
Introduction
The UBA1 (Ubiquitin-like modifier activating enzyme 1) gene, located on the X chromosome, has recently gathered significant interest within the medical community following the 2020 discovery of VEXAS (Vacuoles, E1 enzyme, X-linked, Autoinflammation, Somatic) Syndrome [1]. This novel, difficult-to-treat hemato-inflammatory disorder is caused by three somatic mutations in UBA1, a gene encoding for a key E1 enzyme within the ubiquitin proteasome system (UPS). These mutations, found in hematopoietic stem and progenitor cells predominantly in older males, lead to severe and refractory inflammatory symptoms and loss of mature blood cells (cytopenias). Additionally, a portion of these patients develop hematologic malignancies, including myelodysplastic neoplasms (MDS) and multiple myeloma. Notably, despite the increased risk of acute myeloid leukemia (AML) in MDS patients [2], progression to AML is extremely rare in patients with VEXAS-MDS [3].
VEXAS Syndrome captured the attention of a wide-ranging audience beyond its initial classification as a rare genetic disease, with only 28 described male patients, partly because the genetic mutation was of somatic origin with a cancer association. The perturbation of the UPS is a long-standing cause of inflammation, evidenced by multiple pediatric monogenic autoinflammatory diseases [4]. However, adult-onset genetic inflammatory diseases were not known. It is indeed surprising that loss of function of UBA1 would lead to clonal advantage, as UPS has been the target of multiple anti-cancer drugs [5–9], and UBA1 itself was identified as cancer dependency in multiple studies [10–12]. The paradoxical clonal expansion and the high incidence of MDS yet reduced AML progression in the presence of inflammation presents a unique model for exploring the intersections between inflammation, oncogenesis, and cancer resistance mechanisms.
In the four years since VEXAS was identified, screening efforts have encompassed nearly half a million individuals [13–17], revealing an estimated incidence of 1 in 4,000 among older (predominantly white) males [13]. These screenings have uncovered greater genetic and phenotypic heterogeneity within the syndrome, including variations in inflammation levels and cancer associations. This research perspective aims to delve into the phenotypic diversity of UBA1 mutations, focusing on the impact of loss of ubiquitylation capacity on inflammogenicity, hematologic manifestations, clonality, and oncogenic potential. Based on this knowledge, research can be directed to devise therapeutic strategies tailored to the unique challenges presented by VEXAS Syndrome.
UBA1 loss of function and VEXAS
VEXAS Syndrome results from loss-of-function mutations in UBA1, which encodes for a critical enzyme within the ubiquitylation pathway. UBA1, one of only two E1 enzymes, plays a foundational role in initiating ubiquitylation by activating ubiquitin [18]. This activation is a precursor event for the subsequent transfer of ubiquitin to target substrates by numerous E2 and E3 enzymes, which impart specificity to the process. Positioned at the apex of the ubiquitylation cascade, UBA1’s functionality is indispensable for the ubiquitylation of many protein substrates, implicating virtually all cellular processes in the event of its dysfunction. In fact, the consequences of UBA1 loss-of-function mutations are profound, include embryonic lethality [19], premature death [1] and developmental defects [20] in model organisms as well as growth impairments in cell lines [21–25]. These outcomes underscore the essential role of UBA1 in cellular regulation and development.
UBA1 loss-of-function mutations in VEXAS result in distinct phenotypes not observed in model organisms, including inflammation, cytopenias, thrombotic tendencies, clonality, and blood cancer associations [1]. These differences arise on the one hand from VEXAS being caused by adult-acquired somatic mutations in immune and blood cell progenitors, leading to a tissue-specific, post-developmental partial loss of function. On the other hand, VEXAS mutations are not complete loss-of-function, and the effect of partial loss of function mutations can be various (Figure 1). It has been assumed that partial loss of function mutations only affect E2 and E3 enzymes with greater reliance on UBA1 activity [24, 26, 27], which likely shifts the balance of regulatory proteins, as they ubiquitylate each other in a context-dependent way. For instance, studies in Drosophila showed that complete UBA1 loss led to apoptosis, while partial loss resulted in proliferation, due to the differential effect on the degradation of pro-apoptotic and anti-apoptotic factors [28, 29]. In HEK293T cells partial reduction of UBA1 function paradoxically increased ubiquitin-dependent import of peroxisomal proteins via a partial loss of function of a specific E2 enzyme UBE2D [30]. Research to identify the E2 and E3 enzymes most impacted by VEXAS is ongoing [31].
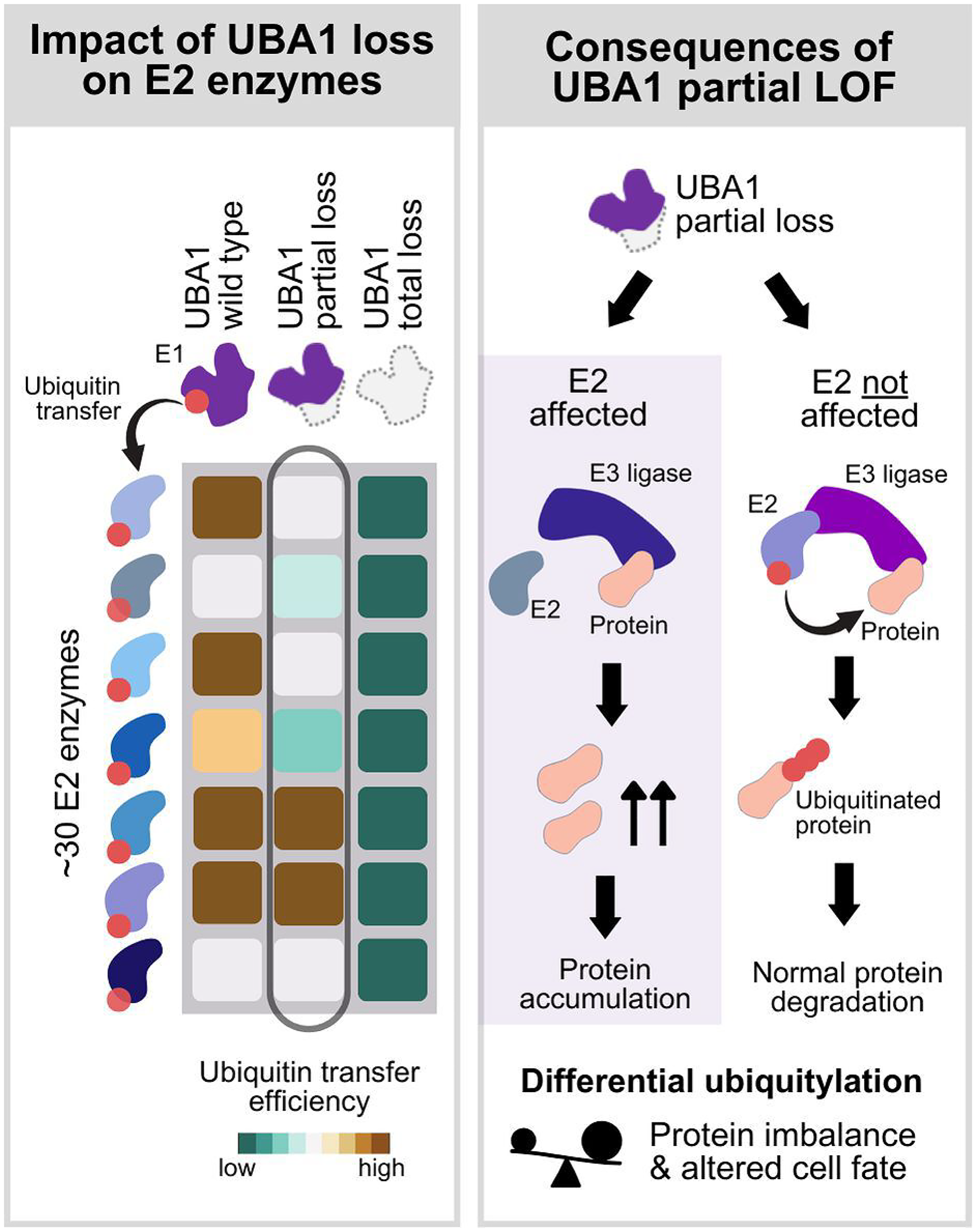
Figure 1: Conceptual representation of the differential effect of UBA1 mutations based on the degree of loss of function of ubiquitin E1 enzyme UBA1.
(left panel) UBA1, an E1 enzyme, activates ubiquitin and subsequently transfers the activated ubiquitin to up to approximately 30 E2 enzymes with various efficiency. The displayed heatmap illustrates the variability in ubiquitin transfer efficiency (dark green: low efficiency, brown: high efficiency) of UBA1 wild type (first column), UBA1 partial loss of function (second column) and UBA1 total loss of function (third column). Wild type UBA1 and partial loss of function mutations affect the ubiquitin transfer efficiency of a subset of E2 enzymes, whereas a total loss of function of UBA1 leads to a complete loss of loading of ubiquitin to E2 enzymes solely dependent on UBA1. (right panel) At the E2/E3-substrate transfer step, the effect of UBA1 loss of function is mediated by the decrease of available ubiquitin-loaded E2 enzymes. In the case of partial loss of function mutations, ubiquitylation of substrates can be variable due to the differential impairment of ubiquitin transfer to the E2 enzymes, which may result in imbalance of regulator proteins and altered cell fate.
In addition, VEXAS mutations uniquely cause a cytoplasm-specific loss of UBA1 function by altering the M41 start codon of its cytoplasmic isoform [1] (Figure 2). Two protein isoforms, UBA1a and UBA1b, are produced from a single mRNA through alternative translation initiated at different start codons [32, 33]. UBA1a, starting from the M1 codon, contains a nuclear localization signal (NLS) and predominantly resides in the nucleus [34]. UBA1b, initiated from the second start codon M41, remains cytoplasmic. The ratio of UBA1a to UBA1b is physiologically regulated during cell cycle and differentiation [35, 36]. VEXAS mutations at M41 reduce UBA1b translation efficiency, favoring translation from an alternative start codon, M67, producing a catalytically inactive isoform, UBA1c [1]. Despite this, translation from M41 can still occur, with efficiency varying among mutations; M41L and M41T maintain 10-15% of wild-type protein levels, whereas M41V has only 5% [37]. It has been shown that M41V mutation significantly reduces overall poly-ubiquitylation capacity, though the nuclear isoform remains unaffected [1].
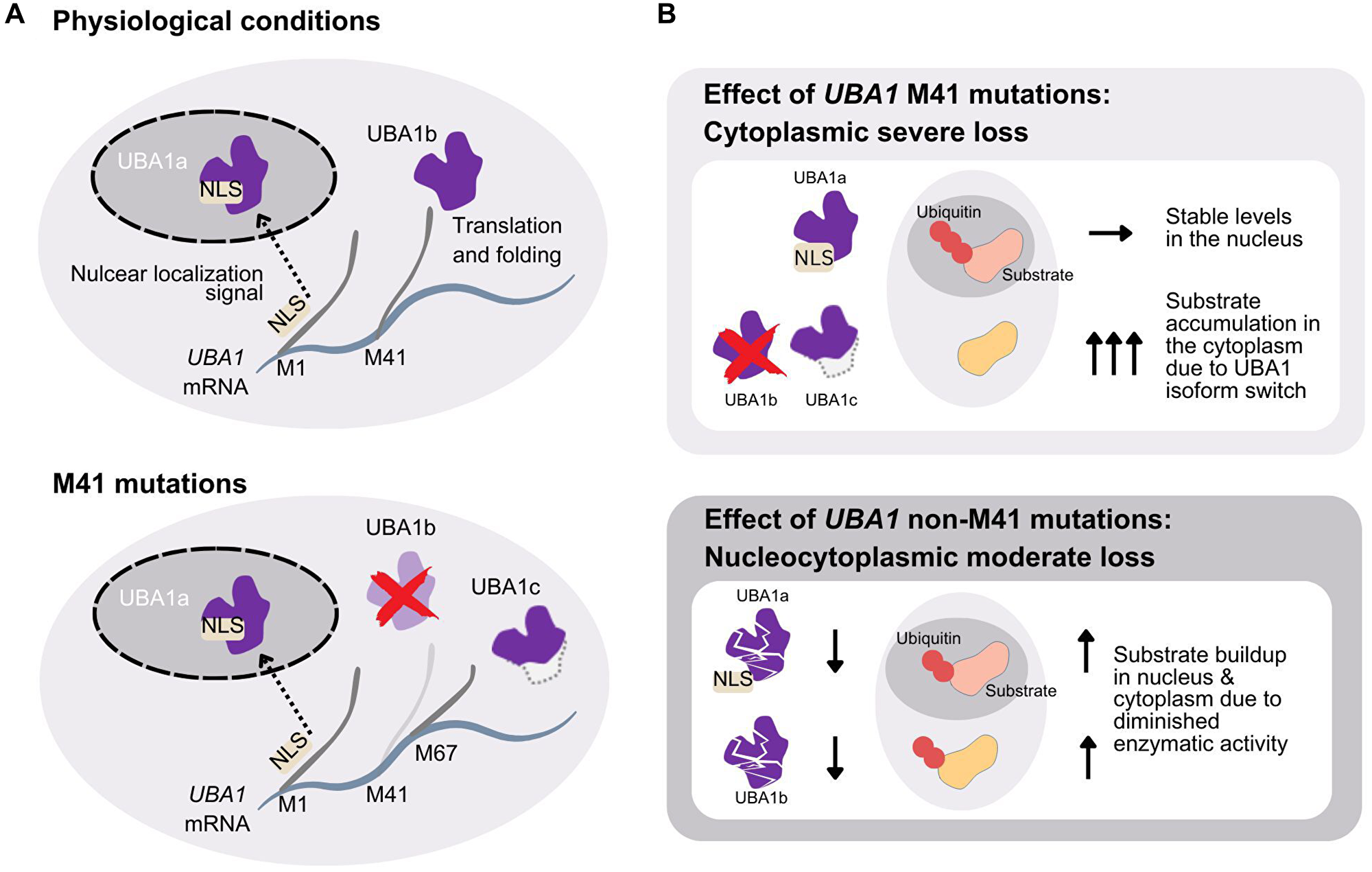
Figure 2: Mechanism of cytoplasmic-specific loss of function mutations and comparison with the non-M41 mutations.
(A) UBA1 mRNA transcript contains three alternative start codons at position M1 (UBA1a), M41 (UBA1b), and M67 (UBA1c). The transcript starting from M1 contains the nuclear localization signal (NLS) and the translated protein is transferred to the nucleus. In physiological conditions UBA1 mRNA is also translated from position M41, lacking the NLS, and the cytoplasmic isoform UBA1b is produced (top panel). Mutations at position M41 greatly reduces the translation efficiency starting at M41 and more transcripts are translated from M67. This results in the translation product, which is the catalytically deficient cytoplasmic isoform UBA1c (bottom panel). The isoform lacks residues from M41 to A65, compared to UBA1b. (B) The effect of M41 mutations result in intact UBA1a in the nucleus and isoform swap in the cytoplasm of the catalytically active UBA1b to the more inactive UBA1c. This results in stable ubiquitylation in the nucleus and substrate accumulation in the cytoplasm (top panel). The effect of non-M41 mutations is equally present in UBA1a and UBA1b, respectively, and substrate accumulation should similarly be seen both in the nucleus and cytoplasm (bottom panel).
Shortly after the discovery of VEXAS, UBA1 mutations not affecting M41 were reported in patients manifesting VEXAS-like inflammation and cytopenias [38]. One type was the splicing variants, which lead to an in-frame deletion of short exonic segments containing M41 [38–40]. The other type was, interestingly, mutations affecting functional sites in the region shared by UBA1a and UBA1b isoforms and led to a partial loss of function of both the nuclear and cytoplasmic isoforms without the appearance of the UBA1c isoform [31, 38, 41] (Figure 2). For example, a recurrent locus mutated in VEXAS patients, Y55 [14, 31, 42], has recently been shown to be the site of phosphorylation by SRC (SRC Proto-Oncogene, Non-Receptor Tyrosine Kinase), which affects ubiquitin activation efficiency [43]. The existence of non-M41 mutations suggests that VEXAS is caused by the decrease of ubiquitin activation in the cytoplasm, and the generation of UBA1c observed in M41 cases or impairment of UBA1a in the non-M41 cases are not required. However, slight phenotypic differences in inflammation, cytopenias, and associations with cancers have been observed between the M41 and non-M41 mutations [14, 44]. Furthermore, phenotypic differences among the M41 variants were also described [37, 45, 46], which suggests that the amount of residual UBA1b may affect the phenotype. The phenotypic differences of UBA1 mutations based on UBA1b amount or defect in UBA1a may identify specific E2 or E3 enzymes responsible for each of the VEXAS symptoms.
VEXAS manifestations and their mechanisms
In the previous section, we provided an overview of UBA1 mutations, the molecular implications of loss of function, and the VEXAS mutations. This section delves into VEXAS manifestations—specifically, inflammogenicity, cytopenias, clonal expansion of the myeloids, and oncogenicity—detailing their clinical and cellular characteristics and their links to reduced ubiquitylation (Figure 3), offering insights into targets of therapeutic intervention.
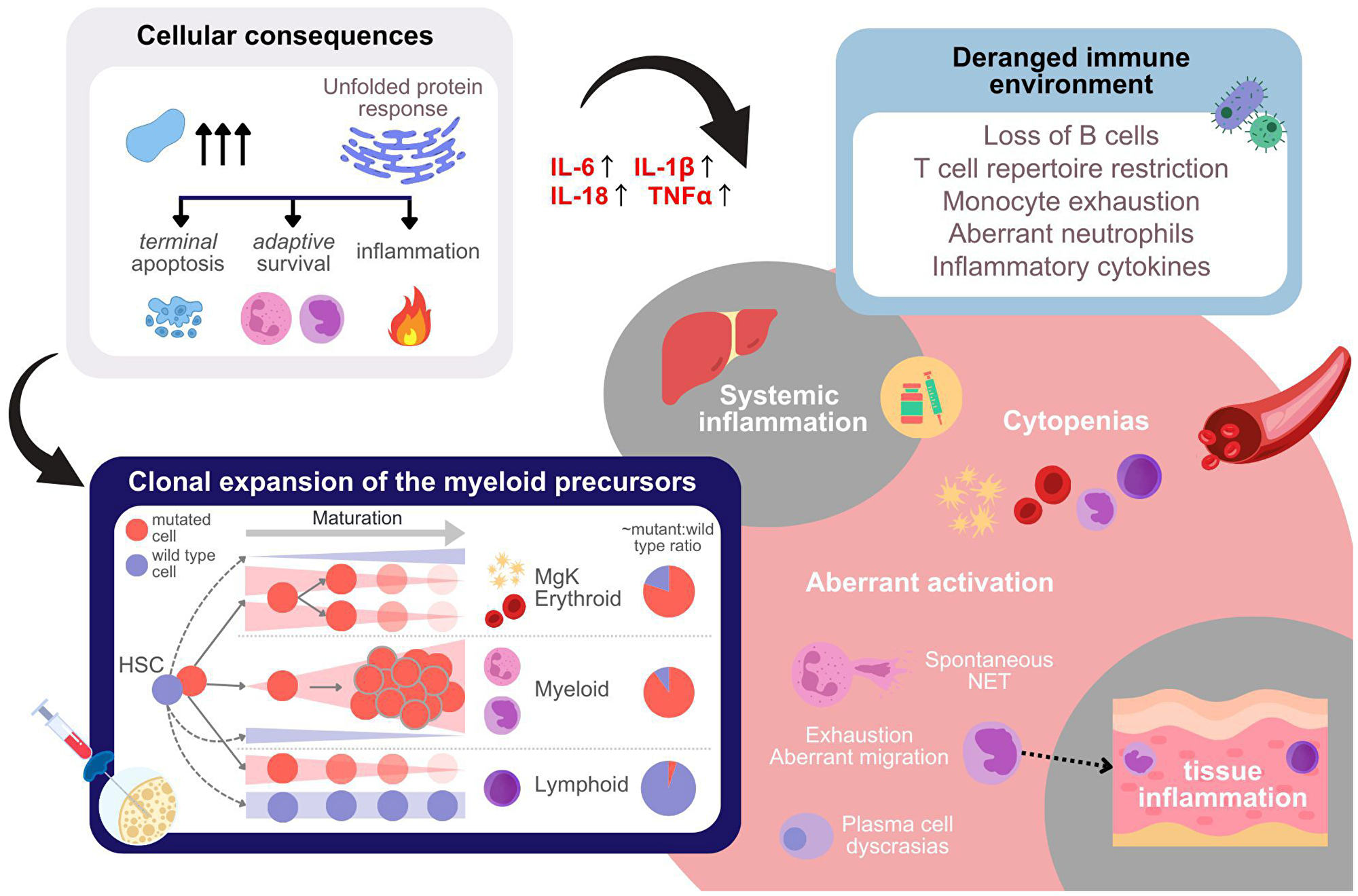
Figure 3: Cellular, tissue-level, immune-environmental, and systemic effect of UBA1 mutations.
UBA1 mutations lead to substrate accumulation, which result in activation of the unfolded protein response (UPR, top left panel). This affects the cell fate of different cell types carrying the mutations in a context dependent way. In addition, UPR results in inflammatory response, including cytokine production. A list of aberrations due to mutations which may lead to altered immune microenvironment is given in the top right panel. The aberrations impair the immuno-competence of the patient. The panel at the bottom left illustrates the alterations in cell type composition within the bone marrow resulting from the cell-type-specific effects of UBA1 mutations in hematopoietic stem cells (HSCs) and progenitor cells. Mutations in UBA1 lead to distinct outcomes depending on the lineage of the mutated cells. Specifically, mutated cells of the lymphoid and likely also erythroid lineages progressively decrease as the cells differentiate, whereas the myeloid cells carrying the mutations undergo clonal expansion. The pie charts on the right side of the panel provide an approximate quantification of the mutant to wild type ratio per lineage observed. In peripheral blood of VEXAS patients cytopenias are observed either as a consequence of differentiation aberrations in the bone marrow, inflammatory environment, or due to cytotoxic anti-inflammatory treatment (bottom right). In addition, aberrant activation of immune cells is observed, which aggravates both systemic and tissue inflammation.
Inflammogenicity
Inflammatory symptoms in VEXAS include non-infectious fever, chondritis, skin rash, and lung infiltrates [1, 46, 47]. Patients show high levels of inflammatory cytokines (IL-1β, IL-18), as well as C-reactive protein, the indicator of inflammation most widely used in the clinics [1, 48, 49]. High-dose corticosteroids are the mainstay for controlling the inflammation long-term, though their adverse effects contribute to mortality in VEXAS. Alternative anti-inflammatory treatments often fail [39], but about 30% of patients respond to JAK inhibitors like ruxolitinib and IL-6 inhibitors [50]. T-cell targeting therapies seem not as effective [39], and the inflammation seems to stem from the aberrant activation of myeloid cells, which is the predominant population carrying the mutation in the bone marrow.
Neutrophils of VEXAS patients spontaneously release neutrophil extracellular trap (NET) [1], which is inflammogenic, and monocytes of VEXAS patients aberrantly express chemokine receptors that may facilitate migration of immune cells and inflammogenicity in the skin [48]. Although the precise mechanism linking ubiquitylation impairment to the reported aberrant myeloid activation is not known, a consistent observation in VEXAS cells or cells treated with UBA1 inhibitors is the upregulation of the unfolded protein response (UPR), likely due to the decrease in the efficiency of endoplasmic reticulum-associated degradation and the consequent accumulation of misfolded proteins [4]. UPR can trigger inflammation by myriads of mechanisms, including the activation of NF-κB pathway and the inflammasome, and its dysregulation is associated with multitudes of phenotypically diverse autoimmune and autoinflammatory diseases [51, 52].
Interestingly, patients with VEXAS syndrome who have mutations other than M41 often experience less severe inflammatory symptoms [16, 44]. In fact, the effects of the UPR are not limited to inflammation but also include cell death, stress responses like reduced protein production and increased autophagy, and changes in cell differentiation [53, 54]. The phenotypic diversity observed might be linked to the different levels of proteotoxic stress caused by M41 and non-M41 mutations. Nonetheless, if the inflammation in both groups is due to the UPR in myeloid cells, focusing on the affected myeloid cells or adjusting the UPR might be more effective than targeting the wide array of cytokines and chemokines individually.
Cytopenias
In VEXAS syndrome, there is a noticeable reduction in various blood cells [37, 47, 55], including red blood cells (98% of cases), platelets (33–54%), neutrophils (23–29%), monocytes (73%), and lymphocytes, especially B cells (91%). Anemia that requires regular blood transfusions is linked to a shorter lifespan [37], and a decrease in lymphocytes can lead to more infections among VEXAS patients, which is a leading cause of death [1]. Therefore, managing hematologic symptoms is a key component of effectively treating VEXAS patients.
Bone marrow examinations of VEXAS patients typically show a hyperplastic bone marrow with increase in myeloid progenitors and decrease in erythroid progenitors. These progenitor cells, both myeloid and erythroid, typically show characteristic vacuoles, which are likely autophagic vacuoles indicating stress [56]. Megakaryocytes (platelet progenitors) also show characteristic dysplasia [55]. About 80% of progenitor cells of all lineages carried mutations, and among mature cells, neutrophils and monocytes showed these mutations, with none found in B and T cells [1]. Single-cell studies confirmed that these mutations are present in progenitors of both lymphoid and erythro-megakaryocytic lineages [45, 57, 58]. However, there is a noticeable reduction of lymphoid cells as they develop, while the trend in erythro-megakaryocytic lineage is less evident, possibly because mature cells in this lineage don’t have a nucleus and weren’t examined, but they showed a similar pattern. In summary, the cytopenias of the lymphoid and erythro-megakaryocytic lineages seem to be due to the preferential differentiation of the hematopoietic stem cells to the myeloid lineage and/or negative selection during lymphoid and erythro-megakaryocytic differentiation (Figure 3). In contrast, neutropenia and monocytopenia are likely due to the spontaneous inflammogenic death or migration into the tissues in the periphery, as mentioned earlier.
The bone marrow differentiation bias and blood cell composition in VEXAS have been quite comprehensively described, but the UBA1 mutation-specific molecular mechanisms that might provide insights into treatment of cytopenias are not fully investigated, partly due to the confounding hematologic side-effect of some anti-inflammatory treatment [59] and due to the general rule that controlling inflammation improves the hematologic symptoms in inflammatory diseases [60]. In fact, inflammation is known to increase granulopoiesis and decrease erythropoiesis in a multifactorial way [61, 62], and in some cases controlling inflammation improved cytopenias even without changes in UBA1-mutated clone size [63]. However, some patients experience worsening of their low blood cell counts during periods when inflammation is not active [64], and those with VEXAS who have mutations other than M41 may have more severe anemia but only mild signs of inflammation [44]. Interestingly, patients carrying the non-M41 mutations often have increased erythropoiesis in the bone marrow [14, 38, 42], which is unusual for anemia caused by inflammation. Thus, the intrinsic mechanism of cytopenias is plausible and worthy of investigation.
Erythropoiesis, megakaryopoiesis, and lymphopoiesis are all regulated by ubiquitylation. For instance, the receptor for erythropoietin is broken down by the E3 ligase β-TRCP [65] and RNF41 [66], both the receptor for thrombopoietin (MPL) [67] and lymphoid development factor receptor IL7R [68] by E3 enzyme CBL, and plasma cell differentiation requires UPR [69]. Most importantly, p53 is degraded by E3 enzyme MDM2, and in mice, uninhibited p53 activity by loss of Mdm2 is known to lead to bone marrow aplasia [70]. Such is relevant in VEXAS, because UBA1 inhibition has been shown to increase P53 protein level, both by chemical inhibition [9] and mutagenesis [71]. Investigation of the stability of protein regulators of hematopoiesis in VEXAS patients is an underexplored area of research, which may open new therapeutic strategies to control cytopenias in VEXAS.
Clonal expansion of the myeloid precursors
Most treatments for VEXAS syndrome currently focus on targeting the inflammatory pathways. However, due to the short duration of success of these treatments [39], there’s an increasing interest in therapies that target the disease-causing cells themselves. By understanding what gives these abnormal cells a growth advantage in VEXAS, it might be possible to identify new druggable targets.
The variant allele fraction (VAF) of a somatic mutation can be used as a surrogate to assess clonal expansion and the VAF of UBA1 can exceed 90% in both bone marrow and peripheral blood. As mentioned, the lymphoid lineage does not contribute to the population of mutated clones nor does the erythro-megakaryocytic lineage since they are known to progressively decrease [55]. Additionally, the proportion of myeloblasts in VEXAS is usually less than 5% [47]. Efforts to create VEXAS-like cells from induced pluripotent stem (iPS) cells have been unsuccessful unless the mutation is introduced at a later stage of myeloid cell development [58]. This suggests that primarily later-stage myeloid progenitors and mature cells contribute to the clonality. However, in lab cultures, UBA1-mutated knock-in cell lines of the myeloid lineage do not grow well and die spontaneously [49, 58, 72], suggesting that VEXAS clonality may depend on the environment.
Recently, VEXAS patients were reported not only to be inflammatory but also immunodeficient, even after controlling for immunosuppressive treatment, either due to loss of lymphocytes or exhausted monocytes [73]. Many years before, a transposon-mediated mutagenesis experiment found transposon insertion in the intron 1 of UBA1 to be one of the few insertion hotspots that were found in mice developing leukemia in immune-deficient but not immunocompetent mice [74]. Thus, the immunological environment created by UBA1 mutations in the myeloid cells may favor the expansion of the mutant clones. The extrinsic aspect of clonal expansion is further supported by cases of clonal mosaicism or multiple independently arising clones. There is a case report of a patient who showed three independent UBA1 M41-mutated clones [75], and almost every large screening attempt of symptomatic persons found at least one patient with multiple independent UBA1-mutated clones [16, 45], suggesting that UBA1-mutated clones gain advantage from the extrinsic inflammatory or immunodeficient environment. The most recent single-cell study [58] suggests that the outcome of UPR in mutated myeloid cells is the activation of an anti-apoptosis pathway, which may be one mechanism that allow the preferential survival of the mutated cells in the inflammatory milieu.
Oncogenicity
Patients with VEXAS syndrome often receive a concurrent diagnosis of MDS and, to a lesser extent, multiple myeloma [1, 45, 55]. Understanding oncogenicity of UBA1 mutations is crucial, especially regarding treatment strategies, because modulating ubiquitylation and inflammation can shift the balance between cell death and survival in different ways [28, 29], offering insights into potentially severe side effects. Moreover, setting aside considerations of quality of life, the median survival for VEXAS patients can reach 10 years from the first appearance of symptoms [37]. Therefore, the diagnosis of cancer would significantly affect the patient’s prognosis and overall health trajectory.
Initially MDS was reported in approximately half of VEXAS patients [46], but strict morphological evaluation of VEXAS bone marrow slides found the co-occurrence to be only 4% [55]. The confusion stems from the difficulty in distinguishing pre-malignant dysplasia from morphological changes secondary to inflammation or other non-malignant causes [76]. Other criteria for the diagnosis of neoplasms are the presence of oncogenic mutations. However, two large studies [16, 45] showed that VEXAS patients rarely harbor co-mutations other than DNMT3A or TET2, which are often also detected in healthy elderly individuals [77]. Thus, the exact prevalence of MDS among VEXAS patients, in the strictest definition, remains uncertain. Additionally, the presence of an MDS diagnosis alongside VEXAS does not appear to influence patient survival rates [37]. This observation challenges the conventional understanding of malignancy, as one would expect a cancer diagnosis to affect survival outcomes. Consequently, the link between MDS and VEXAS does not advocate for the oncogenicity of UBA1 mutations.
The co-diagnosis of multiple myeloma is more difficult to interpret. Due to the absence of UBA1 mutations in lymphocytes and the demographic overlap, some believe that multiple myeloma develops independently of UBA1 mutations [55]. However, the incidence of multiple myeloma in European males over 50 years is approximately 0.03% [78]. The prevalence would be no more than 0.3%, whereas in VEXAS the co-diagnosis is 3–8%. Further research in plasma cells is necessary to understand this association.
Currently, the association between UBA1 mutations and cancer remains uncertain, yet there is a growing body of literature on the connection between non-M41 UBA1 mutations and various cancers. UBA1 is a known orchestrator of DNA damage response [79, 80], and coinciding with the discovery of VEXAS syndrome, UBA1 mutations were implicated as potential key factors in the development of lung cancer among non-smokers, identified through advanced bioinformatics methods [81]. The patients were all females. None of them harbored the M41 mutations, and instead frameshift, nonsense, and non-M41 missense mutations. In addition, we reported that somatic non-M41 variants are detected in various hematologic neoplasms, including lymphoid malignancies [14]. The pathogenicity of the variants is not confirmed, but the possibility that different degrees of loss of function mutations of UBA1 may have oncogenic potential is worth exploring to design safe therapy.
Modifiers of the phenotype – age, sex, and cell type
In the previous section, we mentioned that frameshift and non-sense mutations in lung cancer were exclusively observed in female patients. UBA1 is a known X chromosome escape gene, and studies consistently find UBA1 to be expressed approximately 1.2-fold higher in the peripheral blood of females than males [82–85]. UBA1 pathogenic mutations show a clear sex bias in VEXAS [1, 14, 46] and X-linked spinal muscular atrophy (XL-SMA), a congenital neuromuscular disease caused by germline UBA1 mutations [86]. The functional impairment of UBA1 caused by mutations associated with XL-SMA is relatively minor when compared to VEXAS syndrome [31]. Female carriers of XL-SMA pathogenic UBA1 mutations are asymptomatic, but a female child with UBA1 gene deletion is affected [87]. Thus, the baseline UBA1 expression and the extent to which mutations impair its function appear to influence the resulting phenotype. Moreover, UBA1 protein expression decreases with age in mouse brains [88], and researchers of neurodegenerative diseases propose a threshold hypothesis where disease onset occurs once ubiquitylation capacity falls below a critical functional threshold [89]. Age might also play a role in VEXAS syndrome, as evidenced by the identification of several younger, asymptomatic patients carrying the UBA1 M41 mutations, who exhibited lower VAFs [15]. It appears that both age and sex influence the overall capacity for ubiquitylation, which may potentially impact the manifestation and progression of the disease [14]. Furthermore, the neuron-specific phenotype linked to germline mutations suggests varying thresholds of UBA1 functional deficiency across cell types. Protein aggregates, a known cause of neurodegenerative diseases, indicate neurons’ particular vulnerability to UBA1 dysfunction, potentially exacerbated by impaired direct interactions with proteins crucial for neural development (e.g., SMN1 [90, 91], Gigaxonin [92]).
Novel treatment strategies for VEXAS
The standard approach to treating VEXAS syndrome starts with administering high-dose corticosteroids, followed by a range of anti-inflammatory medications to gradually reduce the corticosteroid dosage. Additionally, supportive care is provided to manage cytopenias, infection, and thrombotic tendency. However, our review of research on the molecular and cellular impacts of UBA1 mutations highlights critical vulnerabilities in VEXAS pathogenesis (Figure 4). Therapies aimed directly at targeting the disease-causing clones could potentially offer more effective relief from all the VEXAS symptoms. Below, we detail several clone-targeting drugs and their mechanisms of action.
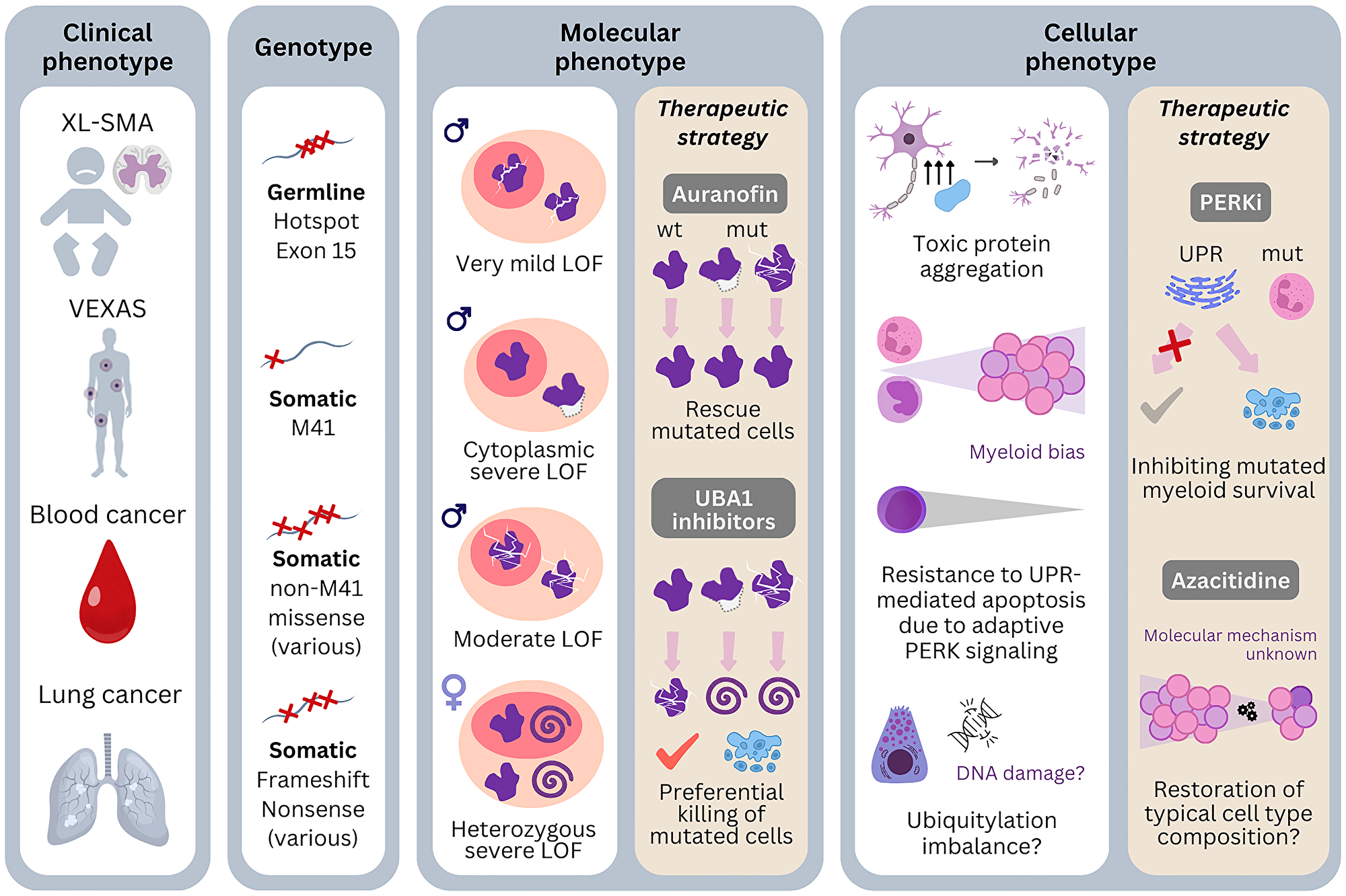
Figure 4: Genotype-phenotype associations of pathogenic UBA1 mutations and possibilities of therapeutic targeting.
Four different clinical phenotypes of pathogenic UBA1 mutations are known: X-linked spinal muscular atrophy (XL-SMA) is caused by germline mutations which have hotspot in exon 15. VEXAS is caused by somatic mutations in the cells of the bone marrow. M41 is the most frequent genotype but recurrent non-M41 mutations are also reported. Blood cancers are associated with both M41 and non-M41 mutations. Lung cancer is also reported with UBA1 mutations in females, which include frameshift and nonsense mutations. Molecularly, the degree of the enzymatic dysfunction and alteration of localization are different by genotypes. Therapies targeting this level: Auranofin tries to ameliorate phenotype by improving enzymatic dysfunction, whereas UBA1 inhibitors targets to tip the balance of survival to apoptosis by preferentially in cells with severe dysfunction. The cellular phenotypes of the mutations not only depend on the nature of the mutations but also on the affected cell type. Neuronal cells are particularly sensitive to protein aggregates and the mutations may be toxic with only slight enzymatic dysfunction. Other phenotypic mechanisms may be binding defects to proteins important in neural development. Concerning VEXAS, myeloid cells seem to be more resistant to UPR-mediated apoptosis due to activation of the PERK arm of the UPR. Therapies targeting of this level: PERK inhibitors try to prevent the preferential escape from apoptosis of the myeloid cells. Azacitidine likely also restore the cell type composition, but the exact mechanism is not known. The diverse clinical phenotypes are likely associated with the variety of mutations and their nature of loss of function and affected cell types. Abbreviations: wt: wild type; mut: mutated; LOF: loss of function; UPR: unfolded protein response; i: inhibitors.
Azacitidine – immunomodulatory effects?
Azacitidine emerged as a first candidate for effective treatment, supported by prior evidence of some success in managing hematoinflammatory symptoms in patients with a co-diagnosis of MDS and systemic inflammatory diseases [93, 94]. Given its approval for MDS, more VEXAS patients with a co-diagnosis of MDS receive Azacitidine, with some studies indicating that patients who respond to the treatment often have concurrent mutations in DNMT3A [95]. Beyond its primary effects, hypomethylating agents are noted for their immunomodulatory properties, such as diversifying T cell repertoire [96, 97]. VEXAS T cells are clonally restricted [57], and this ability to modify the cellular environment, potentially hindering the clonal expansion of UBA1 mutations, represents another avenue through which it may exert its therapeutic effects. Early results from a Phase II clinical trial involving VEXAS patients have been promising [98] and a case has been reported in which Azacitidine effect extends after cessation of therapy [99].
UBA1 inhibitors – synthetic lethality
UBA1 inhibitors were originally developed for cancer treatment, based on the premise that cancer cells require more ubiquitylation compared to normal cells [12]. In healthy physiological conditions, activated ubiquitin exists in abundance, suggesting that UBA1 inhibition might not significantly impact normal cellular functions [18]. However, in the context of VEXAS syndrome, the situation is different. There is an observed decrease of about 90% in UBA1b protein levels, potentially making these cells more vulnerable to UBA1 inhibition. Building on this theory, Chiaramida et al. [49] administered a UBA1 inhibitor TAK-243 to a cell line model knocked-in with M41L mutation and showed that VEXAS cells are killed at a lower concentration than the parent cell line, indicating a potential therapeutic window.
The question of whether there are differences in response to UBA1 inhibitors between M41 variants and non-M41 variants remains unanswered. Variability in response to UBA1 inhibitors has been noted among cell lines even within the same cancer. In squamous cell carcinoma, cell lines with lower expression of UBA1 responded better to TAK-243 [100], whereas in glioblastoma cell lines with lower expression of the ER chaperone GRP78 and not UBA1 expression responded better to TAK-243 [101]. The factors predicting treatment response in VEXAS syndrome have yet to be identified, though sex, age, and specific genetic mutations are potential sources of variability. Particularly, the non-M41 variants may respond differently due to their impact on the nuclear isoform of UBA1. The initial study in TAK-243 [9] indicates that its cytotoxic effects are mediated through several mechanisms, including DNA damage response and the impaired degradation of key proteins such as p53. These proteins play a crucial role in triggering cell cycle arrest and apoptosis in the presence of irreversible DNA damage [9], which seems more relevant in the nucleus.
The nuclear isoform is particularly prominent in G1 and G2 phases in HeLa cells [36], so the cell types which are often cycling are likely to be more sensitive. Further research is necessary to understand the effect of UBA1 inhibitors in the bone marrow, comparing the different M41 variants as well as the non-M41 variants. Combination therapies have been attempted in other cancers, such as radiotherapy and PARP1 inhibitors [102], which can be another direction of investigation. An additional note is that one of the UBA1 inhibitors, PYR-41 activates sumoylation at the same time, because ubiquitylation and sumoylation target the same residues of overlapping target substrate [103].
PERK inhibitors – UPR modulation
UPR modulation is one of the mechanisms that may alter survival advantages of UBA1-mutated myeloid cells. Ganesan et al. showed that UBA1-mutated myeloid cells gain survival advantage over wild type cells by activating the PERK-ATF4 arm of UPR [58]. In a M41V knock-in iPSC model, they showed that the M41V cells were more sensitive to PERK inhibitor GSK2606414 than the wild type cells. More preclinical studies are awaited to develop this promising target.
Auranofin – improving defective UBA1 function
A novel strategy in VEXAS therapy came from an observation that UBA1c can be reactivated by Auranofin, a long-established drug for rheumatoid arthritis. Auranofin was found to enhance UBA1 binding to 20 out of 36 E2 enzymes tested and improved polyubiquitylation of multiple substrates [104]. Importantly, the effective dose was 4.5 to 73 times lower than the approved maximum therapeutic concentration for rheumatoid arthritis. Auranofin shows cytotoxic effect to chronic lymphocytic leukemia [105] as well as chronic myeloid leukemia [106], and its effect in VEXAS cells needs to be investigated.
Conclusions
VEXAS is a disease caused by UBA1 mutations in hematopoietic stem and progenitor cells. VEXAS phenotypes include inflammation, cytopenias, thrombotic tendency, clonality and potential oncogenicity. These diverse clinical features arise from the effects of UBA1 mutations across distinct cell types within the bone marrow and peripheral blood. The relationship between specific UBA1 genotypes and the resultant phenotype appears to be modulated by factors including age, sex, and the cellular context. Future studies clarifying how the genotype and host factors, which determine the severity and localization of the loss of function of UBA1, change the immune environment and shape the clinical phenotypes will be crucial. This, in turn, is expected to inform the development of targeted therapeutic interventions. The advent of clone-targeting therapies offers a promising avenue, yet a more detailed understanding of the specific E2/E3 enzymes involved and the differential impact of the UPR across cell types may identify novel therapeutic targets. Ultimately, a thorough grasp of the pathogenesis of VEXAS, from genetic mutations to clinical manifestations, will be pivotal in devising safe and effective therapeutic strategies to fight this challenging disease.
CONFLICTS OF INTEREST
TH declares part ownership of the MLL Munich Leukemia Laboratory. MS and WW are employed by the MLL Munich Leukemia Laboratory.
FUNDING
This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie Grant Agreement No. 953407.
References
1. Beck DB, Ferrada MA, Sikora KA, Ombrello AK, Collins JC, Pei W, Balanda N, Ross DL, Ospina Cardona D, Wu Z, Patel B, Manthiram K, Groarke EM, et al. Somatic Mutations in UBA1 and Severe Adult-Onset Autoinflammatory Disease. N Engl J Med. 2020; 383:2628–38. https://doi.org/10.1056/NEJMoa2026834. [PubMed].
2. Menssen AJ, Walter MJ. Genetics of progression from MDS to secondary leukemia. Blood. 2020; 136:50–60. https://doi.org/10.1182/blood.2019000942. [PubMed].
3. Battipaglia G, Vincenzi A, Falconi G, Fiore A, D'Agostino F, Iannotta R, Grimaldi F, Gurnari C, Galossi E, Crisà E, Bonello F, Scalia G, Izzo B, et al. New scenarios in Vacuoles, E1 enzyme, X linked, Autoinflammatory, Somatic (VEXAS) syndrome: Evolution from myelodysplastic syndrome to acute myeloid leukemia. Curr Res Transl Med. 2023; 71:103386. https://doi.org/10.1016/j.retram.2023.103386. [PubMed].
4. Beck DB, Werner A, Kastner DL, Aksentijevich I. Disorders of ubiquitylation: unchained inflammation. Nat Rev Rheumatol. 2022; 18:435–47. https://doi.org/10.1038/s41584-022-00778-4. [PubMed].
5. Csizmar CM, Kim DH, Sachs Z. The role of the proteasome in AML. Blood Cancer J. 2016; 6:e503. https://doi.org/10.1038/bcj.2016.112. [PubMed].
6. Deng L, Meng T, Chen L, Wei W, Wang P. The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduct Target Ther. 2020; 5:11. https://doi.org/10.1038/s41392-020-0107-0. [PubMed].
7. Manasanch EE, Orlowski RZ. Proteasome inhibitors in cancer therapy. Nat Rev Clin Oncol. 2017; 14:417–33. https://doi.org/10.1038/nrclinonc.2016.206. [PubMed].
8. Barghout SH, Patel PS, Wang X, Xu GW, Kavanagh S, Halgas O, Zarabi SF, Gronda M, Hurren R, Jeyaraju DV, MacLean N, Brennan S, Hyer ML, et al. Preclinical evaluation of the selective small-molecule UBA1 inhibitor, TAK-243, in acute myeloid leukemia. Leukemia. 2019; 33:37–51. https://doi.org/10.1038/s41375-018-0167-0. [PubMed].
9. Hyer ML, Milhollen MA, Ciavarri J, Fleming P, Traore T, Sappal D, Huck J, Shi J, Gavin J, Brownell J, Yang Y, Stringer B, Griffin R, et al. A small-molecule inhibitor of the ubiquitin activating enzyme for cancer treatment. Nat Med. 2018; 24:186–93. https://doi.org/10.1038/nm.4474. [PubMed].
10. Schlabach MR, Luo J, Solimini NL, Hu G, Xu Q, Li MZ, Zhao Z, Smogorzewska A, Sowa ME, Ang XL, Westbrook TF, Liang AC, Chang K, et al. Cancer proliferation gene discovery through functional genomics. Science. 2008; 319:620–24. https://doi.org/10.1126/science.1149200. [PubMed].
11. Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, Meyers RM, Ali L, Goodale A, et al. Defining a Cancer Dependency Map. Cell. 2017; 170:564–76.e16. https://doi.org/10.1016/j.cell.2017.06.010. [PubMed].
12. Barghout SH, Schimmer AD. E1 Enzymes as Therapeutic Targets in Cancer. Pharmacol Rev. 2021; 73:1–58. https://doi.org/10.1124/pharmrev.120.000053. [PubMed].
13. Beck DB, Bodian DL, Shah V, Mirshahi UL, Kim J, Ding Y, Magaziner SJ, Strande NT, Cantor A, Haley JS, Cook A, Hill W, Schwartz AL, et al. Estimated Prevalence and Clinical Manifestations of UBA1 Variants Associated With VEXAS Syndrome in a Clinical Population. JAMA. 2023; 329:318–24. https://doi.org/10.1001/jama.2022.24836. [PubMed].
14. Sakuma M, Blombery P, Meggendorfer M, Haferlach C, Lindauer M, Martens UM, Kern W, Haferlach T, Walter W. Novel causative variants of VEXAS in UBA1 detected through whole genome transcriptome sequencing in a large cohort of hematological malignancies. Leukemia. 2023; 37:1080–91. https://doi.org/10.1038/s41375-023-01857-5. [PubMed].
15. Corty RW, Brogan J, Byram K, Springer J, Grayson PC, Bick AG. VEXAS-Defining UBA1 Somatic Variants in 245,368 Diverse Individuals in the NIH All Of Us Cohort. Arthritis Rheumatol. 2024; 76:942–48. https://doi.org/10.1002/art.42802. [PubMed].
16. Sirenko M, Bernard E, Creignou M, Domenico D, Farina A, Arango Ossa JE, Kosmider O, Hasserjian RP, Jädersten M, Germing U, Sanz GF, van de Loosdrecht AA, Gurnari C, et al. Molecular and clinical presentation of UBA1-mutated myelodysplastic syndromes. Blood. 2024. [Epub ahead of print]. https://doi.org/10.1182/blood.2023023723. [PubMed].
17. Poulter J, Morgan A, Cargo C, Savic S, and UKGCA/VEXAS Consortium. A High-Throughput Amplicon Screen for Somatic UBA1 Variants in Cytopenic and Giant Cell Arteritis Cohorts. J Clin Immunol. 2022; 42:947–51. https://doi.org/10.1007/s10875-022-01258-w. [PubMed].
18. Schulman BA, Harper JW. Ubiquitin-like protein activation by E1 enzymes: the apex for downstream signalling pathways. Nat Rev Mol Cell Biol. 2009; 10:319–31. https://doi.org/10.1038/nrm2673. [PubMed].
19. Kulkarni M, Smith HE. E1 ubiquitin-activating enzyme UBA-1 plays multiple roles throughout C. elegans development. PLoS Genet. 2008; 4:e1000131. https://doi.org/10.1371/journal.pgen.1000131. [PubMed].
20. Chang TK, Shravage BV, Hayes SD, Powers CM, Simin RT, Wade Harper J, Baehrecke EH. Uba1 functions in Atg7- and Atg3-independent autophagy. Nat Cell Biol. 2013; 15:1067–78. https://doi.org/10.1038/ncb2804. [PubMed].
21. Hyodo M, Suzuki K. A temperature-sensitive mutant isolated from mouse FM3A cells defective in DNA replication at a non-permissive temperature. Exp Cell Res. 1982; 137:31–38. https://doi.org/10.1016/0014-4827(82)90004-0. [PubMed].
22. Tsuji H, Matsudo Y, Tsuji S, Hanaoka F, Hyodo M, Hori T. Isolation of temperature-sensitive CHO-K1 cell mutants exhibiting chromosomal instability and reduced DNA synthesis at nonpermissive temperature. Somat Cell Mol Genet. 1990; 16:461–76. https://doi.org/10.1007/BF01233196. [PubMed].
23. Eki T, Enomoto T, Miyajima A, Miyazawa H, Murakami Y, Hanaoka F, Yamada M, Ui M. Isolation of temperature-sensitive cell cycle mutants from mouse FM3A cells. Characterization of mutants with special reference to DNA replication. J Biol Chem. 1990; 265:26–33. [PubMed].
24. Ayusawa D, Kaneda S, Itoh Y, Yasuda H, Murakami Y, Sugasawa K, Hanaoka F, Seno T. Complementation by a cloned human ubiquitin-activating enzyme E1 of the S-phase-arrested mouse FM3A cell mutant with thermolabile E1. Cell Struct Funct. 1992; 17:113–22. https://doi.org/10.1247/csf.17.113. [PubMed].
25. Kulka RG, Raboy B, Schuster R, Parag HA, Diamond G, Ciechanover A, Marcus M. A Chinese hamster cell cycle mutant arrested at G2 phase has a temperature-sensitive ubiquitin-activating enzyme, E1. J Biol Chem. 1988; 263:15726–31. [PubMed].
26. Mayer A, Gropper R, Schwartz AL, Ciechanover A. Purification, characterization, and rapid inactivation of thermolabile ubiquitin-activating enzyme from the mammalian cell cycle mutant ts85. J Biol Chem. 1989; 264:2060–68. [PubMed].
27. Nishitani H, Goto H, Kaneda S, Yamao F, Seno T, Handley P, Schwartz AL, Nishimoto T. tsBN75 and tsBN423, temperature-sensitive x-linked mutants of the BHK21 cell line, can be complemented by the ubiquitin-activating enzyme E1 cDNA. Biochem Biophys Res Commun. 1992; 184:1015–21. https://doi.org/10.1016/0006-291x(92)90692-e. [PubMed].
28. Pfleger CM, Harvey KF, Yan H, Hariharan IK. Mutation of the gene encoding the ubiquitin activating enzyme ubal causes tissue overgrowth in Drosophila. Fly (Austin). 2007; 1:95–105. https://doi.org/10.4161/fly.4285. [PubMed].
29. Lee TV, Ding T, Chen Z, Rajendran V, Scherr H, Lackey M, Bolduc C, Bergmann A. The E1 ubiquitin-activating enzyme Uba1 in Drosophila controls apoptosis autonomously and tissue growth non-autonomously. Development. 2008; 135:43–52. https://doi.org/10.1242/dev.011288. [PubMed].
30. Hunt LC, Pagala V, Stephan A, Xie B, Kodali K, Kavdia K, Wang YD, Shirinifard A, Curley M, Graca FA, Fu Y, Poudel S, Li Y, et al. An adaptive stress response that confers cellular resilience to decreased ubiquitination. Nat Commun. 2023; 14:7348. https://doi.org/10.1038/s41467-023-43262-7. [PubMed].
31. Collins JC, Magaziner SJ, English M, Hassan B, Chen X, Balanda N, Anderson M, Lam A, Fernandez-Pol S, Kwong B, Greenberg PL, Terrier B, Likhite ME, et al. Shared and distinct mechanisms of UBA1 inactivation across different diseases. EMBO J. 2024; 43:1919–46. https://doi.org/10.1038/s44318-024-00046-z. [PubMed].
32. Dever TE, Ivanov IP, Hinnebusch AG. Translational regulation by uORFs and start codon selection stringency. Genes Dev. 2023; 37:474–89. https://doi.org/10.1101/gad.350752.123. [PubMed].
33. Handley-Gearhart PM, Stephen AG, Trausch-Azar JS, Ciechanover A, Schwartz AL. Human ubiquitin-activating enzyme, E1. Indication of potential nuclear and cytoplasmic subpopulations using epitope-tagged cDNA constructs. J Biol Chem. 1994; 269:33171–78. [PubMed].
34. Stephen AG, Trausch-Azar JS, Handley-Gearhart PM, Ciechanover A, Schwartz AL. Identification of a region within the ubiquitin-activating enzyme required for nuclear targeting and phosphorylation. J Biol Chem. 1997; 272:10895–903. https://doi.org/10.1074/jbc.272.16.10895. [PubMed].
35. Nouspikel T, Hanawalt PC. Impaired nucleotide excision repair upon macrophage differentiation is corrected by E1 ubiquitin-activating enzyme. Proc Natl Acad Sci U S A. 2006; 103:16188–93. https://doi.org/10.1073/pnas.0607769103. [PubMed].
36. Grenfell SJ, Trausch-Azar JS, Handley-Gearhart PM, Ciechanover A, Schwartz AL. Nuclear localization of the ubiquitin-activating enzyme, E1, is cell-cycle-dependent. Biochem J. 1994; 300:701–8. https://doi.org/10.1042/bj3000701. [PubMed].
37. Ferrada MA, Savic S, Cardona DO, Collins JC, Alessi H, Gutierrez-Rodrigues F, Kumar DBU, Wilson L, Goodspeed W, Topilow JS, Paik JJ, Poulter JA, Kermani TA, et al. Translation of cytoplasmic UBA1 contributes to VEXAS syndrome pathogenesis. Blood. 2022; 140:1496–506. https://doi.org/10.1182/blood.2022016985. [PubMed].
38. Poulter JA, Collins JC, Cargo C, De Tute RM, Evans P, Ospina Cardona D, Bowen DT, Cunnington JR, Baguley E, Quinn M, Green M, McGonagle D, Beck DB, et al. Novel somatic mutations in UBA1 as a cause of VEXAS syndrome. Blood. 2021; 137:3676–81. https://doi.org/10.1182/blood.2020010286. [PubMed].
39. Bourbon E, Heiblig M, Gerfaud Valentin M, Barba T, Durel CA, Lega JC, Barraco F, Sève P, Jamilloux Y, Sujobert P. Therapeutic options in VEXAS syndrome: insights from a retrospective series. Blood. 2021; 137:3682–84. https://doi.org/10.1182/blood.2020010177. [PubMed].
40. Templé M, Duroyon E, Croizier C, Rossignol J, Huet T, Friedrich C, Zalmai L, Priollet P, Hayem G, Tournillhac O, Le Guenno G, Hermine O, Terrier B, Kosmider O. Atypical splice-site mutations causing VEXAS syndrome. Rheumatology (Oxford). 2021; 60:e435–37. https://doi.org/10.1093/rheumatology/keab524. [PubMed].
41. Stiburkova B, Pavelcova K, Belickova M, Magaziner SJ, Collins JC, Werner A, Beck DB, Balajkova V, Salek C, Vostry M, Mann H, Vencovsky J. Novel Somatic UBA1 Variant in a Patient With VEXAS Syndrome. Arthritis Rheumatol. 2023; 75:1285–90. https://doi.org/10.1002/art.42471. [PubMed].
42. Allison DR, Dholaria B, Kishtagari A, Mohan S, Steigelfest E, Shaver AC, Mason EF. Distinct bone marrow findings associated with a noncanonical UBA1 variant in VEXAS syndrome. Am J Hematol. 2024; 99:1400–2. https://doi.org/10.1002/ajh.27320. [PubMed].
43. Weng Y, Chen W, Kong Q, Wang R, Zeng R, He A, Liu Y, Mao Y, Qin Y, Ngai WSC, Zhang H, Ke M, Wang J, et al. DeKinomics pulse-chases kinase functions in living cells. Nat Chem Biol. 2024; 20:615–23. https://doi.org/10.1038/s41589-023-01497-x. [PubMed].
44. Al-Hakim A, Kulasekararaj A, Norouzi M, Medlock R, Patrick F, Cargo C, Savic S. S56F UBA1 variant is associated with haematological predominant subtype of VEXAS. Br J Haematol. 2023; 203:331–35. https://doi.org/10.1111/bjh.19021. [PubMed].
45. Gutierrez-Rodrigues F, Kusne Y, Fernandez J, Lasho T, Shalhoub R, Ma X, Alessi H, Finke C, Koster MJ, Mangaonkar A, Warrington KJ, Begna K, Xie Z, et al. Spectrum of clonal hematopoiesis in VEXAS syndrome. Blood. 2023; 142:244–59. https://doi.org/10.1182/blood.2022018774. [PubMed].
46. Georgin-Lavialle S, Terrier B, Guedon AF, Heiblig M, Comont T, Lazaro E, Lacombe V, Terriou L, Ardois S, Bouaziz JD, Mathian A, Le Guenno G, Aouba A, et al, and French VEXAS group, and GFEV, GFM, CEREMAIA, MINHEMON. Further characterization of clinical and laboratory features in VEXAS syndrome: large-scale analysis of a multicentre case series of 116 French patients. Br J Dermatol. 2022; 186:564–74. https://doi.org/10.1111/bjd.20805. [PubMed].
47. Obiorah IE, Patel BA, Groarke EM, Wang W, Trick M, Ombrello AK, Ferrada MA, Wu Z, Gutierrez-Rodrigues F, Lotter J, Wilson L, Hoffmann P, Cardona DO, et al. Benign and malignant hematologic manifestations in patients with VEXAS syndrome due to somatic mutations in UBA1. Blood Adv. 2021; 5:3203–15. https://doi.org/10.1182/bloodadvances.2021004976. [PubMed].
48. Kosmider O, Possémé C, Templé M, Corneau A, Carbone F, Duroyon E, Breillat P, Chirayath TW, Oules B, Sohier P, Luka M, Gobeaux C, Lazaro E, et al. VEXAS syndrome is characterized by inflammasome activation and monocyte dysregulation. Nat Commun. 2024; 15:910. https://doi.org/10.1038/s41467-024-44811-4. [PubMed].
49. Chiaramida A, Obwar SG, Nordstrom AEH, Ericsson M, Saldanha A, Ivanova EV, Griffin GK, Khan DH, Belizaire R. Sensitivity to targeted UBA1 inhibition in a myeloid cell line model of VEXAS syndrome. Blood Adv. 2023; 7:7445–56. https://doi.org/10.1182/bloodadvances.2023010531. [PubMed].
50. Hadjadj J, Nguyen Y, Mouloudj D, Bourguiba R, Heiblig M, Aloui H, McAvoy C, Lacombe V, Ardois S, Campochiaro C, Maria A, Coustal C, Comont T, et al, and FRENVEX. Efficacy and safety of targeted therapies in VEXAS syndrome: retrospective study from the FRENVEX. Ann Rheum Dis. 2024. [Epub ahead of print]. https://doi.org/10.1136/ard-2024-225640. [PubMed].
51. Di Conza G, Ho PC, Cubillos-Ruiz JR, Huang SC. Control of immune cell function by the unfolded protein response. Nat Rev Immunol. 2023; 23:546–62. https://doi.org/10.1038/s41577-023-00838-0. [PubMed].
52. Grootjans J, Kaser A, Kaufman RJ, Blumberg RS. The unfolded protein response in immunity and inflammation. Nat Rev Immunol. 2016; 16:469–84. https://doi.org/10.1038/nri.2016.62. [PubMed].
53. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012; 13:89–102. https://doi.org/10.1038/nrm3270. [PubMed].
54. Hetz C, Papa FR. The Unfolded Protein Response and Cell Fate Control. Mol Cell. 2018; 69:169–81. https://doi.org/10.1016/j.molcel.2017.06.017. [PubMed].
55. Olteanu H, Patnaik M, Koster MJ, Herrick JL, Chen D, He R, Viswanatha D, Warrington KJ, Go RS, Mangaonkar AA, Kourelis T, Hines A, Gibson SE, et al. Comprehensive morphologic characterization of bone marrow biopsy findings in a large cohort of patients with VEXAS syndrome: A single-institution longitudinal study of 111 bone marrow samples from 52 patients. Am J Clin Pathol. 2024; 161:609–24. https://doi.org/10.1093/ajcp/aqad186. [PubMed].
56. Lacombe V, Hadjadj J, Georgin-Lavialle S, Lavigne C, Geneviève F, Kosmider O. Vacuoles in bone marrow progenitors: VEXAS syndrome and beyond. Lancet Haematol. 2024; 11:e160–67. https://doi.org/10.1016/S2352-3026(23)00375-7. [PubMed].
57. Wu Z, Gao S, Gao Q, Patel BA, Groarke EM, Feng X, Manley AL, Li H, Ospina Cardona D, Kajigaya S, Alemu L, Quinones Raffo D, Ombrello AK, et al. Early activation of inflammatory pathways in UBA1-mutated hematopoietic stem and progenitor cells in VEXAS. Cell Rep Med. 2023; 4:101160. https://doi.org/10.1016/j.xcrm.2023.101160. [PubMed].
58. Ganesan S, Murray RM, Sotelo J, Eton EO, Takashima K, Botella T, Beattie K, Indart AC, Chraiki N, Croizier C, Izzo F, Potenski C, Marro S, et al. Single-cell genotype-phenotype mapping identifies therapeutic vulnerabilities in VEXAS syndrome. bioRxiv. 2024. https://doi.org/10.1101/2024.05.19.594376.
59. Koster MJ, Lasho TL, Olteanu H, Reichard KK, Mangaonkar A, Warrington KJ, Patnaik MM. VEXAS syndrome: Clinical, hematologic features and a practical approach to diagnosis and management. Am J Hematol. 2024; 99:284–99. https://doi.org/10.1002/ajh.27156. [PubMed].
60. Weiss G, Ganz T, Goodnough LT. Anemia of inflammation. Blood. 2019; 133:40–50. https://doi.org/10.1182/blood-2018-06-856500. [PubMed].
61. Marques O, Weiss G, Muckenthaler MU. The role of iron in chronic inflammatory diseases: from mechanisms to treatment options in anemia of inflammation. Blood. 2022; 140:2011–23. https://doi.org/10.1182/blood.2021013472. [PubMed].
62. Romano L, Seu KG, Papoin J, Muench DE, Konstantinidis D, Olsson A, Schlum K, Chetal K, Chasis JA, Mohandas N, Barnes BJ, Zheng Y, Grimes HL, et al. Erythroblastic islands foster granulopoiesis in parallel to terminal erythropoiesis. Blood. 2022; 140:1621–34. https://doi.org/10.1182/blood.2022015724. [PubMed].
63. Heiblig M, Ferrada MA, Koster MJ, Barba T, Gerfaud-Valentin M, Mékinian A, Coelho H, Fossard G, Barraco F, Galicier L, Bienvenu B, Hirsch P, Vial G, et al. Ruxolitinib is more effective than other JAK inhibitors to treat VEXAS syndrome: a retrospective multicenter study. Blood. 2022; 140:927–31. https://doi.org/10.1182/blood.2022016642. [PubMed].
64. Sakuma M, Tanimura A, Yasui S, Ishiguro K, Kobayashi T, Ohshiro Y, Miyazaki H, Minamimoto R, Okafuji T, Shimozawa K, Ogura G, Miwa A, Yamashita H, Kaneko H. A Case of polychondritis-onset refractory organizing pneumonia with cytopaenia diagnosed as VEXAS syndrome: the disease course of 7 years. Rheumatology (Oxford). 2021; 60:e356–59. https://doi.org/10.1093/rheumatology/keab349. [PubMed].
65. Meyer L, Deau B, Forejtníková H, Duménil D, Margottin-Goguet F, Lacombe C, Mayeux P, Verdier F. beta-Trcp mediates ubiquitination and degradation of the erythropoietin receptor and controls cell proliferation. Blood. 2007; 109:5215–22. https://doi.org/10.1182/blood-2006-10-055350. [PubMed].
66. Basiorka AA, McGraw KL, De Ceuninck L, Griner LN, Zhang L, Clark JA, Caceres G, Sokol L, Komrokji RS, Reuther GW, Wei S, Tavernier J, List AF. Lenalidomide Stabilizes the Erythropoietin Receptor by Inhibiting the E3 Ubiquitin Ligase RNF41. Cancer Res. 2016; 76:3531–40. https://doi.org/10.1158/0008-5472.CAN-15-1756. [PubMed].
67. Saur SJ, Sangkhae V, Geddis AE, Kaushansky K, Hitchcock IS. Ubiquitination and degradation of the thrombopoietin receptor c-Mpl. Blood. 2010; 115:1254–63. https://doi.org/10.1182/blood-2009-06-227033. [PubMed].
68. Rathinam C, Flavell RA. c-Cbl deficiency leads to diminished lymphocyte development and functions in an age-dependent manner. Proc Natl Acad Sci U S A. 2010; 107:8316–21. https://doi.org/10.1073/pnas.0914496107. [PubMed].
69. Iwakoshi NN, Lee AH, Vallabhajosyula P, Otipoby KL, Rajewsky K, Glimcher LH. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat Immunol. 2003; 4:321–29. https://doi.org/10.1038/ni907. [PubMed].
70. Pant V, Quintás-Cardama A, Lozano G. The p53 pathway in hematopoiesis: lessons from mouse models, implications for humans. Blood. 2012; 120:5118–27. https://doi.org/10.1182/blood-2012-05-356014. [PubMed].
71. Chowdary DR, Dermody JJ, Jha KK, Ozer HL. Accumulation of p53 in a mutant cell line defective in the ubiquitin pathway. Mol Cell Biol. 1994; 14:1997–2003. https://doi.org/10.1128/mcb.14.3.1997-2003.1994. [PubMed].
72. Adachi S, Kirino Y, Higashitani K, Hirahara L, Maeda A, Horita N, Takase-Minegishi K, Yoshimi R, Nakajima H. Targeting enhanced cell death represents a potential therapeutic strategy for VEXAS syndrome. Rheumatol Adv Pract. 2024; 8:rkae065. https://doi.org/10.1093/rap/rkae065. [PubMed].
73. de Valence B, Delaune M, Nguyen Y, Jachiet V, Heiblig M, Jean A, Riescher Tuczkiewicz S, Henneton P, Guilpain P, Schleinitz N, Le Guenno G, Lobbes H, Lacombe V, et al, and French VEXAS Group. Serious infections in patients with VEXAS syndrome: data from the French VEXAS registry. Ann Rheum Dis. 2024; 83:372–81. https://doi.org/10.1136/ard-2023-224819. [PubMed].
74. Rogers LM, Olivier AK, Meyerholz DK, Dupuy AJ. Adaptive immunity does not strongly suppress spontaneous tumors in a Sleeping Beauty model of cancer. J Immunol. 2013; 190:4393–99. https://doi.org/10.4049/jimmunol.1203227. [PubMed].
75. Podvin B, Cleenewerck N, Nibourel O, Marceau-Renaut A, Roynard P, Preudhomme C, Duployez N, Terriou L. Three UBA1 clones for a unique VEXAS syndrome. Rheumatology (Oxford). 2024; 63:e48–50. https://doi.org/10.1093/rheumatology/kead472. [PubMed].
76. Malcovati L. VEXAS: walking on the edge of malignancy. Blood. 2023; 142:214–15. https://doi.org/10.1182/blood.2023020772. [PubMed].
77. Jaiswal S, Ebert BL. Clonal hematopoiesis in human aging and disease. Science. 2019; 366:eaan4673. https://doi.org/10.1126/science.aan4673. [PubMed].
78. Huang J, Chan SC, Lok V, Zhang L, Lucero-Prisno DE 3rd, Xu W, Zheng ZJ, Elcarte E, Withers M, Wong MCS, and Non-communicable Disease Global Health Research Group, and Association of Pacific Rim Universities. The epidemiological landscape of multiple myeloma: a global cancer registry estimate of disease burden, risk factors, and temporal trends. Lancet Haematol. 2022; 9:e670–77. https://doi.org/10.1016/S2352-3026(22)00165-X. [PubMed].
79. Moudry P, Lukas C, Macurek L, Hanzlikova H, Hodny Z, Lukas J, Bartek J. Ubiquitin-activating enzyme UBA1 is required for cellular response to DNA damage. Cell Cycle. 2012; 11:1573–82. https://doi.org/10.4161/cc.19978. [PubMed].
80. Kumbhar R, Vidal-Eychenié S, Kontopoulos DG, Larroque M, Larroque C, Basbous J, Kossida S, Ribeyre C, Constantinou A. Recruitment of ubiquitin-activating enzyme UBA1 to DNA by poly(ADP-ribose) promotes ATR signalling. Life Sci Alliance. 2018; 1:e201800096. https://doi.org/10.26508/lsa.201800096. [PubMed].
81. Zhang T, Joubert P, Ansari-Pour N, Zhao W, Hoang PH, Lokanga R, Moye AL, Rosenbaum J, Gonzalez-Perez A, Martínez-Jiménez F, Castro A, Muscarella LA, Hofman P, et al. Genomic and evolutionary classification of lung cancer in never smokers. Nat Genet. 2021; 53:1348–59. https://doi.org/10.1038/s41588-021-00920-0. [PubMed].
82. Haupt S, Caramia F, Herschtal A, Soussi T, Lozano G, Chen H, Liang H, Speed TP, Haupt Y. Identification of cancer sex-disparity in the functional integrity of p53 and its X chromosome network. Nat Commun. 2019; 10:5385. https://doi.org/10.1038/s41467-019-13266-3. [PubMed].
83. Oliva M, Muñoz-Aguirre M, Kim-Hellmuth S, Wucher V, Gewirtz ADH, Cotter DJ, Parsana P, Kasela S, Balliu B, Viñuela A, Castel SE, Mohammadi P, Aguet F, et al, and GTEx Consortium. The impact of sex on gene expression across human tissues. Science. 2020; 369:eaba3066. https://doi.org/10.1126/science.aba3066. [PubMed].
84. So J, Tai AK, Lichtenstein AH, Wu D, Lamon-Fava S. Sexual dimorphism of monocyte transcriptome in individuals with chronic low-grade inflammation. Biol Sex Differ. 2021; 12:43. https://doi.org/10.1186/s13293-021-00387-y. [PubMed].
85. Navarro-Cobos MJ, Balaton BP, Brown CJ. Genes that escape from X-chromosome inactivation: Potential contributors to Klinefelter syndrome. Am J Med Genet C Semin Med Genet. 2020; 184:226–38. https://doi.org/10.1002/ajmg.c.31800. [PubMed].
86. Ramser J, Ahearn ME, Lenski C, Yariz KO, Hellebrand H, von Rhein M, Clark RD, Schmutzler RK, Lichtner P, Hoffman EP, Meindl A, Baumbach-Reardon L. Rare missense and synonymous variants in UBE1 are associated with X-linked infantile spinal muscular atrophy. Am J Hum Genet. 2008; 82:188–93. https://doi.org/10.1016/j.ajhg.2007.09.009. [PubMed].
87. Liu J, Wang K, Li B, Yang X. A novel Xp11.22-22.33 deletion suggesting a possible mechanism of congenital cervical spinal muscular atrophy. Mol Genet Genomic Med. 2021; 9:e1606. https://doi.org/10.1002/mgg3.1606. [PubMed].
88. Wade BE, Wang CE, Yan S, Bhat K, Huang B, Li S, Li XJ. Ubiquitin-activating enzyme activity contributes to differential accumulation of mutant huntingtin in brain and peripheral tissues. J Neurosci. 2014; 34:8411–22. https://doi.org/10.1523/JNEUROSCI.0775-14.2014. [PubMed].
89. Groen EJN, Gillingwater TH. UBA1: At the Crossroads of Ubiquitin Homeostasis and Neurodegeneration. Trends Mol Med. 2015; 21:622–32. https://doi.org/10.1016/j.molmed.2015.08.003. [PubMed].
90. Wishart TM, Mutsaers CA, Riessland M, Reimer MM, Hunter G, Hannam ML, Eaton SL, Fuller HR, Roche SL, Somers E, Morse R, Young PJ, Lamont DJ, et al. Dysregulation of ubiquitin homeostasis and β-catenin signaling promote spinal muscular atrophy. J Clin Invest. 2014; 124:1821–34. https://doi.org/10.1172/JCI71318. [PubMed].
91. Powis RA, Karyka E, Boyd P, Côme J, Jones RA, Zheng Y, Szunyogova E, Groen EJ, Hunter G, Thomson D, Wishart TM, Becker CG, Parson SH, et al. Systemic restoration of UBA1 ameliorates disease in spinal muscular atrophy. JCI Insight. 2016; 1:e87908. https://doi.org/10.1172/jci.insight.87908. [PubMed].
92. Allen E, Ding J, Wang W, Pramanik S, Chou J, Yau V, Yang Y. Gigaxonin-controlled degradation of MAP1B light chain is critical to neuronal survival. Nature. 2005; 438:224–28. https://doi.org/10.1038/nature04256. [PubMed].
93. Mekinian A, Grignano E, Braun T, Decaux O, Liozon E, Costedoat-Chalumeau N, Kahn JE, Hamidou M, Park S, Puéchal X, Toussirot E, Falgarone G, Launay D, et al. Systemic inflammatory and autoimmune manifestations associated with myelodysplastic syndromes and chronic myelomonocytic leukaemia: a French multicentre retrospective study. Rheumatology (Oxford). 2016; 55:291–300. https://doi.org/10.1093/rheumatology/kev294. [PubMed].
94. Fraison JB, Mekinian A, Grignano E, Kahn JE, Arlet JB, Decaux O, Denis G, Buchdahl AL, Omouri M, Maigne G, Aouba A, Leon N, Berthier S, et al. Efficacy of Azacitidine in autoimmune and inflammatory disorders associated with myelodysplastic syndromes and chronic myelomonocytic leukemia. Leuk Res. 2016; 43:13–7. https://doi.org/10.1016/j.leukres.2016.02.005. [PubMed].
95. Raaijmakers MHG, Hermans M, Aalbers A, Rijken M, Dalm VAS, van Daele P, Valk PJM. Azacytidine Treatment for VEXAS Syndrome. Hemasphere. 2021; 5:e661. https://doi.org/10.1097/HS9.0000000000000661. [PubMed].
96. Fozza C, Corda G, Barraqueddu F, Virdis P, Contini S, Galleu A, Isoni A, Dore F, Angelucci E, Longinotti M. Azacitidine improves the T-cell repertoire in patients with myelodysplastic syndromes and acute myeloid leukemia with multilineage dysplasia. Leuk Res. 2015; 39:957–63. https://doi.org/10.1016/j.leukres.2015.06.007. [PubMed].
97. Grimm J, Simnica D, Jäkel N, Paschold L, Willscher E, Schulze S, Dierks C, Al-Ali HK, Binder M. Azacitidine-induced reconstitution of the bone marrow T cell repertoire is associated with superior survival in AML patients. Blood Cancer J. 2022; 12:19. https://doi.org/10.1038/s41408-022-00615-7. [PubMed].
98. Mekinian A, Zhao LP, Chevret S, Desseaux K, Pascal L, Comont T, Maria A, Peterlin P, Terriou L, D'Aveni Piney M, Gourin MP, Vey N, Rauzy OB, et al. A Phase II prospective trial of azacitidine in steroid-dependent or refractory systemic autoimmune/inflammatory disorders and VEXAS syndrome associated with MDS and CMML. Leukemia. 2022; 36:2739–42. https://doi.org/10.1038/s41375-022-01698-8. [PubMed].
99. Aalbers AM, van Daele PLA, Dalm VAS, Valk PJM, Raaijmakers MHG. Long-term genetic and clinical remissions after cessation of azacitidine treatment in patients with VEXAS syndrome. Hemasphere. 2024; 8:e129. https://doi.org/10.1002/hem3.129. [PubMed].
100. McHugh A, Fernandes K, South AP, Mellerio JE, Salas-Alanís JC, Proby CM, Leigh IM, Saville MK. Preclinical comparison of proteasome and ubiquitin E1 enzyme inhibitors in cutaneous squamous cell carcinoma: the identification of mechanisms of differential sensitivity. Oncotarget. 2018; 9:20265–81. https://doi.org/10.18632/oncotarget.24750. [PubMed].
101. Liu G, Yu J, Wu R, Shi L, Zhang X, Zhang W, Zhong X, Wang Y, Li H, Shen Y, Wu C, Yu R, Niu M, Liu X. GRP78 determines glioblastoma sensitivity to UBA1 inhibition-induced UPR signaling and cell death. Cell Death Dis. 2021; 12:733. https://doi.org/10.1038/s41419-021-04023-w. [PubMed] Retraction in: Cell Death Dis. 2024; 15:188. https://doi.org/10.1038/s41419-024-06574-0. [PubMed].
102. Majeed S, Aparnathi MK, Nixon KCJ, Venkatasubramanian V, Rahman F, Song L, Weiss J, Barayan R, Sugumar V, Barghout SH, Pearson JD, Bremner R, Schimmer AD, et al. Targeting the Ubiquitin-Proteasome System Using the UBA1 Inhibitor TAK-243 is a Potential Therapeutic Strategy for Small-Cell Lung Cancer. Clin Cancer Res. 2022; 28:1966–78. https://doi.org/10.1158/1078-0432.CCR-21-0344. [PubMed].
103. Yang Y, Kitagaki J, Dai RM, Tsai YC, Lorick KL, Ludwig RL, Pierre SA, Jensen JP, Davydov IV, Oberoi P, Li CC, Kenten JH, Beutler JA, et al. Inhibitors of ubiquitin-activating enzyme (E1), a new class of potential cancer therapeutics. Cancer Res. 2007; 67:9472–81. https://doi.org/10.1158/0008-5472.CAN-07-0568. [PubMed].
104. Yan W, Zhong Y, Hu X, Xu T, Zhang Y, Kales S, Qu Y, Talley DC, Baljinnyam B, LeClair CA, Simeonov A, Polster BM, Huang R, et al. Auranofin targets UBA1 and enhances UBA1 activity by facilitating ubiquitin trans-thioesterification to E2 ubiquitin-conjugating enzymes. Nat Commun. 2023; 14:4798. https://doi.org/10.1038/s41467-023-40537-x. [PubMed].
105. Fiskus W, Saba N, Shen M, Ghias M, Liu J, Gupta SD, Chauhan L, Rao R, Gunewardena S, Schorno K, Austin CP, Maddocks K, Byrd J, et al. Auranofin induces lethal oxidative and endoplasmic reticulum stress and exerts potent preclinical activity against chronic lymphocytic leukemia. Cancer Res. 2014; 74:2520–32. https://doi.org/10.1158/0008-5472.CAN-13-2033. [PubMed].
106. Chen X, Shi X, Zhao C, Li X, Lan X, Liu S, Huang H, Liu N, Liao S, Zang D, Song W, Liu Q, Carter BZ, et al. Anti-rheumatic agent auranofin induced apoptosis in chronic myeloid leukemia cells resistant to imatinib through both Bcr/Abl-dependent and -independent mechanisms. Oncotarget. 2014; 5:9118–32. https://doi.org/10.18632/oncotarget.2361. [PubMed].