
Malignant peripheral nerve sheath tumors (MPNSTs) are deadly sarcomas of the myelinating nerve sheath. They arise sporadically in 50% of all patients but are highly associated with a tumor predisposition syndrome, called Neurofibromatosis Type I (NF1). In NF1 patients, MPNSTs are the leading cause of mortality and they arise from the malignant transformation of benign plexiform neurofibromas (pNFs) [1]. As depicted in Figure 1, inactivation of the NF1 tumor suppressor gene is the initiating event in all MPNSTs, which increases RAS-MAPK signaling and MEK activation [2]. The next most frequent alteration (in nearly 90% of MPNSTs) is loss of the CDKN2A tumor suppressor, which results in hyperactivation of oncogenic Cyclin-Dependent Kinases 4 and 6 (CDK4/6) [2–7].
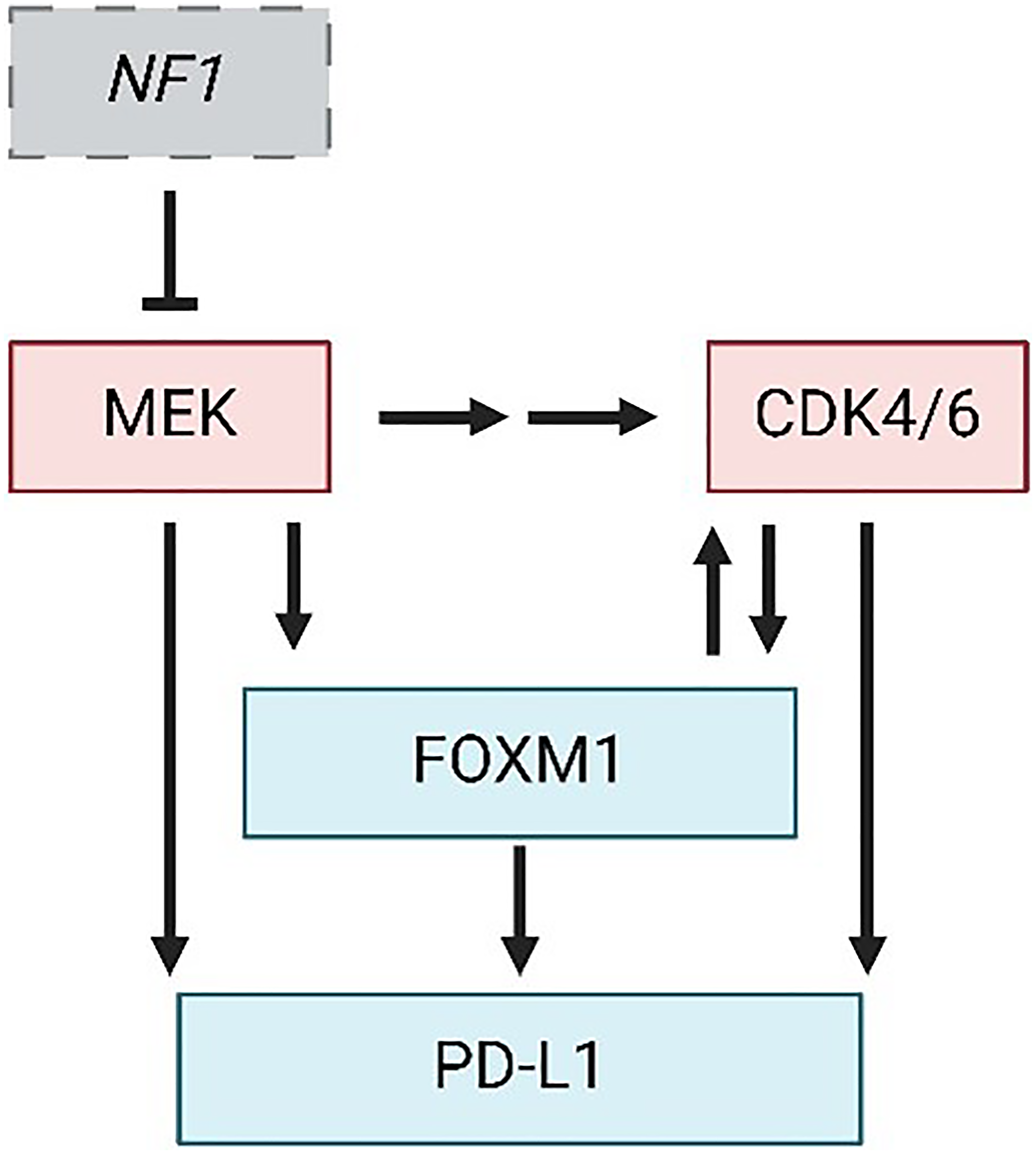
Figure 1: Central mechanisms of MPNST progression and therapy resistance.
Pathway diagram depicting NF1 loss and hyperactivation of MEK and CDK4/6 kinases as defining events in MPNST formation. FOXM1 and PD-L1 are downstream effectors of MEK and CDK4/6 whose upregulation likely mediates resistance to inhibitors of those kinases. Perpendicular bar, inhibition; Arrow, activation. Figure made with https://www.biorender.com/.
MPNSTs unfortunately lack effective therapies. The only curative treatment is complete surgical resection with clean, negative margins, but this is often not possible due to tumor size, location, and/or presence of metastatic disease [1]. Radiation and chemotherapy have limited efficacy and significant toxicity, making them poor options for treatment. To date, there still exists limited clinical trial data for immune checkpoint blockade (ICB) therapy in MPNST, although in most other types of sarcomas treatment with ICB agents failed to meet RECIST criteria [8]. Uniformly, the field has recognized a need for improved, more directed therapeutics to treat MPNSTs and many recently identified drivers of the disease are considered relevant clinical targets. For this commentary, we will discuss evidence supporting combination therapies against CDK4/6 and MEK for treating MPNSTs and their contribution toward anti-tumor immunity. We will also consider likely mediators of acquired resistance to such therapies, particularly Forkhead box protein M1 (FOXM1) and programmed death-ligand 1 (PD-L1) (Figure 1), whose simultaneous inhibition may be needed to achieve sustained, and possibly curative, anti-tumor activity.
We previously reported that de novo MPNSTs initiated by Nf1 and Cdkn2a inactivation in the sciatic nerve are sensitive to CDK4/6 inhibitors; however, drug resistance emerged rapidly in all tumors [6]. MEK is well known to promote resistance to CDK4/6 inhibitors by enhancing the transcription of CDK4 and CDK6 as well as their regulatory partners, the D type cyclins [9, 10]. As such, other groups explored combinations of CDK4/6 and MEK inhibitors in Ras-driven lung [11] and pancreatic cancers [12], where remarkable success of the combination was observed [13]. Those successes, as well as CDK4/6 and MEK hyperactivation in patient MPNSTs, propelled our examination of combined CDK4/6 and MEK inhibition in our de novo MPNST model [14]. As anticipated, single agent CDK4/6 or MEK inhibitors provided a modest survival benefit reflecting slowed tumor growth whereas dual CDK4/6-MEK inhibition induced tumor regression and greatly extended survival. Shrinkage of the tumors by CDK4/6-MEK targeting was significant in both magnitude and duration. Nonetheless, despite early tumor regression the effects were transient as 100% of tumors eventually regrew due to acquired drug resistance.
To determine the mechanisms underlying MPNST response to CDK4/6-MEK inhibitor therapy, gene expression profiling of drug sensitive versus drug resistant tumors was performed [14]. The most prominent difference was an immune activation profile in drug sensitive tumors that was lost in resistant tumors. Activation of anti-tumor immunity was expected in sensitive tumors because they regressed with drug treatment. Moreover, combined CDK4/6-MEK therapy was previously shown to activate CD8+ T cell or natural killer (NK) cell-mediated immune responses in other tumor types [11, 12]. What was surprising in the MPNST analyses, however, was a dominant signature for B and/or plasma cell activation within the drug sensitive tumor [14]. Histopathological evaluation of the tumors verified an accumulation of plasma cells, not B cells, in tumors that shrunk upon CDK4/6-MEK inhibition. Enhanced clustering of CD8+ T cells, an indicator of their activation, was also observed. Conversely, resistant tumors adopted a highly immunosuppressive environment enriched with M2 tumor-associated macrophages (TAMs) and elevated expression of PD-L1 in the tumor cells (Figure 2).
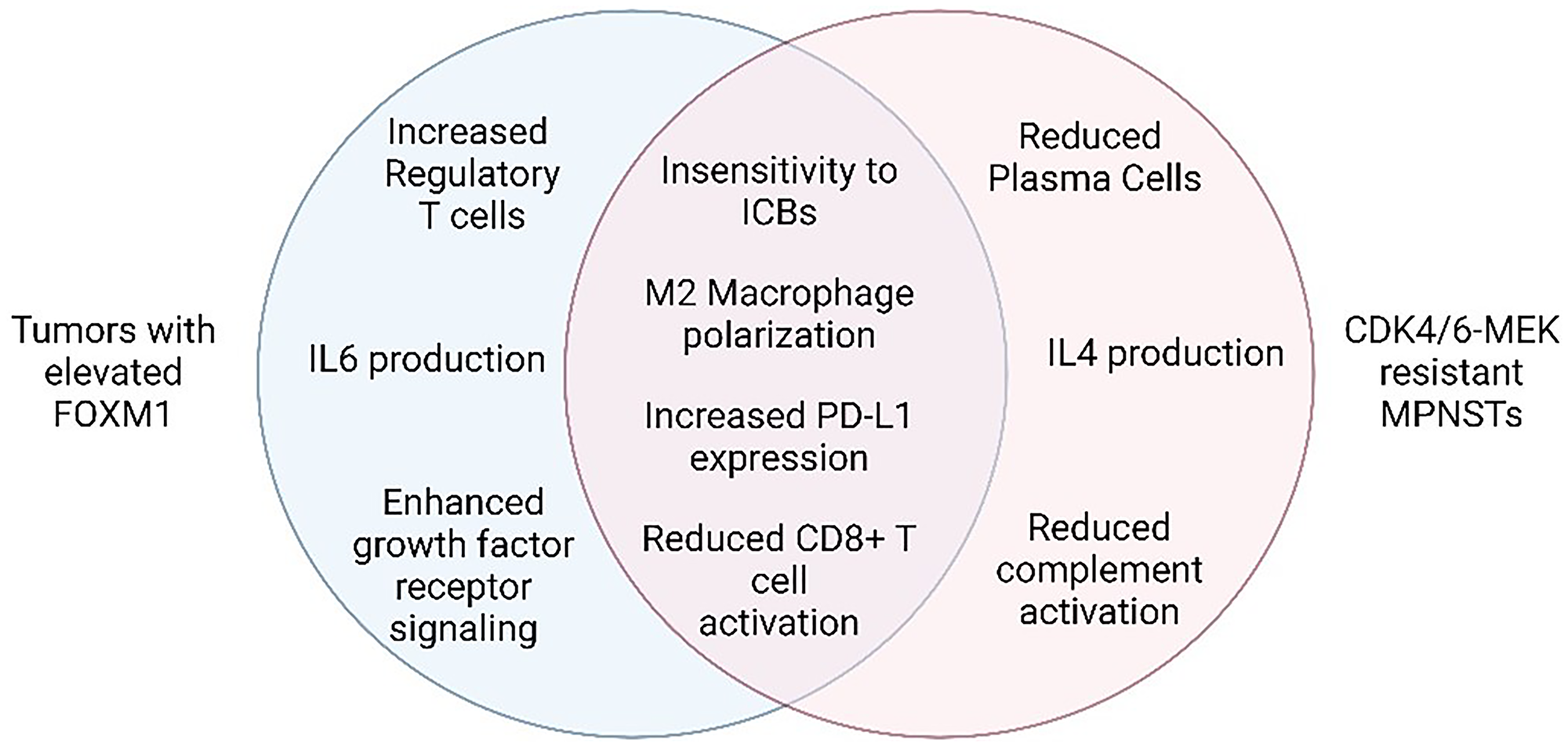
Figure 2: Hallmark features of CDK4/6 and MEK inhibitor resistance and FOXM1 elevation in tumors.
Venn diagram indicating the shared (middle) and unique (left and right) features of each setting. Figure made with https://www.biorender.com/.
The plasma cell discovery was exciting because those immune cells are increasingly appreciated to connote enhanced patient survival and improved response to ICBs in many other types of human cancers [15–19]. Such associations have been seen in a few types of sarcoma, but so far analyses have not extended to include MPNSTs [15, 19]. Our mouse MPNST model findings predicted that CDK4/6-MEK inhibitor therapy would sensitize MPNSTs to ICB therapy, not only because of increased plasma cells but also given the upregulation of PD-L1 in drug resistant MPNSTs. That possibility was tested by combining PD-L1 antibody therapy with CDK4/6-MEK inhibition, which did indeed elicit impressive anti-tumor effects [14]. The triple combination not only led to sustained tumor regression and improved animal survival relative to individual drugs, but it was also curative (complete disease ablation) in 12% of mice. These results support the conclusion that PD-L1 upregulation promotes resistance to CDK4/6-MEK inhibition therapy. Moreover, we speculate that the best responders to therapy (those that were cured) may have had the highest accumulation of plasma cells since they are thought to be critical orchestrators of anti-tumor immunity [19, 20]. However, the necessity and role of plasma cells in mediating response to therapy and anti-tumor immunity, in any tumor type or treatment setting, remains to be tested.
Although CDK4/MEK/PD-L1 combination therapy proved highly effective in MPNSTs, the majority of tumors still eventually became resistant and resumed growth [14]. Given the increasing clinical use of inhibitors to CDK4/6, MEK, and PD-L1 in cancer patients, either as monotherapies or in combination strategies, finding new targets to combat resistance to these agents is of significant interest. Many potential mediators of resistance to CDK4/6 and MEK inhibitors have already been defined [10, 21]. High on that list may be the protein tyrosine phosphatase, SHP2, a positive regulator of many oncogenic signaling pathways that functionally interacts with RAS-MEK-ERK, CDK4/6-RB, and PD-1/PD-L1 pathways [22]. The Pratilas lab showed that SHP2 inhibitors act synergistically with MEK inhibitors [21] and more so with CDK4/6 inhibitors [23] to suppress MPNST growth. The excitement about dual CDK4/6-SHP2 inhibition has even led to clinical testing of ribociclib (CDK4/6 inhibitor) plus TNO155 (SHP2 inhibitor) in patients with NF1-deficient cancers (NCT04000529). The combination of CDK4/6-SHP2 inhibitors with ICB therapy would also merit testing as it may have greater potential for durable antitumor activity.
Another likely mediator of therapy resistance in CDK4/MEK/PD-L1 targeted MPNSTs is the FOXM1 oncoprotein [7]. As a transcription factor, FOXM1 controls the expression of numerous genes important for cellular proliferation, survival, and metastasis, making it a powerful and promising target in many human cancers [24]. It has been minimally studied in MPNSTs but, for several reasons, may play a key role in the disease and therapy resistance. First, FOXM1 expression is elevated in patient MPNSTs, along with CDK4, which prognosed worse survival [25]. In agreement, Aimaier et al demonstrated a direct role for FOXM1 in promoting MPNST pathogenesis through knockdown and overexpression studies [26] while we have discovered that FOXM1 expression and transcriptional activity are greatly increased as benign human precursor pNFs transform into MPNSTs (unpublished data). Second, FOXM1 is phosphorylated at many sites by CDK4/6 during the G1/S phase of the cell cycle and these modifications activate FOXM1 transcriptionally [27] (Figure 1). In turn, FOXM1 represses expression of FOXO1, a transcription factor, that normally promotes expression of the CDK inhibitors p16, p21, and p27 [28, 29]. Third, ERK1/2 can phosphorylate FOXM1 at sites that promote its translocation into the nucleus and increase its transcriptional activity, indicating that MEK inhibition (which acts upstream of ERK) could effectively prevent this interaction [30, 31]. Fourth, in breast cancer studies, CDK4/6 inhibitors synergized with novel compounds that target FOXM1 [32, 33]. Finally, FOXM1 has been shown to promote resistance to a broad range of therapies including DNA damaging agents [34, 35], radiation [36], and several targeted agents including inhibitors of CDK4/6 [10, 27], PI3K [10], and EGFR [37].
Notably, tumors with elevated FOXM1 share many features with MPNSTs that are resistant to CDK4/6-MEK inhibition, most of which reflect extra-tumoral changes that suppress anti-cancer immunity [38, 39] (Figure 2). For instance, FOXM1 directly binds to the PD-L1 promoter (CD274 gene) to activate its transcription [40] (see Figure 1), which aligns with our observation of upregulated PD-L1 protein in MPNSTs that overcame CDK4/6 and MEK inhibition. In agreement, PROTAC degraders of FOXM1 protein, as well as knockdown of FOXM1, decrease PD-L1 expression [41]. As for specific effects of FOXM1 on immune cell populations, one study in esophageal adenocarcinoma suggested FOXM1 inhibits CD8+ T cell chemotaxis, tumor infiltration, and tumor cell killing, in part through regulation of Th1 chemokine expression [42]. In cholangiocarcinoma, FOXM1 promoted FoxP3+ Treg cell tumor infiltration, thereby suppressing CD8+ T cell activity [43]. In lung adenocarcinoma, phosphorylated FOXM1 was shown to recruit monocytes and promote M2 macrophage polarization when tumor cells were co-cultured with macrophages [44]. MPNSTs resistant to CDK4/6-MEK inhibition likewise displayed reduced CD8+ T cell activation and increased M2 TAMs (Figure 2). FOXM1 can also prevent the maturation of bone marrow-derived dendritic cells in pancreatic ductal and colorectal adenocarcinomas [45]. Several of the pro-tumorigenic immune changes induced by FOXM1 in tumors reflect activated transcription of cytokine genes, such as IL-6, IL1β, and CCL4 [38, 39]. Of note, heightened FOXM1 expression in osteosarcoma correlates with decreased response to immunotherapy [46], bolstering the possibility that inhibition of FOXM1 may be key to preventing ICB therapy resistance in our sarcoma model of MPNST.
In sum, the findings discussed herein suggest that FOXM1 inhibition may block or effectively delay acquired resistance of MPNSTs to sustained ICB immunotherapy and/or CDK4/6-MEK inhibition. There is in fact growing enthusiasm for targeting FOXM1 in cancer using newly developed FOXM1 inhibitors [7], particularly for controlling drug resistance as part of combination therapies [33]. More pre-clinical work is warranted to move those drugs into the clinic, but other potential mediators of resistance to CDK4/6-MEK inhibitors and ICB agents, such as SHP2 inhibitors, are already being tested clinically. Moving forward, this represents an exciting time of discovery that should guide better treatment options for patients with MPNSTs as well as other solid Ras-driven cancers.
ACKNOWLEDGMENTS
Figures created with https://www.biorender.com/.
CONFLICTS OF INTEREST
Authors have no conflicts of interest to declare.
FUNDING
This research was funded by grants from the National Institutes of Health including R01 NS119322, P30 CA086862, and F31 CA281312.
References
1. Kim A, Stewart DR, Reilly KM, Viskochil D, Miettinen MM, Widemann BC. Malignant Peripheral Nerve Sheath Tumors State of the Science: Leveraging Clinical and Biological Insights into Effective Therapies. Sarcoma. 2017; 2017:7429697. https://doi.org/10.1155/2017/7429697. [PubMed].
2. Ratner N, Miller SJ. A RASopathy gene commonly mutated in cancer: the neurofibromatosis type 1 tumour suppressor. Nat Rev Cancer. 2015; 15:290–1. https://doi.org/10.1038/nrc3911. [PubMed].
3. Pemov A, Li H, Presley W, Wallace MR, Miller DT. Genetics of human malignant peripheral nerve sheath tumors. Neurooncol Adv. 2019; 2:i50–61. https://doi.org/10.1093/noajnl/vdz049. [PubMed].
4. Lee W, Teckie S, Wiesner T, Ran L, Prieto Granada CN, Lin M, Zhu S, Cao Z, Liang Y, Sboner A, Tap WD, Fletcher JA, Huberman KH, et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat Genet. 2014; 46:1227–32. https://doi.org/10.1038/ng.3095. [PubMed].
5. Kohlmeyer JL, Gordon DJ, Tanas MR, Monga V, Dodd RD, Quelle DE. CDKs in Sarcoma: Mediators of Disease and Emerging Therapeutic Targets. Int J Mol Sci. 2020; 21:3018. https://doi.org/10.3390/ijms21083018. [PubMed].
6. Kohlmeyer JL, Kaemmer CA, Pulliam C, Maharjan CK, Samayoa AM, Major HJ, Cornick KE, Knepper-Adrian V, Khanna R, Sieren JC, Leidinger MR, Meyerholz DK, Zamba KD, et al. RABL6A Is an Essential Driver of MPNSTs that Negatively Regulates the RB1 Pathway and Sensitizes Tumor Cells to CDK4/6 Inhibitors. Clin Cancer Res. 2020; 26:2997–11. https://doi.org/10.1158/1078-0432.CCR-19-2706. [PubMed].
7. Voigt E, Quelle DE. FOXM1, MEK, and CDK4/6: New Targets for Malignant Peripheral Nerve Sheath Tumor Therapy. Int J Mol Sci. 2023; 24:13596. https://doi.org/10.3390/ijms241713596. [PubMed].
8. D’Angelo SP, Mahoney MR, Van Tine BA, Atkins J, Milhem MM, Jahagirdar BN, Antonescu CR, Horvath E, Tap WD, Schwartz GK, Streicher H. Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): two open-label, non-comparative, randomised, phase 2 trials. Lancet Oncol. 2018; 19:416–26. https://doi.org/10.1016/S1470-2045(18)30006-8. [PubMed].
9. Rodriguez-Puebla ML, Robles AI, Conti CJ. ras activity and cyclin D1 expression: an essential mechanism of mouse skin tumor development. Mol Carcinog. 1999; 24:1–6. https://doi.org/10.1002/(SICI)1098-2744(199901)24:1<1::AID-MC1>3.0.CO;2-E. [PubMed].
10. Álvarez-Fernández M, Malumbres M. Mechanisms of Sensitivity and Resistance to CDK4/6 Inhibition. Cancer Cell. 2020; 37:514–29. https://doi.org/10.1016/j.ccell.2020.03.010. [PubMed].
11. Ruscetti M, Leibold J, Bott MJ, Fennell M, Kulick A, Salgado NR, Chen CC, Ho YJ, Sanchez-Rivera FJ, Feucht J, Baslan T, Tian S, Chen HA, et al. NK cell-mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science. 2018; 362:1416–22. https://doi.org/10.1126/science.aas9090. [PubMed].
12. Ruscetti M, Morris JP 4th, Mezzadra R, Russell J, Leibold J, Romesser PB, Simon J, Kulick A, Ho YJ, Fennell M, Li J, Norgard RJ, Wilkinson JE, et al. Senescence-Induced Vascular Remodeling Creates Therapeutic Vulnerabilities in Pancreas Cancer. Cell. 2020; 181:424–41.e21. https://doi.org/10.1016/j.cell.2020.03.008. [PubMed].
13. Kohlmeyer JL, Gordon DJ, Tanas MR, Dodd RD, Monga V, Darbro BW, Quelle DE. Combination therapies for MPNSTs targeting RABL6A-RB1 signaling. Oncotarget. 2021; 12:10–14. https://doi.org/10.18632/oncotarget.27862. [PubMed].
14. Kohlmeyer JL, Lingo JJ, Kaemmer CA, Scherer A, Warrier A, Voigt E, Raygoza Garay JA, McGivney GR, Brockman QR, Tang A, Calizo A, Pollard K, Zhang X, et al. CDK4/6-MEK Inhibition in MPNSTs Causes Plasma Cell Infiltration, Sensitization to PD-L1 Blockade, and Tumor Regression. Clin Cancer Res. 2023; 29:3484–97. https://doi.org/10.1158/1078-0432.CCR-23-0749. [PubMed].
15. Petitprez F, de Reyniès A, Keung EZ, Chen TW, Sun CM, Calderaro J, Jeng YM, Hsiao LP, Lacroix L, Bougoüin A, Moreira M, Lacroix G, Natario I, et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature. 2020; 577:556–60. https://doi.org/10.1038/s41586-019-1906-8. [PubMed].
16. Meylan M, Petitprez F, Becht E, Bougoüin A, Pupier G, Calvez A, Giglioli I, Verkarre V, Lacroix G, Verneau J, Sun CM, Laurent-Puig P, Vano YA, et al. Tertiary lymphoid structures generate and propagate anti-tumor antibody-producing plasma cells in renal cell cancer. Immunity. 2022; 55:527–41.e5. https://doi.org/10.1016/j.immuni.2022.02.001. [PubMed].
17. Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, Yizhak K, Sade-Feldman M, Blando J, Han G, Gopalakrishnan V, Xi Y, Zhao H, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature. 2020; 577:549–55. https://doi.org/10.1038/s41586-019-1922-8. [PubMed].
18. Kroeger DR, Milne K, Nelson BH. Tumor-Infiltrating Plasma Cells Are Associated with Tertiary Lymphoid Structures, Cytolytic T-Cell Responses, and Superior Prognosis in Ovarian Cancer. Clin Cancer Res. 2016; 22:3005–15. https://doi.org/10.1158/1078-0432.CCR-15-2762. [PubMed].
19. Laumont CM, Banville AC, Gilardi M, Hollern DP, Nelson BH. Tumour-infiltrating B cells: immunological mechanisms, clinical impact and therapeutic opportunities. Nat Rev Cancer. 2022; 22:414–30. https://doi.org/10.1038/s41568-022-00466-1. [PubMed].
20. Sautès-Fridman C, Petitprez F, Calderaro J, Fridman WH. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat Rev Cancer. 2019; 19:307–25. https://doi.org/10.1038/s41568-019-0144-6. [PubMed].
21. Wang J, Pollard K, Allen AN, Tomar T, Pijnenburg D, Yao Z, Rodriguez FJ, Pratilas CA. Combined Inhibition of SHP2 and MEK Is Effective in Models of NF1-Deficient Malignant Peripheral Nerve Sheath Tumors. Cancer Res. 2020; 80:5367–79. https://doi.org/10.1158/0008-5472.CAN-20-1365. [PubMed].
22. Wang LN, Yang YL, Zhao LY. TFAP-2: A Special Regulator with Bidirectional Effect in Human Cancer. J Cancer Immunol. Journal of Cancer Immunology. 2021; 3:1–5. https://doi.org/10.33696/cancerimmunol.3.035.
23. Wang J, Calizo A, Zhang L, Pino JC, Lyu Y, Pollard K, Zhang X, Larsson AT, Conniff E, Llosa NJ, Wood DK, Largaespada DA, Moody SE, et al. CDK4/6 inhibition enhances SHP2 inhibitor efficacy and is dependent upon RB function in malignant peripheral nerve sheath tumors. Sci Adv. 2023; 9:eadg8876. https://doi.org/10.1126/sciadv.adg8876. [PubMed].
24. Liao GB, Li XZ, Zeng S, Liu C, Yang SM, Yang L, Hu CJ, Bai JY. Regulation of the master regulator FOXM1 in cancer. Cell Commun Signal. 2018; 16:57. https://doi.org/10.1186/s12964-018-0266-6. [PubMed].
25. Yu J, Deshmukh H, Payton JE, Dunham C, Scheithauer BW, Tihan T, Prayson RA, Guha A, Bridge JA, Ferner RE, Lindberg GM, Gutmann RJ, Emnett RJ, et al. Array-based comparative genomic hybridization identifies CDK4 and FOXM1 alterations as independent predictors of survival in malignant peripheral nerve sheath tumor. Clin Cancer Res. 2011; 17:1924–34. https://doi.org/10.1158/1078-0432.CCR-10-1551. [PubMed].
26. Aimaier R, Chung MH, Gu Y, Yu Q, Wei C, Li H, Guo Z, Long M, Li Y, Wang W, Li Q, Wang Z. FOXM1 promotes neurofibromatosis type 1-associated malignant peripheral nerve sheath tumor progression in a NUF2-dependent manner. Cancer Gene Ther. 2023; 30:1390–402. https://doi.org/10.1038/s41417-023-00645-8. [PubMed].
27. Anders L, Ke N, Hydbring P, Choi YJ, Widlund HR, Chick JM, Zhai H, Vidal M, Gygi SP, Braun P, Sicinski P. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell. 2011; 20:620–34. https://doi.org/10.1016/j.ccr.2011.10.001. [PubMed].
28. Chand V, Liao X, Guzman G, Benevolenskaya E, Raychaudhuri P. Hepatocellular carcinoma evades RB1-induced senescence by activating the FOXM1-FOXO1 axis. Oncogene. 2022; 41:3778–90. https://doi.org/10.1038/s41388-022-02394-8. [PubMed].
29. Diep CH, Charles NJ, Gilks CB, Kalloger SE, Argenta PA, Lange CA. Progesterone receptors induce FOXO1-dependent senescence in ovarian cancer cells. Cell Cycle. 2013; 12:1433–49. https://doi.org/10.4161/cc.24550. [PubMed].
30. Ma RY, Tong TH, Cheung AM, Tsang AC, Leung WY, Yao KM. Raf/MEK/MAPK signaling stimulates the nuclear translocation and transactivating activity of FOXM1c. J Cell Sci. 2005; 118:795–806. https://doi.org/10.1242/jcs.01657. [PubMed].
31. Kruiswijk F, Hasenfuss SC, Sivapatham R, Baar MP, Putavet D, Naipal KA, van den Broek NJ, Kruit W, van der Spek PJ, van Gent DC, Brenkman AB, Campisi J, Burgering BM, et al. Targeted inhibition of metastatic melanoma through interference with Pin1-FOXM1 signaling. Oncogene. 2016; 35:2166–77. https://doi.org/10.1038/onc.2015.282. [PubMed].
32. Ziegler Y, Laws MJ, Sanabria Guillen V, Kim SH, Dey P, Smith BP, Gong P, Bindman N, Zhao Y, Carlson K, Yasuda MA, Singh D, Li Z, et al. Suppression of FOXM1 activities and breast cancer growth in vitro and in vivo by a new class of compounds. NPJ Breast Cancer. 2019; 5:45. https://doi.org/10.1038/s41523-019-0141-7. [PubMed].
33. Katzenellenbogen BS, Guillen VS, Katzenellenbogen JA. Targeting the oncogenic transcription factor FOXM1 to improve outcomes in all subtypes of breast cancer. Breast Cancer Res. 2023; 25:76. https://doi.org/10.1186/s13058-023-01675-8. [PubMed].
34. Zhang N, Wu X, Yang L, Xiao F, Zhang H, Zhou A, Huang Z, Huang S. FoxM1 inhibition sensitizes resistant glioblastoma cells to temozolomide by downregulating the expression of DNA-repair gene Rad51. Clin Cancer Res. 2012; 18:5961–71. https://doi.org/10.1158/1078-0432.CCR-12-0039. [PubMed].
35. Li X, Qiu W, Liu B, Yao R, Liu S, Yao Y, Liang J. Forkhead box transcription factor 1 expression in gastric cancer: FOXM1 is a poor prognostic factor and mediates resistance to docetaxel. J Transl Med. 2013; 11:204. https://doi.org/10.1186/1479-5876-11-204. [PubMed].
36. Kim SH, Joshi K, Ezhilarasan R, Myers TR, Siu J, Gu C, Nakano-Okuno M, Taylor D, Minata M, Sulman EP, Lee J, Bhat KP, Salcini AE, Nakano I. EZH2 protects glioma stem cells from radiation-induced cell death in a MELK/FOXM1-dependent manner. Stem Cell Reports. 2015; 4:226–38. https://doi.org/10.1016/j.stemcr.2014.12.006. [PubMed].
37. Nilsson MB, Sun H, Robichaux J, Pfeifer M, McDermott U, Travers J, Diao L, Xi Y, Tong P, Shen L, Hofstad M, Kawakami M, Le X, et al. A YAP/FOXM1 axis mediates EMT-associated EGFR inhibitor resistance and increased expression of spindle assembly checkpoint components. Sci Transl Med. 2020; 12:eaaz4589. https://doi.org/10.1126/scitranslmed.aaz4589. [PubMed].
38. Xue J, Zhou A, Tan C, Wu Y, Lee HT, Li W, Xie K, Huang S. Forkhead Box M1 Is Essential for Nuclear Localization of Glioma-associated Oncogene Homolog 1 in Glioblastoma Multiforme Cells by Promoting Importin-7 Expression. J Biol Chem. 2015; 290:18662–70. https://doi.org/10.1074/jbc.M115.662882. [PubMed].
39. Kopanja D, Chand V, O’Brien E, Mukhopadhyay NK, Zappia MP, Islam ABM, Frolov MV, Merrill BJ, Raychaudhuri P. Transcriptional Repression by FoxM1 Suppresses Tumor Differentiation and Promotes Metastasis of Breast Cancer. Cancer Res. 2022; 82:2458–71. https://doi.org/10.1158/0008-5472.CAN-22-0410. [PubMed].
40. Madhi H, Lee JS, Choi YE, Li Y, Kim MH, Choi Y, Goh SH. FOXM1 Inhibition Enhances the Therapeutic Outcome of Lung Cancer Immunotherapy by Modulating PD-L1 Expression and Cell Proliferation. Adv Sci (Weinh). 2022; 9:e2202702. https://doi.org/10.1002/advs.202202702. [PubMed].
41. Wang K, Dai X, Yu A, Feng C, Liu K, Huang L. Peptide-based PROTAC degrader of FOXM1 suppresses cancer and decreases GLUT1 and PD-L1 expression. J Exp Clin Cancer Res. 2022; 41:289. https://doi.org/10.1186/s13046-022-02483-2. [PubMed].
42. Ziman B, Yang Q, Zheng Y, Sheth M, Nam C, Zhao H, Zhang L, Hu B, Bhowmick NA, Sinha UK, Lin DC. Epigenomic analyses identify FOXM1 as a key regulator of anti-tumor immune response in esophageal adenocarcinoma. Cell Death Dis. 2024; 15:152. https://doi.org/10.1038/s41419-024-06488-x. [PubMed].
43. Ma K, Sun Z, Li X, Guo J, Wang Q, Teng M. Forkhead box M1 recruits FoxP3+ Treg cells to induce immune escape in hilar cholangiocarcinoma. Immun Inflamm Dis. 2022; 10:e727. https://doi.org/10.1002/iid3.727. [PubMed].
44. Xu R, Lee YJ, Kim CH, Min GH, Kim YB, Park JW, Kim DH, Kim JH, Yim H. Invasive FoxM1 phosphorylated by PLK1 induces the polarization of tumor-associated macrophages to promote immune escape and metastasis, amplified by IFITM1. J Exp Clin Cancer Res. 2023; 42:302. https://doi.org/10.1186/s13046-023-02872-1. [PubMed].
45. Zhou Z, Chen H, Xie R, Wang H, Li S, Xu Q, Xu N, Cheng Q, Qian Y, Huang R, Shao Z, Xiang M. Epigenetically modulated FOXM1 suppresses dendritic cell maturation in pancreatic cancer and colon cancer. Mol Oncol. 2019; 13:873–93. https://doi.org/10.1002/1878-0261.12443. [PubMed].
46. Shi S, Wang Q, Du X. Comprehensive bioinformatics analysis reveals the oncogenic role of FoxM1 and its impact on prognosis, immune microenvironment, and drug sensitivity in osteosarcoma. J Appl Genet. 2023; 64:779–96. https://doi.org/10.1007/s13353-023-00785-5. [PubMed].