRC1 in immunosurveillance in CRC. The flow cytometry gating strategy for these populations are described in Supplementary Figure 3D, 3E.
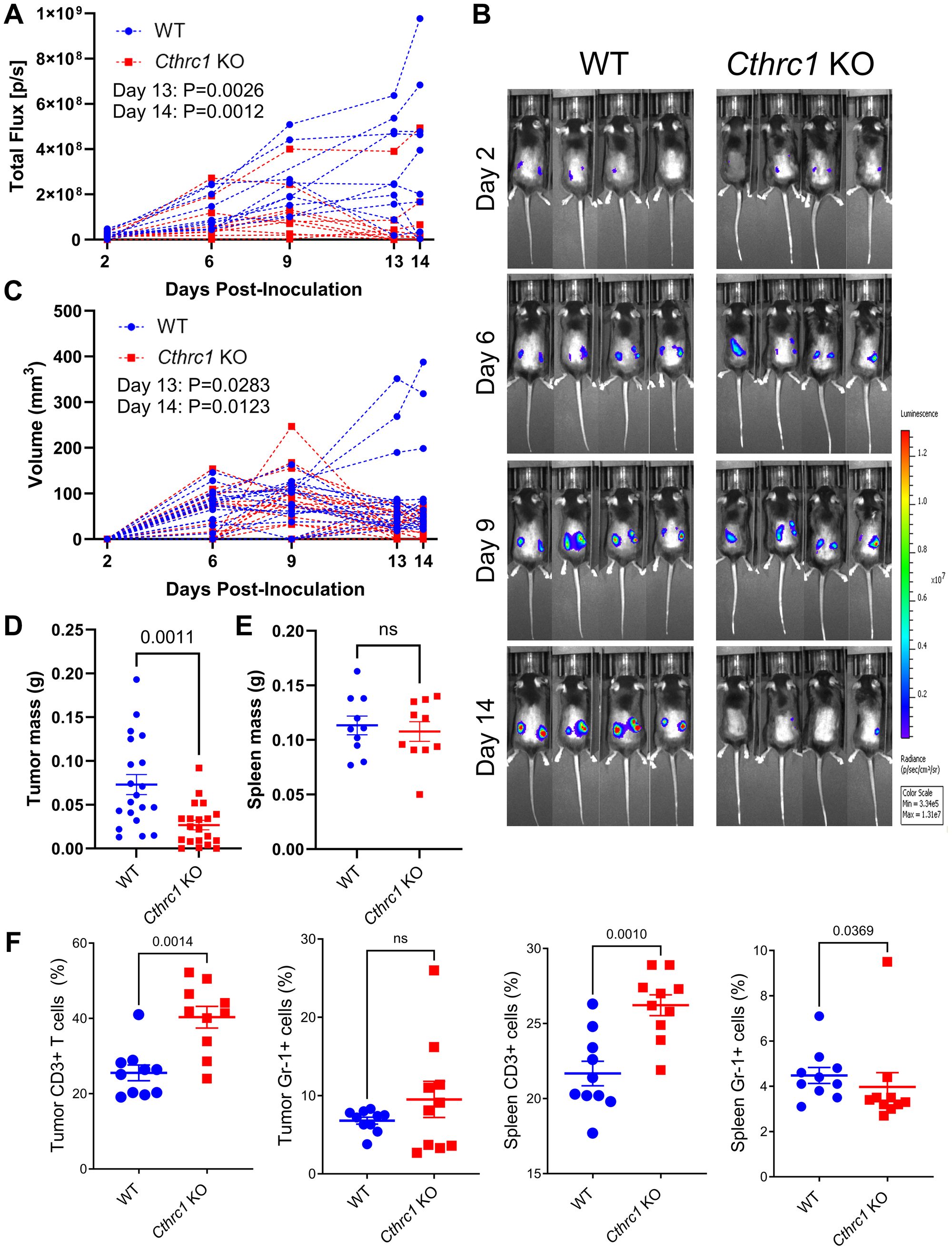
Figure 3: Knocking out Cthrc1 in mice leads to decreased CRC tumor burden and reduces immunosuppression.
(A) Shows the longitudinal total flux (p/s) by BLI of individual WT and Cthrc1 KO mice, each line representing the average total flux between the two tumors on individual mice. Statistics determined by two-way ANOVA. Panel (B) shows representative BLI images of WT and Cthrc1 KO mice at Days 2, 6, 9, and 14 post-inoculation, with a luminescence scale of 3.34 × 105–1.31 × 107. Representative mice were chosen based on the smallest deviation from cohort averages. (C) Depicts individual tumor volumes over time as measured by electronic calipers, with left and right tumors graphed individually. Panel (D) shows individual tumors weights post-mortem. Statistics determined by Welch's correction for unpaired t-tests. Graph (E) depicts spleen mass at endpoint, with statistics determined by unpaired t-test. Panel (F) shows immune cell analyses, specifically the percentage of CD3+ T cells and Gr-1+ myeloid cells from tumors and spleens of WT and Cthrc1 KO mice, analyzed by Mann-Whitney tests. All data represent mean ± SEM. (n = 10 per group).
To confirm for a third time that Cthrc1 KO mice are protected from CRC, we repeated the endpoint study using an additional independent cohort (Supplementary Figure 4), which again demonstrated that Cthrc1 KO mice have reduced tumor burden compared to WT and exhibit tumor regression between Days 7–11 (Supplementary Figure 4A–4D). No significant differences were detected between groups in spleen weights, either post-mortem or longitudinally (Supplementary Figure 4E, 4F), nor in sex-matched body weights throughout the study (Supplementary Figure 4G).
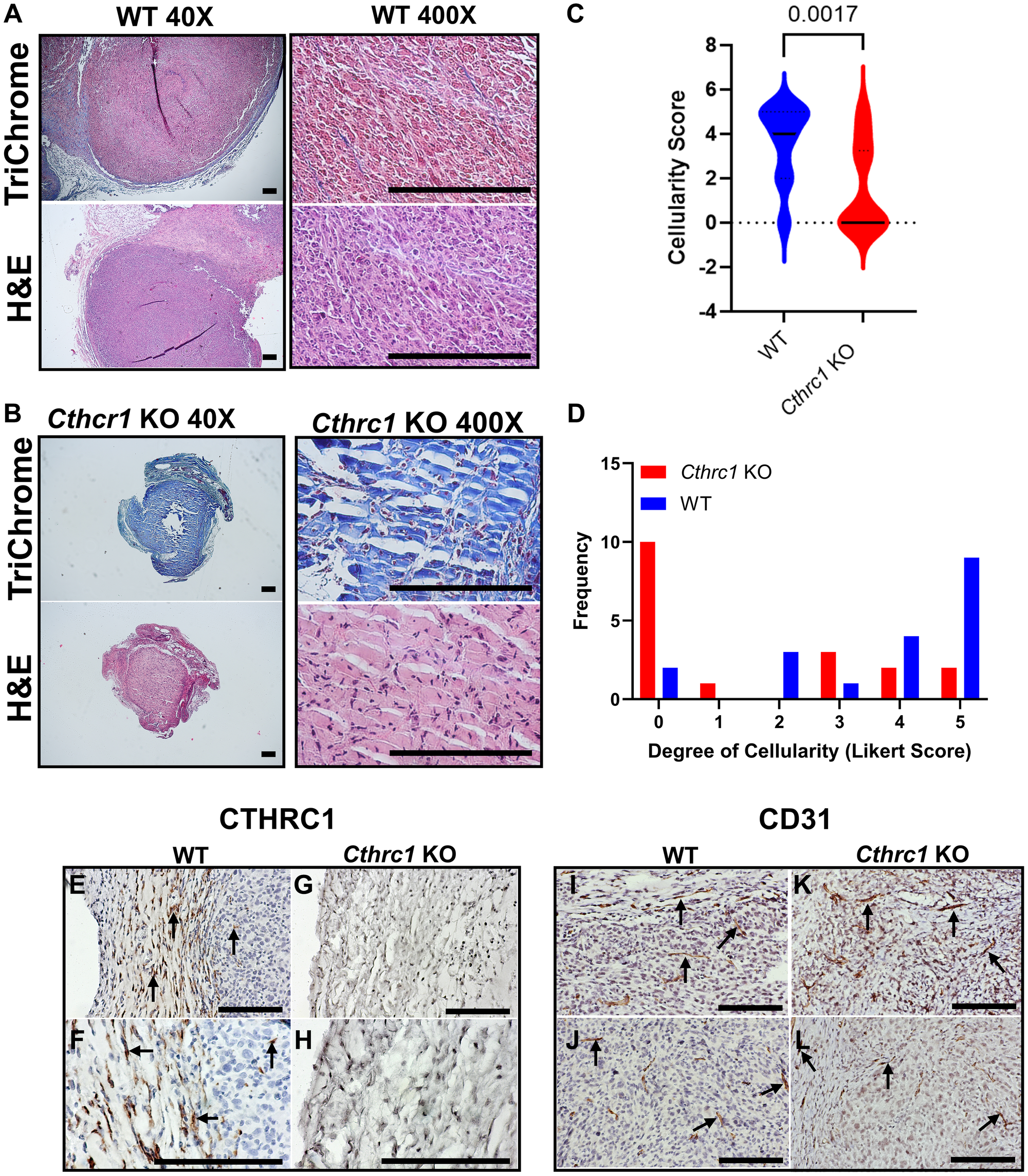
Figure 4: Histological staining reveals Cthrc1 KO alters tumor microarchitecture and cellularity.
Histological sections of mouse tumors from the endpoint study are shown. Tumors from WT and Cthrc1 KO mice at Day 14 post-inoculation were stained with Trichrome (top) and Hematoxylin and Eosin (H&E; bottom). Representative tumors from (A) WT and (B) Cthrc1 KO mice are shown at 40X (left) and 400X (right) magnification. Scale bar, 200 μm. Tumor cellularity per histological sample was scored using a Likert scale as described in Methods and are shown as (C) violin and (D) histogram plots (n = 18 WT, n = 19 Cthrc1 KO). Because the sample size is small and the scores are not normally distributed, differences in cellularity between WT and Cthrc1 KO tumor samples were assessed using a Mann-Whitney U-test. Vli55 immunohistochemical staining of tumor sections at 200X and 400X magnification for (E, F) WT and (G) Cthrc1 KO mice shows the fibrous capsule and tumor tissue, with positive staining for CTHRC1 only in WT tumors. (H) A stained regressed tumor from a Cthrc1 KO mouse shows residual matrix, few fibroblasts, and no CTHRC1. Scale bar, 200 μm. CD31 immunostaining of tumor sections at 200X magnification shows positive vessel-like structures in stroma and tumors of both (I, J) WT and (K, L) Cthrc1 KO mice. Positive staining (dark brown) is marked by arrows.
Histological staining reveals Cthrc1 KO alters tumor microarchitecture and cellularity
To further evaluate tumor morphology in Cthrc1 KO and WT mice, we employed Trichrome and Hematoxylin and Eosin (H&E) histological staining and IHC analysis of CTHRC1 and CD31 expression on subcutaneous tumors collected at Day 14 post-injection (Figure 4). Trichrome and H&E staining of representative WT (Figure 4A) and Cthrc1 KO (Figure 4B) tumors showed abundant collagen matrix and paucity of cells in the tumors from Cthrc1 KO mice. Presumably, the residual acellular extracellular matrix reflects the prior presence of tumor cells. Likert scale quantification of hematoxylin-stained cells revealed significantly reduced cellularity in tumors from Cthrc1 KO mice compared to WT (Figure 4C, 4D). CTHRC1 staining with Vli55 demonstrated CTHRC1 mainly in the tumor stroma of WT mice, with some positive staining of presumed fibroblasts within the tumor mass close to the fibrous capsule (Figure 4E, 4F). The absence of CTHRC1 in both stroma and tumors of Cthrc1 KO mice supports the hypothesis that host-derived CTHRC1 is responsible for tumor progression (Figure 4G, 4H). Vascularization of tumors was assessed by CD31 immunostaining (Figure 4I–4L) and no apparent difference between Cthrc1 KO mice and WT mice was noted.
DISCUSSION
The studies herein demonstrate a specific role of non-tumor cell (microenvironmental) CTHRC1 in the development of CRC. Mined clinical data had previously associated CTHRC1 with certain cancers [12–15], including CRC, which has a dependence on stroma [17], where CTHRC1 is expressed. Using highly specific CTHRC1 antibodies, we found that tumor cells in human CRC samples and the MC38 CRC cells in vitro do not produce CTHRC1 protein. Immunohistochemical staining for CTHRC1 in KO mice was negative in all samples indicating that CTHRC1 is not made by MC38 tumor cells in vivo and is host-derived. Comparing subcutaneous CRC growth between immunocompetent C57BL/6 and global Cthrc1 null mice revealed significant tumor regression in Cthrc1 KO mice across three independent cohorts. Immune profiling revealed that Cthrc1 KO mice have increased T cell percentages in both tumors and spleens, and decreased splenic Gr-1+ myeloid cells, suggesting both stromal and circulating CTHRC1 may contribute to immunosuppression in WT mice. Because the Gr-1+ population includes myeloid-derived suppressor cells (MDSCs), which inhibit T cell activation and proliferation [18], reduced systemic Gr-1+ myeloid cells in Cthrc1 KO mice may reduce myeloid-mediated T cell suppression and contribute to CRC protection. Variability in tumor progression/regression in individual mice could be related to innate differences in splenic immune composition, which warrants further study.
The exact mechanism by which Cthrc1 KO mice are protected from CRC is unknown and likely multifaceted, and a more detailed investigation into the effects of CTHRC1 on immune cell regulation is an important next step to understanding this protection. We have a very good understanding of the timing of CTHRC1 expression in response to fibroblast activation occurring after myocardial infarction, skin incisional wounding, and arterial injury in mice. In these acute injury models, fibroblasts start to express CTHRC1 as determined by immunostaining within two days, with CTHRC1 expression increasing over the following days until a gradual decline in expression is observable from two weeks on with very few fibroblasts expressing CTHRC1 four weeks post-injury [5, 10]. In all cohorts of the present study, there was initial growth of tumors, but significant tumor regression was seen after approximately 9 days in the Cthrc1 KO mice, with most tumors in WT mice continuing to grow. If induction of CTHRC1 by the presence of tumor cells follows a similar timeline, this would explain the timing of immunosuppression and promotion of tumor growth in WT mice. Although universal tissue repair processes that result in complete repair likely follow a similar time course of CTHRC1 expression, in the context of expanding tumor growth, we anticipate that stromal fibroblast activation is ongoing, resulting in sustained CTHRC1 expression. We were not able to study the exact timing of CTHRC1 expression in these mice, a limitation to our study. Future time course studies, along with inducible Cthrc1 KO models (either global or cell type specific), will help determine at what time, and in what cells, the CTHRC1 silencing should occur for the anti-tumor effects.
Another important limitation of the study herein is the subcutaneous nature of this model, in part due to differences in TME immune composition between subcutaneous and orthotopic tumors in colon tissues. Zhao and colleagues [19] demonstrated that, compared to subcutaneous models, orthotopic CRC tumors have stronger anti-tumor immune responses (e.g., increased presence of T cell and B cells, increased NK-cell infiltration, and higher expression of cytokines IL2, IL6, IFNγ and granzyme B), whereas subcutaneous CRC tumors exhibit increased levels of immunosuppressive MDSCs. These differences in immune composition between models have important implications for translation to human patients, particularly in the context of immunotherapies, as immune cell infiltration is known to impact patient outcomes [20]. Given the relatively sparse immune landscape and immunosuppressive TME of the subcutaneous CRC model used in the present study, it is possible that the protection from CRC progression observed in Cthrc1 KO mice may be even more pronounced in a more immune-infiltrated orthotopic environment. Further considerations when working with a subcutaneous model of CRC include differences in the ECM, which can influence mechanical properties, angiogenesis, and stromal cell interactions [21]. Future studies should therefore assess CTHRC1 function in orthotopic models of CRC.
Many studies have suggested that CTHRC1 promotes angiogenesis [2, 11]. Although there do not appear to be overt differences in the presence of CD31+ vessels in tumors between WT and Cthrc1 KO mice, solid tumors often have aberrant angiogenesis that influences the tumor microenvironment and tumor immune cells [22]. Further study of the mechanisms and timing of CTHRC1’s action on the TME will be critical to fully understanding its role in tumor promotion. In humans, targeting CTHRC1 may hold great potential as a novel strategy in oncology, but first, we purport, the receptor(s) for it must be identified, and inhibitors to these designed. Overall, we find that elevated CTHRC1 expression in cancer contributes to a dysregulated TME, leading to tumor growth.
Materials and Methods
Cell culture
MC38Luc+RFP+ murine colon cancer cells (ALSTEM; Cat# LRL04) were cultured in DMEM with 4.5 g/L glucose, L-glutamine, and sodium pyruvate (Corning, Cat # 50-003-PB) supplemented with 3.7 g/L sodium bicarbonate (Sigma, Cat # S5761), 10% fetal bovine serum (Gibco, Cat # A52568-01), and 1% penicillin-streptomycin (Anti-Anti; Gibco, Cat # 15240-062). Cells were maintained by weekly media changes and passaging at 37°C with 5% CO2. For additional details and validation, see Supplementary Methods.
In vivo studies
All experimental procedures involving mice were approved by the Institutional Animal Care and Use Committee (#2406) of MaineHealth. C57BL/6J (wild type, WT) and Cthrc1tm1VliC57BL/6J (Cthrc1 KO) global null mice, previously described [6], were bred within the MHIR animal facility.
In all studies, mice received bilateral subcutaneous injections of 5 × 105 MC38Luc+RFP+ cells in serum-free DMEM and Matrigel (Corning; Cat# 356237). Tumor growth was monitored by bioluminescent imaging (BLI) and electronic calipers (VWR®; Cat# 62379-531) twice weekly. Mice were euthanized if one or both tumors ulcerated or reached 2 cm in diameter, which served as the two defined endpoint criteria.
Three independent cohorts were used: a survival cohort, an endpoint cohort, and a repeated endpoint cohort, each consisting of 20 mice (10 WT and 10 Cthrc1 KO). The survival and endpoint cohorts included 5 males and 5 females per group. The repeated endpoint cohort included 4 male and 6 female WT mice, and 5 male and 5 female Cthrc1 KO mice.
Survival mice were inoculated at approximately 10.5 weeks of age and euthanized individually upon reaching an endpoint criterion. Mice that survived to Day 69 post-inoculation were sacrificed at that time. Endpoint mice were inoculated at approximately 8.5 weeks of age and euthanized on Day 16 post-inoculation, when most had reached an endpoint criterion. The repeated endpoint cohort was inoculated at approximately 9 weeks of age and euthanized on Days 18–19 post-inoculation, when most had reached an endpoint criterion. In this cohort, two male Cthrc1 KO mice and four WT mice (3 male and 1 female) did not survive to the endpoint; all remaining mice were euthanized at that time.
Immunohistochemistry and microscopy
Subcutaneous tumor composition and CTHRC1 prevalence were assessed by IHC. Tumors were fixed in 10% neutral buffered formalin (NBF) for 24–48 hours, then preserved in either optimal cutting temperature (OCT) compound for frozen sectioning or 70% ethanol for paraffin embedding. Sections were stained with hematoxylin and eosin (H&E), Masson’s trichrome, or antibodies against CTHRC1 or CD31 (Abcam, ab182981) to identify endothelial cells. IHC for CTHRC1 was performed with extensively validated rabbit monoclonal antibody (Vli55, www.mhir.org/antibody) as previously described [6]. Slides were imaged using an Axioskop 40 microscope (Zeiss). Formalin-fixed RFP+ tumor tissues embedded in OCT were imaged using an EVOS 5000 Imaging System (ThermoFisher Scientific, AMF5000). Maine Medical Center’s BioBank provided us with deidentified, formalin-fixed, paraffin-embedded tissue blocks of breast, lung, and colon cancer on which CTHRC1 immunohistochemistry was performed with Vli55 as described. Images of representative tumors are shown.
Immunology and flow cytometry
Single-cell suspensions were prepared from tumors and spleen samples for immune profiling by flow cytometry. Tumor samples were minced and enzymatically digested with collagenase I/dispase II and DNase I for one hour at 37°C. Tumors and spleens were gently pressed though a 40 μm cell strainer and subjected to red blood cell lysis before resuspension in FACS buffer (PBS containing 0.5% BSA and 2 mM EDTA) for staining and analysis. Additional procedural and antibody details are provided in Supplementary Methods.
Statistical analysis
Single day comparisons were analyzed by unpaired t-tests and data are presented as mean ± SEM unless otherwise stated. Longitudinal comparisons were analyzed by two-way ANOVA followed by Šidák’s post hoc test. Images of tumor H&E-stained sections were evaluated using a Likert scale by a blinded investigator to assess cellular density. Scores were assigned as follows: 0 = 0%, 1 = 0–25%, 2 = 25–50%, 3 = 50–75%, 4 = 75–100%, and 5 = 100% dense cellularity.
Data availability statement
Data generated in this study are available from the corresponding authors upon reasonable request.
ACKNOWLEDGMENTS
We greatly appreciate the unidentified cancer patients who have donated their tumor tissues for this work. We also express our deepest appreciation to Joel Miller, David Campbell, Lucy Liaw, Larisa Ryzhova, Diane Gallagher, Patrick Reagan, Jeanette and George Miller, Linda and Allan Fairfield, and Peg and Jim Campbell for supporting our families as we pursued this work.
CONFLICTS OF INTEREST
VL holds patents on CTHRC1 antibodies. All others declare no CoIs.
ETHICAL STATEMENT
All protocols involving animals were approved by the Institutional Animal Care and Use Committee (#2406) of MaineHealth in compliance with all applicable regulations and guidelines including the National Institutes of Health Guide for Care and Use of Laboratory Animals. The animal facility of MaineHealth is an AAALAC accredited facility. Paraffin embedded formalin fixed tissue sections of human tissue blocks were obtained under a protocol approved (#3657) by the Institutional Review Board (IRB) of Maine Medical Center. The specimens were de-identified, and the IRB had approved the use of the archival material for immunohistochemistry.
FUNDING
This work was supported by the American Cancer Society (Research Grant RSG-19-037-01-LIB), the NIH (R50CA265331, R37CA245330, R24DK092759-01, and U54GM115516), the Arthur Gary Family, and the Kane Foundation. Dr. Reagan is a Scholar of the Leukemia and Lymphoma Society. The Histopathology and Microscopy Core is supported by NIH COBRE grant P20GM121301. This project also utilized the Flow Core Facility, funded by NIH COBRE award P20GM106391 (R. Friesel, PI), and samples from the MaineHealth Biobank. We further acknowledge NIH grant R01 HL146504 (V. Lindner and S. Ryzhov, PIs) and the MaineHealth Institute for Research.
References
1. Siegel RL, Kratzer TB, Giaquinto AN, Sung H, Jemal A. Cancer statistics, 2025. CA Cancer J Clin. 2025; 75:10–45. https://doi.org/10.3322/caac.21871. [PubMed].
2. Toomey BH, Mitrovic SA, Lindner-Liaw M, Leon Vazquez RG, Kacer D, Ryzhov S, Prudovsky I, Lindner V. Activated CTHRC1 promotes glycolysis in endothelial cells: Implications for metabolism and angiogenesis. Vascul Pharmacol. 2023; 153:107246. https://doi.org/10.1016/j.vph.2023.107246. [PubMed].
3. Duarte CW, Stohn JP, Wang Q, Emery IF, Prueser A, Lindner V. Elevated plasma levels of the pituitary hormone Cthrc1 in individuals with red hair but not in patients with solid tumors. PLoS One. 2014; 9:e100449. https://doi.org/10.1371/journal.pone.0100449. [PubMed].
4. Pyagay P, Heroult M, Wang Q, Lehnert W, Belden J, Liaw L, Friesel RE, Lindner V. Collagen triple helix repeat containing 1, a novel secreted protein in injured and diseased arteries, inhibits collagen expression and promotes cell migration. Circ Res. 2005; 96:261–8. https://doi.org/10.1161/01.RES.0000154262.07264.12. [PubMed].
5. Ruiz-Villalba A, Romero JP, Hernández SC, Vilas-Zornoza A, Fortelny N, Castro-Labrador L, San Martin-Uriz P, Lorenzo-Vivas E, García-Olloqui P, Palacio M, Gavira JJ, Bastarrika G, Janssens S, et al. Single-Cell RNA Sequencing Analysis Reveals a Crucial Role for CTHRC1 (Collagen Triple Helix Repeat Containing 1) Cardiac Fibroblasts After Myocardial Infarction. Circulation. 2020; 142:1831–47. https://doi.org/10.1161/CIRCULATIONAHA.119.044557. [PubMed].
6. Stohn JP, Perreault NG, Wang Q, Liaw L, Lindner V. Cthrc1, a novel circulating hormone regulating metabolism. PLoS One. 2012; 7:e47142. https://doi.org/10.1371/journal.pone.0047142. [PubMed].
7. Stohn JP, Wang Q, Siviski ME, Kennedy K, Jin YR, Kacer D, DeMambro V, Liaw L, Vary CP, Rosen CJ, Prudovsky I, Lindner V. Cthrc1 controls adipose tissue formation, body composition, and physical activity. Obesity (Silver Spring). 2015; 23:1633–42. https://doi.org/10.1002/oby.21144. [PubMed].
8. Jin YR, Stohn JP, Wang Q, Nagano K, Baron R, Bouxsein ML, Rosen CJ, Adarichev VA, Lindner V. Inhibition of osteoclast differentiation and collagen antibody-induced arthritis by CTHRC1. Bone. 2017; 97:153–67. https://doi.org/10.1016/j.bone.2017.01.022. [PubMed].
9. Myngbay A, Manarbek L, Ludbrook S, Kunz J. The Role of Collagen Triple Helix Repeat-Containing 1 Protein (CTHRC1) in Rheumatoid Arthritis. Int J Mol Sci. 2021; 22:2426. https://doi.org/10.3390/ijms22052426. [PubMed].
10. Nordquist EM, Dutta P, Kodigepalli KM, Mattern C, McDermott MR, Trask AJ, LaHaye S, Lindner V, Lincoln J. Tgfβ1-Cthrc1 Signaling Plays an Important Role in the Short-Term Reparative Response to Heart Valve Endothelial Injury. Arterioscler Thromb Vasc Biol. 2021; 41:2923–42. https://doi.org/10.1161/ATVBAHA.121.316450. [PubMed].
11. Mei D, Zhu Y, Zhang L, Wei W. The Role of CTHRC1 in Regulation of Multiple Signaling and Tumor Progression and Metastasis. Mediators Inflamm. 2020; 2020:9578701. https://doi.org/10.1155/2020/9578701. [PubMed].
12. Wan H, Ling Z, Xie Y, Jiang H, Ruan Z, Yang D, Yang X, Pei J. Single-cell and transcriptome analyses revealed CTHRC1 a potential therapeutic target mediating invasion and tumor microenvironment in TNBC: experimental validation. Front Immunol. 2025; 16:1534981. https://doi.org/10.3389/fimmu.2025.1534981. [PubMed].
13. Wang Z, Zhang S, Zheng C, Xia K, Sun L, Tang X, Zhou F, Ouyang Y, Tang F. CTHRC1 is a Potential Prognostic Biomarker and Correlated with Macrophage Infiltration in Breast Cancer. Int J Gen Med. 2022; 15:5701–13. https://doi.org/10.2147/IJGM.S366272. [PubMed].
14. Sial N, Ahmad M, Hussain MS, Iqbal MJ, Hameed Y, Khan M, Abbas M, Asif R, Rehman JU, Atif M, Khan MR, Hameed Z, Saeed H, et al. CTHRC1 expression is a novel shared diagnostic and prognostic biomarker of survival in six different human cancer subtypes. Sci Rep. 2021; 11:19873. https://doi.org/10.1038/s41598-021-99321-w. [PubMed].
15. Hu S, Qin J, Gao R, Xiao Q, Liu X, Pan Y, Wang S. Integrated analysis of single cell and bulk RNA sequencing identifies CTHRC1+ INHBA+ CAF as drivers of colorectal cancer progression. Mol Carcinog. 2023; 62:1787–802. https://doi.org/10.1002/mc.23615. [PubMed].
16. Kadomoto S, Izumi K, Mizokami A. Macrophage Polarity and Disease Control. Int J Mol Sci. 2021; 23:144. https://doi.org/10.3390/ijms23010144. [PubMed].
17. Reslinger M, Plateroti M. Stromal cells in normal colon and colon cancers: importance of thyroid hormone signaling. Cell Death Dis. 2025; 16:699. https://doi.org/10.1038/s41419-025-08005-0. [PubMed].
18. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009; 9:162–74. https://doi.org/10.1038/nri2506. [PubMed].
19. Zhao X, Li L, Starr TK, Subramanian S. Tumor location impacts immune response in mouse models of colon cancer. Oncotarget. 2017; 8:54775–87. https://doi.org/10.18632/oncotarget.18423. [PubMed].
20. Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012; 12:298–306. https://doi.org/10.1038/nrc3245. [PubMed].
21. Zhang W, Xiang Y, Lu C, Wang F, Ren H, Wu H, Yan M. The extracellular matrix in inflammation and cancer. Mol Biomed. 2026; 7:21. https://doi.org/10.1186/s43556-026-00415-6. [PubMed].
22. Lamplugh Z, Fan Y. Vascular Microenvironment, Tumor Immunity and Immunotherapy. Front Immunol. 2021; 12:811485. https://doi.org/10.3389/fimmu.2021.811485. [PubMed].