
Introduction
One of the epigenetic modifications is histone acetylation which is catalyzed by histone acetyltransferases. During this process, the N-terminal tails of histones undergo acetylation by adding acetyl groups to the positively charged lysine residues. This modification reduces the interactions between histones and negatively charged DNA, which results in the relaxation of the chromatin structure. Increased transcriptional activation has been associated with relaxed chromatin [1]. Histone deacetylases (HDACs) reverse this process by removing the acetyl group, leading to a condensed, transcriptionally inactive chromatin. The histone acetylation/deacetylation process induces structural alterations in distant chromosome regions, thereby having a broad impact on gene expression and various cellular processes such as DNA replication and cell division. In particular, HDAC overexpression may down-regulate tumor suppressor genes [2]. Various HDAC inhibitors (HDACis) have been developed to induce gene expression, leading to cell differentiation, cell cycle arrest, and apoptosis [3]. Some HDACis have regulatory approval for the treatment of patients with hematologic malignancies, including vorinostat, romidepsin, panobinostat, and belinostat.
Although HDACis have shown promising results in preclinical studies, their clinical efficacy as monotherapy is limited; however, when combined with other anticancer drugs, enhanced anti-tumor activity has been reported [4]. The diverse impact of HDACis on regulating cellular drug transporters should be considered when planning their use in combination with chemotherapy. For example, in human hematologic cancer cell lines, HDACis have been shown to decrease expression of the MRP1 protein and increase expression of the MDR1 protein [5]. Notably, in patients with previously untreated peripheral T-cell lymphoma, the addition of the HDACi romidepsin to cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) did not improve clinical outcomes [6]. The lack of clinical improvement when romidepsin was added to MDR1 ligands (e.g., doxorubicin, vincristine) may be at least partially attributed to the effects of romidepsin on the expressions of MRP1 and MDR1 [6]. The addition of HDACis to chemotherapy may enhance its efficacy by inducing DNA double-strand breaks. Indeed, alterations in chromatin structure induced by HDACis directly trigger activation of the DNA damage response [7].
HDACis can affect the acetylation of proteins involved in different DNA repair mechanisms, thereby influencing the genomic instability displayed by cancer cells. Researchers have extensively studied the effects of HDACis on genomic stability, as well as their interactions with poly (ADP ribose) polymerase (PARP) inhibitors (PARPi), particularly in solid tumors [8–11]. PARPis have a direct effect on protein poly (ADP-ribosyl)ation (PARylation), which is important for DNA repair [12]. PARP enzymes catalyze PARylation and bind to DNA breaks, self-ribosylate, and recruit and PARylate DNA repair proteins [12]. We have previously reported on the synergistic activity of HDACis and PARPis in hematologic malignancies that occurs via enhanced inhibition of protein PARylation and decreased levels and phosphorylation of major proteins involved in DNA repair [13]. Similar synergistic interactions between HDACis and PARPis have been reported in thyroid and breast cancer cells [9, 10]. Published data suggest that the synergism may be partly attributed to blocking chromatin PARylation with histone acetylation [12].
For patients with metastatic pancreatic adenocarcinoma bearing BRCA1/2 germ-line mutations (gBRCAm; BRCA1 prevalence, 0.3% to 2.3% and BRCA2 prevalence, 0.7 to 5.7%) [14–16] who achieve clinical response to first-line treatment, the PARPi olaparib is now offered as an alternative maintenance treatment option. In a randomized study (POLO), 154 patients with gBRCAm metastatic pancreatic adenocarcinoma treated with olaparib had longer progression-free survival (7.4 months) than those treated with placebo (3.8 months) (hazard ratio = 0.53; p = 0.0035) [17]. No statistically significant difference was noted in overall survival [18].
Decitabine (5-aza-2′-deoxycytidine) is a nucleoside cytidine analogue that, when phosphorylated, is incorporated into a growing DNA strand, and inhibits DNA methylation. Decitabine also induces DNA damage by inactivating and trapping DNA methyltransferase on DNA, consequently activating transcriptionally silenced DNA loci [19–21]. KRAS-dependent gene signatures have been reported to be associated with sensitivity to decitabine in selected patients with KRAS-mutated pancreatic cancer [22]. Other nucleoside analogues (e.g., gemcitabine) have established antitumor activity in pancreatic cancer.
Therefore, we explored various combinations of HDACis and PARPis, with or without decitabine, in pancreatic cancer cell lines. In this study, we show that HDACis inhibit protein PARylation and exhibit synergistic cytotoxicity with PARPis and a demethylating agent (decitabine). The results provide novel preclinical data that demonstrate synergism between HDACi- and PARPi-mediated inhibition of DNA repair and decitabine in pancreatic cancer and have implications for the exploitation of therapeutic purposes.
Results
Sensitivity of pancreatic cancer cell lines to HDACis, PARPis, and decitabine
Pancreatic cancer cells were exposed to various concentrations of the individual drugs to determine the differences in their drug sensitivity and the concentrations appropriate for the drug combination experiments. Cellular proliferation is summarized in Figure 1, and differences among all non-0 doses summarized by cell line and drug in Supplementary Table 1. All cell lines showed significantly reduced proliferation at the highest doses when compared with the lowest non-zero dose. The Capan-1 cell line, which has a BRCA2 mutation [23], showed a trend towards greater resistance to panobinostat compared with the BxPC-3 and PL45 cells (Figure 1A); the three cell lines showed similar sensitivity to vorinostat (Figure 1B). The PL45 cell line, which is wild-type for BRCA1 and BRCA2, showed a trend towards more resistance to talazoparib and olaparib (PARPis) compared with the BxPC-3 and Capan-1 cell lines (Figures 1C, 1D). The three cell lines showed similar sensitivity to decitabine (Figure 1E). The differences in drug sensitivities of the three pancreatic cell lines are summarized in Figure 1F.
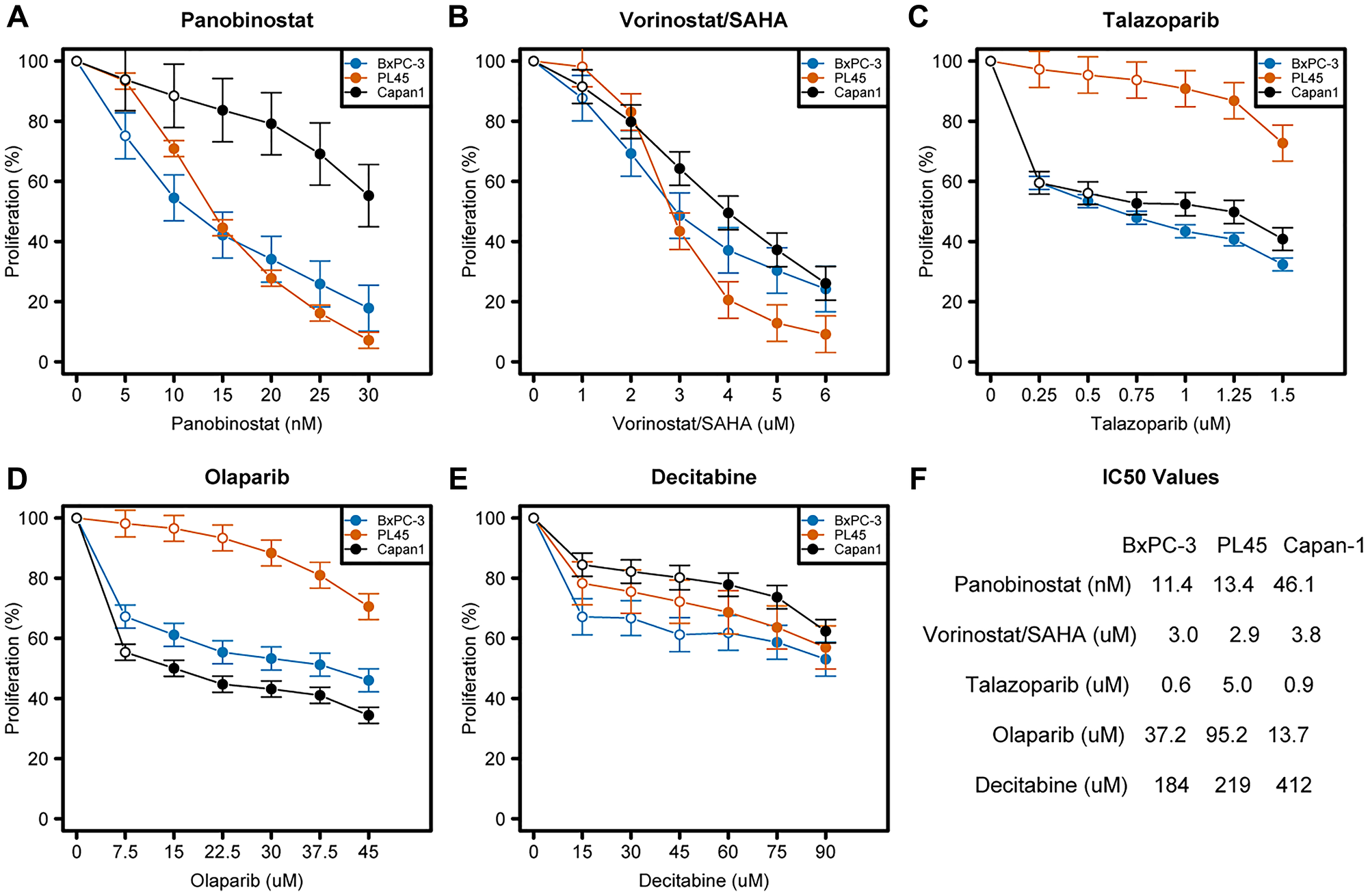
Figure 1: Dose-response curves of various drugs in three pancreatic cancer cell lines.
(A–F) Cells were seeded in 96-well plates overnight and exposed to drugs for 3 days as described in the Materials and Methods. Rate of cell proliferation was determined relative to control by MTT assay. Model-adjusted means are shown with 95% confidence intervals for the non-zero doses modeled, and solid points indicate a significant difference from the first non-zero dose (Supplementary Table 1). Each cell line of each drug was modeled independently.
Synergistic cytotoxicity of HDACis, PARPis, and decitabine
HDACis and PARPis are known to have synergistic interactions in hematologic, thyroid, and breast cancer cells [9, 10, 13]. We, therefore, investigated whether these drugs would provide synergistic cytotoxicity in pancreatic cancer cell lines. Cells were exposed to various concentrations of single agents or combinations of two drugs (HDACi + PARPi), using a constant ratio, followed by the MTT assay. Figure 2 shows the calculated CI at increasing drug effects. Significant synergism was noted between HDACis and PARPis, as evidenced by CI values <1 at fraction affected >0.5 in all cell lines.
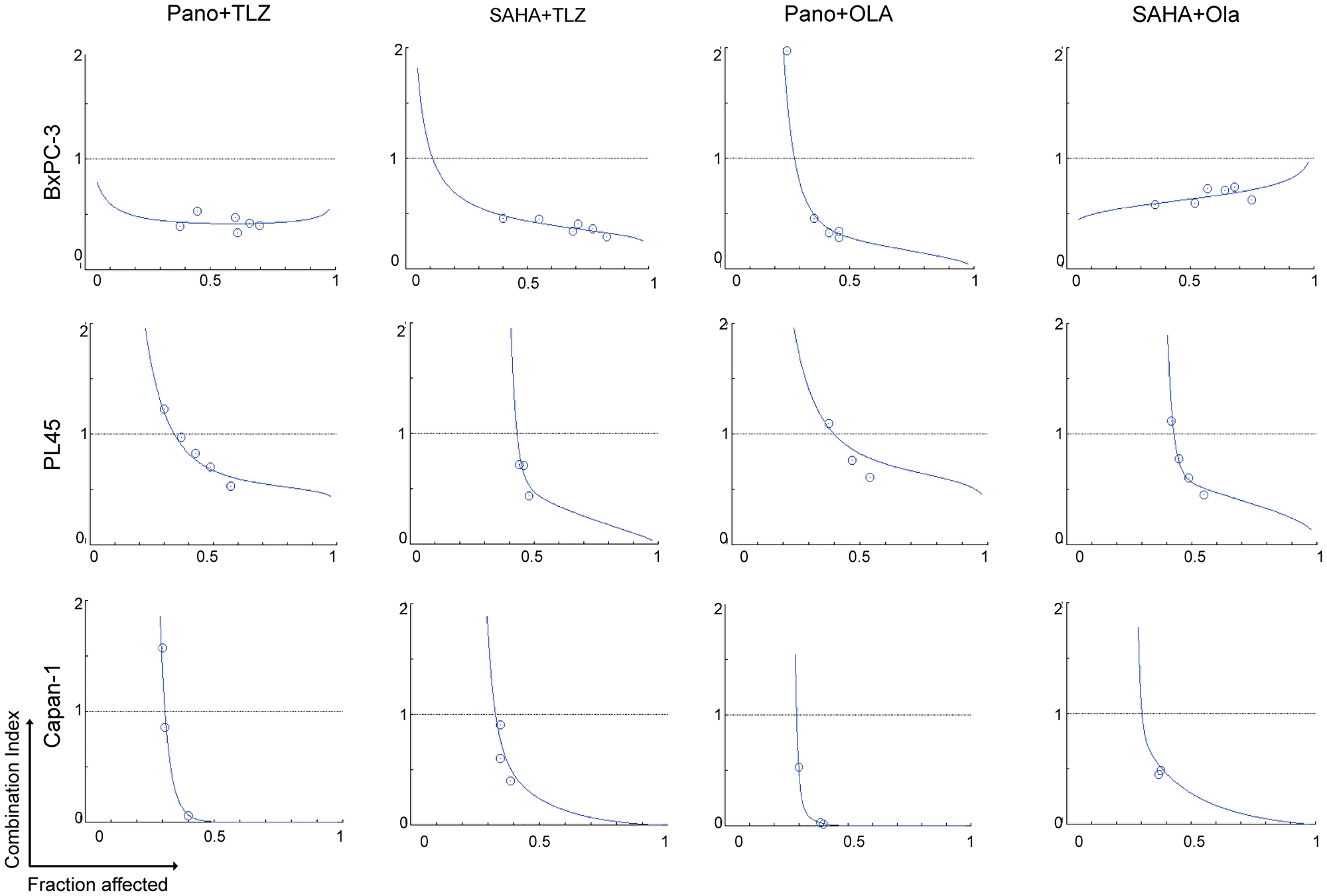
Figure 2: Synergistic cytotoxicity of HDACi and PARPi.
Cells were seeded in 96-well plates overnight and exposed to different concentrations of individual drugs or to the two-drug combinations at a constant concentration ratio, and cell proliferation was analyzed after 3 days. The relationships between the calculated combination indexes (CI, Y-axis) and fractions affected (Fa, X-axis) are shown. CI <1.0 indicates synergism. The graphs are representative of two independent experiments. Abbreviations: Ola: Olaparib; Pano: panobinostat; SAHA: vorinostat; TLZ: talazoparib.
To further determine the synergistic cytotoxicity of HDACi and PARPi in the three pancreatic cell lines, a clonogenic assay was performed. Quantitative analysis of the colony formation (Figure 3A) showed that exposure of BxPC-3 cells to panobinostat + talazoparib, panobinostat + olaparib, vorinostat + talazoparib, and vorinostat + olaparib resulted in ~77%, ~71%, ~65%, and ~71% inhibition of colony formation, respectively (Figure 3B, Table 1). Panobinostat + talazoparib significantly inhibited colony formation compared with either panobinostat (P < 0.0001) or talazoparib (P = 0.0008) alone. Similarly, vorinostat + olaparib significantly inhibited colony formation compared with vorinostat (P < 0.0001). The inhibition of colony formation mediated by vorinostat + talazoparib was not significantly different than that with the individual drugs. The inhibition of colony formation mediated by panobinostat + olaparib was significantly lower than that noted with panobinostat alone (p < 0.0001). The addition of decitabine to each of the two-drug combinations significantly augmented inhibition of colony formation (~83% - 90% inhibition; P values < 0.0001), suggesting synergistic cytotoxicity caused by the three-drug combinations to the BxPC-3 cells. Exposure of PL45 cells to panobinostat + talazoparib, panobinostat + olaparib, vorinostat + talazoparib, and vorinostat + olaparib resulted in ~69%, ~57%, ~44%, and ~42% inhibition of colony formation, respectively (Figure 3B, Table 1, Supplementary Table 2). The addition of decitabine to each of these two-drug combinations significantly increased the inhibition rates to ~90%, ~79%, ~72%, and ~62%, respectively (Figure 3B, Table 1). Similar results were obtained when Capan-1 cells were exposed to the two-drug (~54–62% inhibition) or three-drug (~69–76% inhibition) combinations (Figure 3B, Table 1). The inhibition rates of colony formation mediated by all three-drug combinations in the three pancreatic cell lines were mostly statistically higher than those for the individual drugs (Figure 3B, Table 1). These results suggest drug synergistic cytotoxicity using HDACi + PARPi and HDACi + PARPi + decitabine combinations.
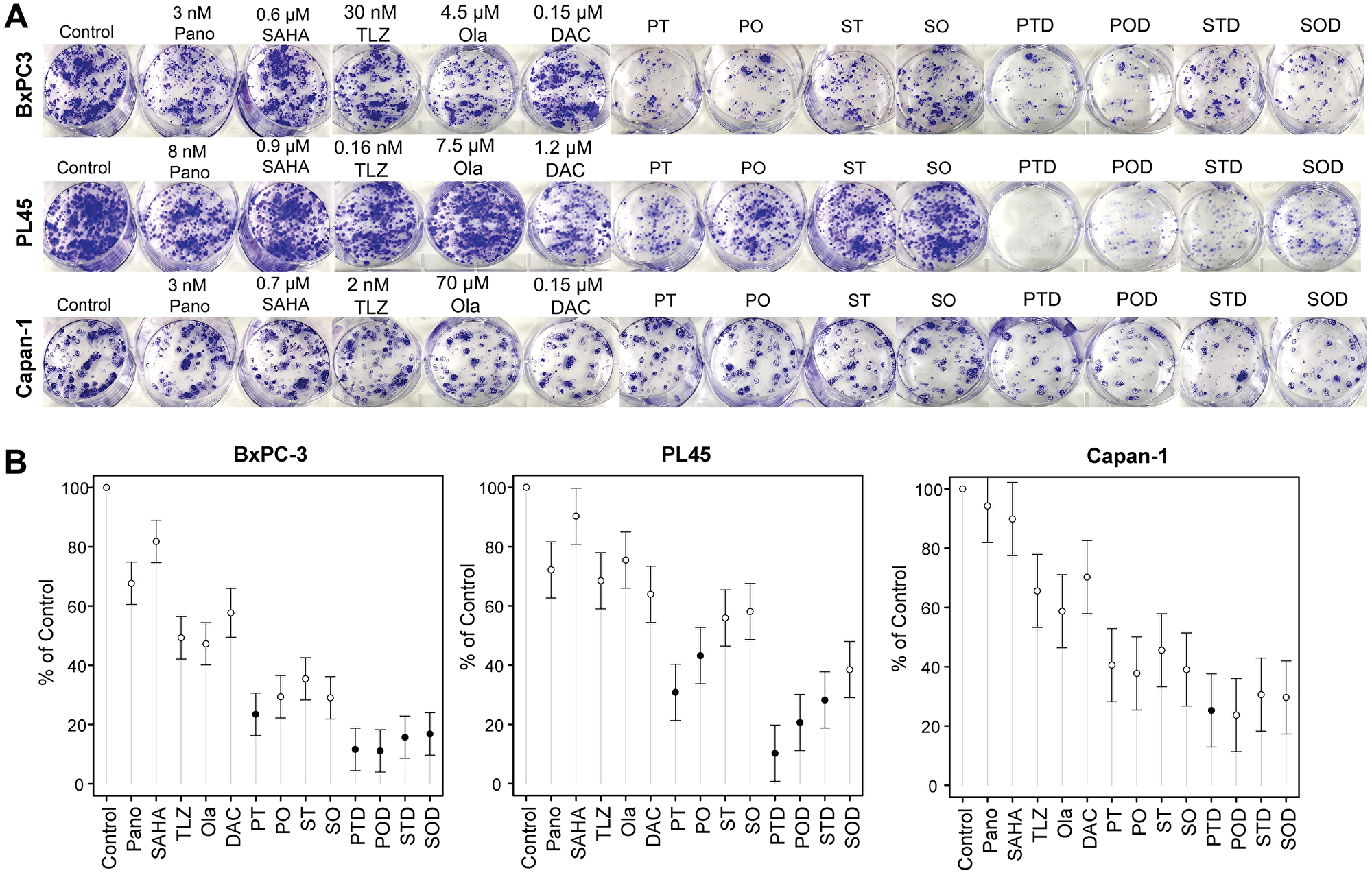
Figure 3: Colony formation assay.
Cells were seeded in 6-well plates overnight and exposed to individual drug or drug-combinations for 1–2 weeks and stained as described in the Materials and Methods (A). Colony formation is presented relative to control (B). Model-adjusted means are shown with 95% confidence intervals, and solid points indicate a significant synergistic difference from all the individual drugs (see Supplementary Table 3). Each cell line of each drug was modeled independently. Abbreviations: DAC/D: decitabine; Ola/O: olaparib; Pano/P: panobinostat; SAHA: vorinostat; TLZ/T: talazoparib.
Table 1: Colony formation and drug-mediated inhibition of cell proliferation in pancreatic cancer cell lines
| Colony formation | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BxPC-3 | PL45 | Capan-1 | ||||||||||||||
| Pano | SAHA | TLZ | Ola | DAC | Pano | SAHA | TLZ | Ola | DAC | Pano | SAHA | TLZ | Ola | DAC | ||
| Pano+TLZ | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | 0.1235 | ||||||||||
| Pano+Ola | <0.0001 | 0.0164 | 0.0015 | 0.0003 | <0.0001 | 0.3544 | ||||||||||
| SAHA+TLZ | <0.0001 | 0.1630 | <0.0001 | 0.7696 | 0.0001 | 0.4327 | ||||||||||
| SAHA+Ola | <0.0001 | 0.0135 | 0.0003 | 0.2393 | <0.0001 | 0.4640 | ||||||||||
| Pano+TLZ+DAC | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | 0.0005 | <0.0001 | |||||||
| Pano+Ola+DAC | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | 0.0041 | 0.0007 | |||||||
| SAHA+TLZ+DAC | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | 0.0041 | 0.0005 | |||||||
| SAHA+Ola+DAC | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | 0.0086 | <0.0001 | 0.0346 | <0.0001 | |||||||
| Drug-mediated inhibition of cell proliferation* | ||||||||||||||||
| Pano+TLZ+DAC | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | 0.0043 | <0.0001 | |||||||
| Pano+Ola+DAC | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | 0.0017 | <0.0001 | |||||||
| SAHA+TLZ+DAC | <0.0001 | 0.0020 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | 0.0007 | <0.0001 | |||||||
| SAHA+Ola+DAC | <0.0001 | 0.0083 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | 0.0003 | <0.0001 | |||||||
The results of the clonogenic assay were consistent with those of the MTT assay for cell proliferation. The addition of the hypomethylating agent decitabine to panobinostat + talazoparib resulted in ~60%, ~85%, and ~57% inhibition of cell proliferation in the BxPC-3, PL45, and Capan-1 cells, respectively; the addition of decitabine to panobinostat + olaparib resulted in ~54%, ~75%, and ~56% inhibition in BxPC-3, PL45, and Capan-1 cells, respectively (Figure 4A). Similar results were obtained when decitabine was combined with vorinostat + talazoparib, which caused ~43%, ~80%, and ~61% inhibition of proliferation in the BxPC-3, PL45, and Capan-1 cells, respectively; the addition of decitabine to vorinostat + olaparib resulted in ~41%, ~70%, and ~61% inhibition of cell proliferation in the BxPC-3, PL45, and Capan-1 cells, respectively (Figure 4A). The three-drug combinations were associated with statistically higher inhibition of proliferation compared to each drug alone (Table 1, Supplementary Table 3).
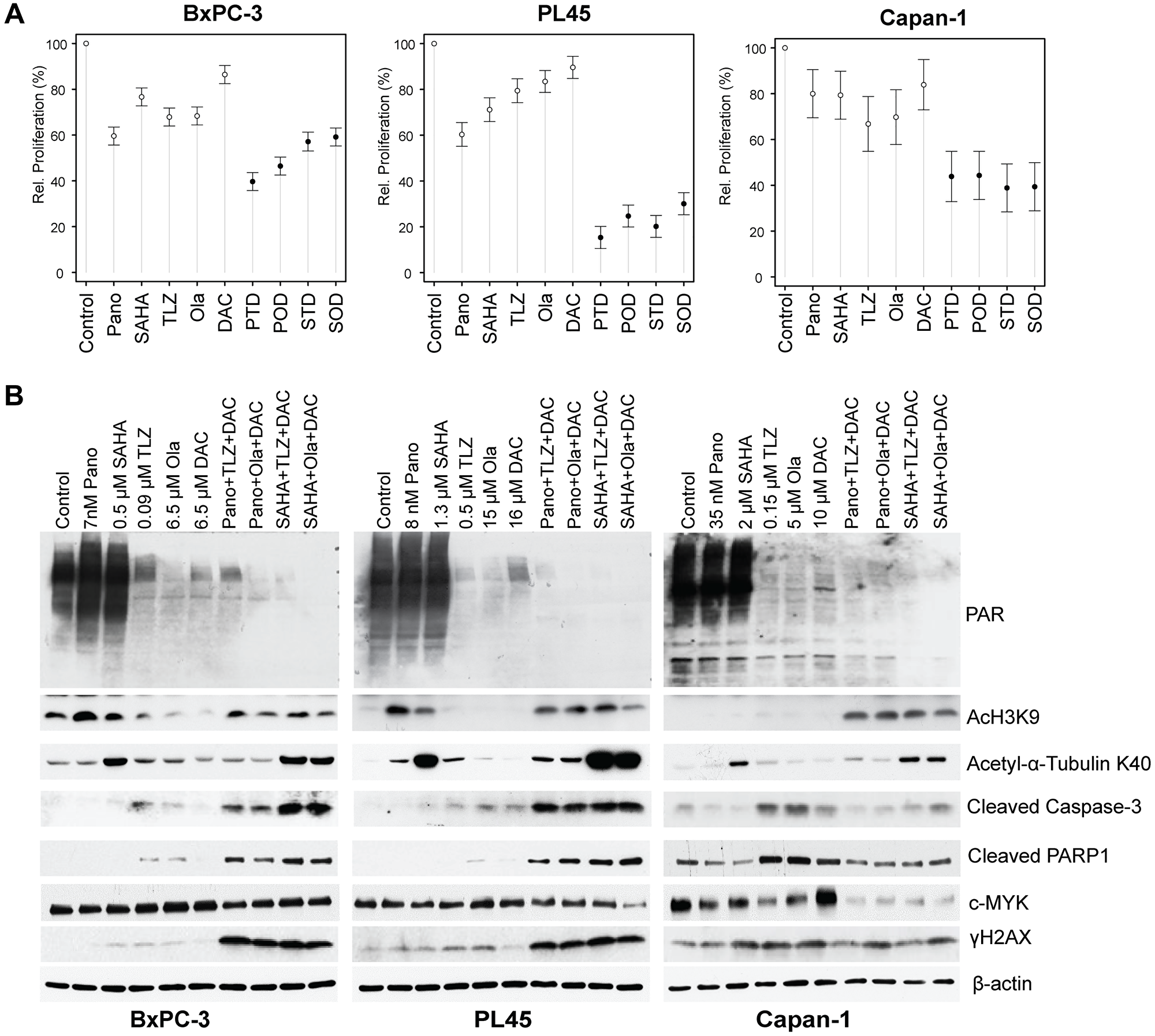
Figure 4: Drug-mediated inhibition of cell proliferation and PARylation, and effects on survival and apoptosis protein markers.
Cells were seeded in T25 flasks overnight and exposed to individual drugs or drug combinations for 3 days, harvested, and analyzed for cell proliferation by MTT assay (A) and Western blotting (B). Model-adjusted means are shown with 95% confidence intervals, and solid points indicate a significant synergistic difference from all the individual drugs (Supplementary Table 2). Each cell line of each drug was modeled independently. Abbreviations: Casp: caspase; DAC/D: decitabine; Ola/O: olaparib; Pano/P: panobinostat; SAHA: vorinostat; TLZ/T: talazoparib.
The three-drug combinations inhibit PARylation and enhance cleavage of caspase 3 and PARP1
Talazoparib and olaparib are potent inhibitors of PARP. We, therefore, sought to determine whether they also inhibit protein PARylation. While the HDACis panobinostat and vorinostat did not inhibit PARylation, talazoparib and olaparib decreased the PARylation levels in BxPC-3, PL45, and Capan-1 cells, and the addition of decitabine enhanced their inhibitory effects (Figure 4B). Surprisingly, decitabine alone also decreased the levels of PARylation in the three cell lines.
To investigate whether the decrease in colony formation (Figure 3A) and cell proliferation (Figure 4A) were associated with apoptosis, we analyzed the cleavage of caspase 3 and PARP1 (indicators of apoptosis). Figure 4B shows that the triple-drug combinations markedly enhanced both caspase 3 and PARP1 cleavage.
Cells exposed to the triple-drug combinations exhibited increased phosphorylation of histone 2AX. This finding indicates DNA damage response (double strand break formation and/or activation), likely attributed to activation of nuclear DNases, mediated by caspases [24]. These observations are consistent with a decreased level of pro-survival c-MYC in cells exposed to HDACI + PARPi + decitabine (Figure 4B).
HDACi, PARPi, and decitabine combinations affect the levels of proteins involved in DNA damage response and repair
Post-translational modifications (acetylation and PARylation) of proteins associated with DNA repair may affect their stability, as previously described for BRCA1 and UHFR1 [8, 11, 12]. The current study was conducted to assess the effect of HDACi, PARPi, and decitabine, as single agents or in combination, on the levels of the proteins associated with DNA damage response in the aforementioned pancreatic cancer cell lines. The findings indicated that when the cells were exposed to the three-drug combinations, there was a decrease in the levels of the ATM (ataxia-telangiectasia mutated) protein, that is responsible for DNA double-strand break repair and cell cycle checkpoint activation. Additionally, there was also a decrease in the level of BRCA1, which is involved in homologous recombination repair and in the level of the ATRX, a chromatin remodeling protein that participates in homologous recombination repair (Figure 5). In cells exposed to the three-drug combinations, a decrease in the non-homologous end joining repair proteins DNAPKcs, Artemis, and DNA ligase 1 levels was also noted. The cellular DNA damage response is regulated by the nucleosome remodeling and deacetylase (NuRD) complex, which plays a crucial role in chromatin remodeling and deacetylation processes [25] and controls DNA damage signaling and repair [26]. Analysis of some of the subunits of the NuRD complex showed decreased levels of the CHD3, CHD4, MTA1, RBAP46, and HDAC1 proteins in all cell lines exposed to the three-drug combinations (Figure 5). Overall, these results demonstrated that the three-drug combinations decreased the levels of proteins involved in DNA damage response.
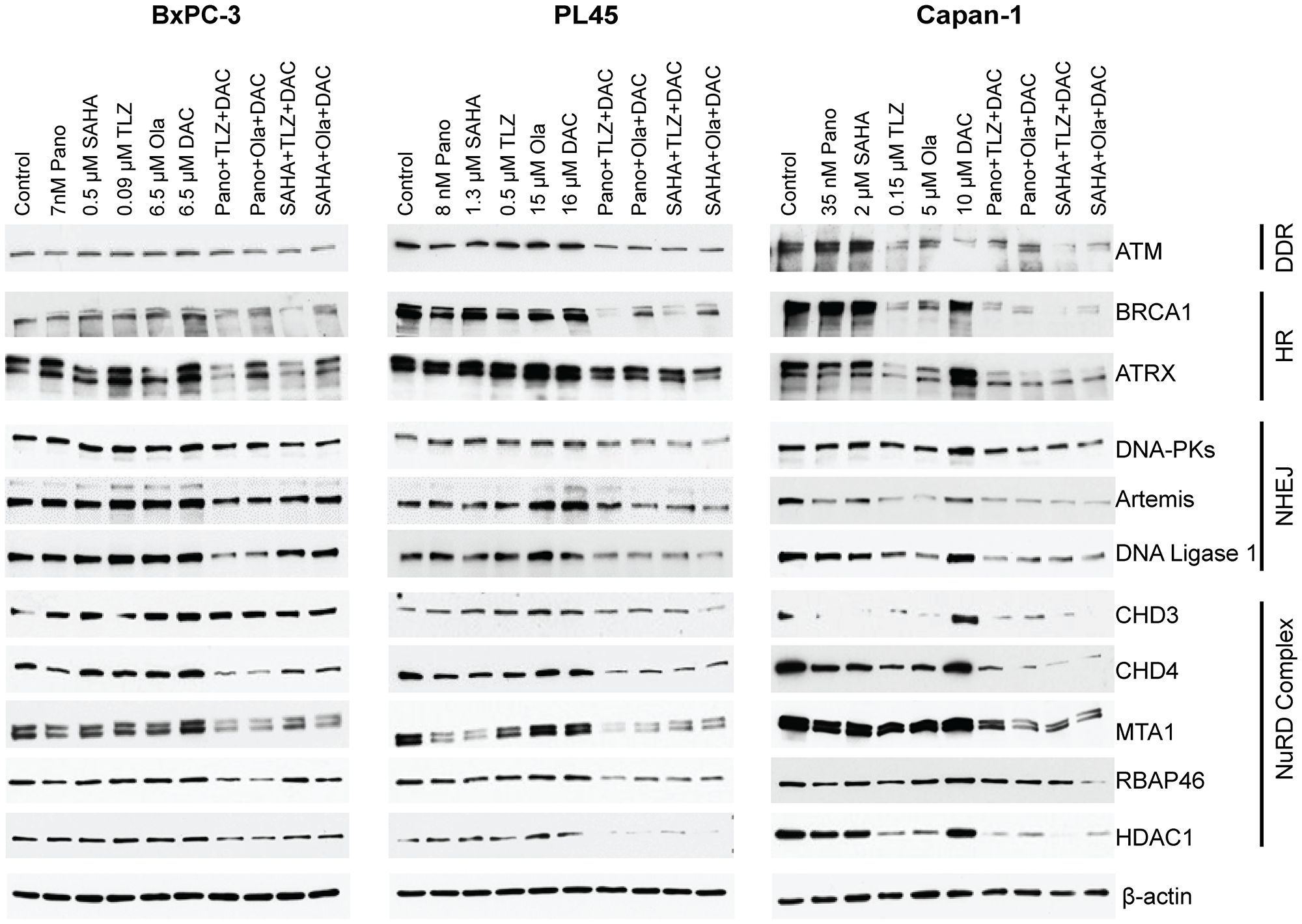
Figure 5: Effects of drugs on the levels of various proteins involved in DNA repair/DNA damage response.
Cells were exposed to the indicated drug concentrations for 3 days prior to analysis by Western blotting. Abbreviations: NuRD: nucleosome remodeling and deacetylase; DAC: decitabine; DDR: DNA damage response; HR: Homologous recombination; NHEJ: Non Homologous End Joining; NuRD: The Nucleosome Remodeling and Deacetylase (NuRD); Ola: olaparib; Pano: panobinostat; SAHA: vorinostat; TLZ: talazoparib.
DISCUSSION
This is the first study to demonstrate synergistic interactions between PARPis (olaparib or talazoparib) and panobinostat, vorinostat, and decitabine in pancreatic cancer cell lines with wild-type BRCA1/2 (BxPC-3 or PL45) or BRCA2 mutation (Capan-1). An intriguing finding of our study was the observation that the cytotoxicity of PARPis was noted across cell lines and not only in the BRCA2-mutated cell line (Capan-1). Synthetic lethality was only expected in Capan-1, but both the BxPC-3 and PL45 cell lines also showed sensitivity to talazoparib and olaparib. The PL45 cell line exhibited weaker sensitivity to these inhibitors compared to the BxPC-3 cell line (Figure 1). This difference in sensitivity to talazoparib and olaparib may be attributed to a non-functional TP53 mutation in the PL45 cell line, as previously reported [27]. Other investigators have reported similar results in a subset of colorectal cancer cells, which was partly attributed to wild-type TP53-mediated suppression of RAD51, a BRCA2-interacting protein [28].
The study demonstrated the following novel findings. The combination of an HDACi (panobinostat or vorinostat) with a PARPi (talazoparib or olaparib) resulted in synergistic cytotoxicity in all pancreatic cell lines tested, as evidenced by a CI of <1.0 for all combinations (Figure 2) and the results of the colony-formation assays (Figure 3A). The addition of the hypomethylating agent decitabine further increased the synergistic cytotoxicity noted with an HDACi (panobinostat or vorinostat) and a PARPi (talazoparib or olaparib) combination (Figure 3B). Several HDACis were investigated, mostly in preclinical studies, for the treatment of pancreatic cancer. M344 (a histone H3 deacetylation inhibitor) has demonstrated antitumor activity as a single agent or combined with gemcitabine in vitro and in vivo [29]. Trichostatin A has shown suppression of pancreatic adenocarcinoma cell growth, inducing G2 phase cell cycle arrest and apoptotic cell death in cell lines with a mutated p53 gene [30]. Vorinostat has exhibited increased gemcitabine-induced pancreatic cancer cell death through G1 cell cycle arrest [31]. In patients with resected pancreatic ductal cancer, the high expression of HDAC1 activity measured using an HDAC1 inhibitor assay in clinical samples was significantly associated with poor progression-free survival, and distant metastasis-free survival, and indicated that HDAC1 inhibitors may have activity in suppressing metastasis through inhibition of the epithelial-mesenchymal transition (EMT) [32]. CG200745 was shown to overcome resistance of pancreatic cancer cells to gemcitabine [33]. The dual inhibitors Metavert (inhibitor of GSK3B and HDACs) or CUDC-907 (Phosphoinositide 3-kinases/HDAC Inhibitor) and the HDAC inhibitor AES-135 have shown antitumor activity in mouse models of pancreatic cancer [34–36]. Other investigators added vorinostat to capecitabine and radiation therapy in patients with non-metastatic pancreatic cancer and reported that the treatment was well tolerated, and the median overall survival was 1.1 years [37]. Overall, these studies indicate the therapeutic potential of HDAC inhibitors in pancreatic cancer, paving the way for novel therapeutic approaches.
We also explored the mechanisms behind this enhanced cytotoxicity. We observed that the combination treatment caused more cell death by triggering apoptosis, as shown by the increased cleavage of caspase 3 and PARP1 (Figure 4B). The three-drug combination treatment (vorinostat, talazoparib, and decitabine; vorinostat, olaparib, and decitabine; panobinostat, talazoparib, and decitabine; or panobinostat, olaparib, and decitabine) also induced more DNA damage (compared to the individual drugs), as evidenced by the increased phosphorylation of histone 2AX. Furthermore, the combination treatment impaired the DNA repair pathways, as indicated by the decreased levels of ATM, BRCA1, and ATRX proteins (Figure 5). Additionally, the three-drug combination treatment altered the epigenetic regulation of gene expression, as suggested by the reduced levels of NuRD complex subunits. Other investigators have shown that in ovarian cancer cells, the addition of panobinostat to olaparib enhanced the efficacy of olaparib by modulating the expression of genes involved in homologous recombination repair and immune response, and the combination reduced tumor growth and increased tumor cell death, DNA damage, and CD8+ T cell infiltration [38].
Similarly, the combination of vorinostat and talazoparib caused substantial inhibition of pancreatic cancer cell proliferation, especially in PL45 and Capan-1 cells (Figure 3). Cleavage of caspase 3 and PARP1 (Figure 4B), which is indicative of apoptotic cell death, accompanied by the modulation of DNA repair proteins and the downregulation of non-homologous end-joining repair proteins (Figure 5), underscores the disruption of DNA repair processes. The modulation of DNA repair proteins and NuRD complex subunits (including CHD3, CHD4, MTA1, RBAP46, and HDAC1) highlights a comprehensive disruption of DNA repair and chromatin remodeling and epigenetic modifications. The phosphorylation of histone 2AX implies the presence of double-strand DNA breaks, possibly attributed to caspase-mediated activation of nuclear DNases, as previously reported [39]. The downregulation of NuRD complex subunits is consistent with previous reports [40–42], emphasizing the NuRD complex’s role in DNA damage response, DNA repair, and chromatin remodeling.
The combination of vorinostat and olaparib demonstrated synergistic effects against pancreatic cancer cells (Figure 2), with increased apoptotic cell death and DNA damage (Figures 4B and 5). Consistent with previous studies in different cancer types, such as triple-negative breast cancer (TNBC) and ovarian cancer, this finding highlights the potential efficacy of this combination [10, 43]. PTEN loss was suggested as a potential biomarker for predicting response to vorinostat and olaparib in patients with TNBC. Although the exact mechanism is not fully understood, it has been suggested that PTEN expression in TNBC cells can modulate the response to vorinostat and olaparib by affecting the PI3K/AKT/mTOR, HRR, p53, BAX, and autophagy pathways. The interaction of these pathways has synergistic anti-tumor effects in TNBC cells that express functional PTEN [10, 43]. Overall, these findings support the clinical evaluation of vorinostat and olaparib in advanced pancreatic cancer.
The sensitivity of BRCA-mutated pancreatic cancer cells to the drug combinations varied. As expected, Capan-1 displayed sensitivity to PARP inhibition, particularly to olaparib. The limited use of HDACis in solid tumors is attributed to pharmacokinetic challenges [44] and resistance mechanisms. In addition, HDACis have been reported to inhibit the functionality of the homologous recombination pathway, leading to a homologous recombination repair-deficient state and, thereby, increasing the effectiveness of PARPis [10]. In addition to olaparib [17], another PARPi, rucaparib, has demonstrated antitumor activity in patients with pancreatic cancer and a known deleterious BRCA mutation [45], but antitumor activity has not been noted with veliparib. Furthermore, azacitidine and oxaliplatin combination therapy demonstrated safety with no dose-limiting toxicity in platinum-resistant cancer [46]. Ongoing clinical trials exploring various PARPis aim to further enhance survival outcomes for pancreatic cancer patients.
Although possible mechanisms for the observed synergistic interactions are provided, some limitations of the study must be considered prior to translating these results into clinical trials. As an in vitro study, this cell line model cannot predict the complexities of potential interactions within a complex organ, including drug toxicities and pharmacodynamics. A three-dimensional cell model may be more effective in predicting drug effects on tumor tissues. Animal studies may be considered as an alternative, but they have their own inherent drawbacks. The effect of the drugs used in the present study on normal cells should also be considered. Nevertheless, the in vitro results presented in this study provide proof of concept for the synergistic interactions of HDACi, PARPi, and decitabine in pancreatic cancer cell lines.
In conclusion, the use of PARPis (olaparib or talazoparib) combined with HDACis (panobinostat, vorinostat), with or without decitabine, demonstrated synergistic cytotoxicity in pancreatic cell lines harboring wild-type or mutated BRCA1/2 genes. Further research is needed to understand the mechanisms underlying the observed synergistic effects and to identify biomarkers that can predict response to treatment. Collectively, our findings suggest that these combinations should be explored in clinical trials in pancreatic cancer.
Materials and Methods
Cell lines and drugs
The pancreatic cancer cell lines BxPC-3 (Catalog # CRL-1687), PL45 (CRL-2558), and Capan-1 (HTB-79) were purchased from American Type Culture Collection (ATCC; Manassas, VA, USA). BxPC-3 and PL45 cells possess wild-type BRCA1 and BRCA2 genes. Capan-1 cells lack a functional BRCA2 allele, with single base deletion at c.6174 resulting in a loss of the C-terminal 1416 amino acids of the proteins [23]. Following ATCC protocols, all cells were cultured in a 5% CO2 humidified incubator at 37°C. PL45 and Capan-1 cells were cultured in Dulbecco’s Modified Eagle Medium, while BxPC-3 cells were grown in Roswell Park Memorial Institute 1640 medium. Both media contained 10% heat-inactivated fetal bovine serum along with antibiotics (100 IU/mL penicillin and 100 μg/mL streptomycin).
The HDACis panobinostat and vorinostat, the PARPis talazoparib and olaparib, and the demethylating agent decitabine were obtained from Selleck Chemicals (Houston, TX, USA). Stock solutions were prepared using dimethyl sulfoxide, of which the final concentration did not exceed 0.1% of the total volume.
Cell proliferation assay and drug synergism
Cell proliferation was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. Briefly, 100 μL of cells (BxPC-3: 3 × 104 cells/mL; PL45: 3.8 × 104 cells/mL; Capan-1: 8 × 104 cells/mL) were seeded per well in a 96-well plate. After 24 hours, the medium was replaced with 100 μL of appropriate medium containing drug(s) aiming for IC10 to IC20 concentrations and incubated for 3 days. These doses are used to assess synergism between drugs, indicating inhibition of 10% to 20% of cell growth, respectively; a higher drug dose does not allow this assessment. The MTT assay was done by adding 30 μL of MTT (2 mg MTT/mL) in phosphate-buffered saline (PBS) per well and incubating for 4 hours at 37oC. The insoluble purple formazan product was dissolved by adding 100 μL of solubilization solution (0.1 N HCl in isopropanol containing 10% Triton X-100) to each well, mixing, and incubating at 37°C overnight. Absorbance at 570 nm was measured using a Victor X3 plate reader (Perkin Elmer Life and Analytical Sciences, Shelton, CT). The rate of cell proliferation was determined relative to the control cells exposed to solvent alone.
To determine drug synergism, cells were seeded in 96-well plates as described above. The medium was changed after 24 hours, and the cells were exposed to various drug combinations aiming for IC10 to IC20 concentrations for 3 days prior to the MTT assay. Fractions affected (Fa) refer to cell death which was determined using the MTT assay. Drug combination effects were estimated based on the combination index (CI) values calculated using CalcuSyn software (ComboSyn, Inc., Paramus, NJ, USA as previously described [47].
Colony formation assay
BxPC-3 (1 × 103 cells/mL), PL45 (1.2 × 103 cells/mL), and Capan-1 (3.3 × 103 cells/mL) cells were seeded (3 mL) onto 6-well plates. The next day, the medium was replaced with fresh medium containing drug(s) and incubated at 37°C for 3 days. Then, the medium was replaced with fresh medium without drugs. After 1–2 weeks, formed colonies were fixed using 4% glutaraldehyde for 20 minutes. The colonies were then washed thrice with PBS and stained using 0.5% crystal violet for 15 minutes. Excess stain was removed by washing twice with PBS. The procedures were done at room temperature, and the experiments were repeated at least three times.
Western blot analysis
Cells (6 mL) were seeded in T25 flasks (BxPC-3: 4.2 × 104 cells; PL45: 5 × 104 cells/mL; Capan-1: 13.3 × 104 cells) overnight. The next day, the old medium was replaced with fresh medium containing drug(s), and the cells were exposed continuously to drug(s) for 3 days. Cells were dissociated from the flask using accutase (MilliporeSigma, St. Louis, MO, USA), harvested, and washed with cold PBS. Cells were lysed with lysis buffer (Cell Signaling Technology, Danvers, MA, USA). Western blot analysis was performed as previously described [13]. Antibodies used for immunoblotting are described in the Supplementary Table 4. The β-actin protein was used as an internal control.
Statistical analysis
Separately for each drug and for each cell line, mixed effect analysis of variance was used to model the association between cellular proliferation percentage (percentage referenced to the zero-dose sample) and dose (6 discrete levels, excluding dose 0). The same cell culture was used as the basis of all doses of two experiments per day, and experiments were conducted on three separate days. Model-adjusted differences between dose levels were assessed by contrasts with Tukey-adjusted p-values using the emmeans package [48], with adjusted means weighted proportionally to covariate marginal frequencies. Mixed effect modeling utilized the nlme package [49, 50]. Cytotoxicity was modeled similarly with relation to drug treatment, separately by cell line, clustering on experiment day, with Hommel-adjusted p-values for comparisons between synergistic 3-way drug combinations and component drugs. Colony formation was modeled by analysis of variance with relation to drug treatment, separately by cell line, with Hommel-adjusted p-values for comparisons between 2-way and 3-way synergistic drug combinations and component drugs. All statistical modeling of proliferation was conducted using R statistical software (version 4.3.1); a 95% level of statistical confidence was assumed.
Abbreviations
ATM: Ataxia-Telangiectasia Mutated protein; ATRX: Alpha Thalassemia/Mental Retardation Syndrome X-Linked protein; BRCA: Breast Cancer gene; CI: Combination Index (used in drug synergism analysis); c-MYC: Cellular Myelocytomatosis oncogene; DAC: Decitabine; DCIS: Ductal carcinoma in situ; DDR: DNA damage response; DNAPKcs: DNA-dependent protein kinase catalytic subunit; Fa: Fractions affected (refers to cell death in drug synergism analysis); FDA: U.S. Food and Drug Administration; HDACis: histone deacetylase inhibitors; HR: Homologous recombination; IC50: Half-maximal inhibitory concentration; MTT: 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyl Tetrazolium Bromide; NHEJ: Non-Homologous End Joining; NuRD: Nucleosome Remodeling and Deacetylase complex; Ola: Olaparib; Pano: Panobinostat; PARPis: Poly(ADP Ribose) Polymerase Inhibitors; PBS: Phosphate-buffered saline; PD-(L)1: Programmed Death (Ligand) 1; SAHA: Vorinostat; TLZ: Talazoparib; TNF: Tumor Necrosis Factor; UHFR1: Ubiquitin-Specific Histone H2A and H2B Deubiquitinase Factor R1; γ-H2AX: Phosphorylated Histone H2AX.
ACKNOWLEDGMENTS
The authors are fully responsible for the content of this manuscript, and the views and opinions described in the manuscript reflect solely those of the authors.
CONFLICTS OF INTEREST
Apostolia M. Tsimberidou declares receipt of clinical trial research funding (to The University of Texas MD Anderson Cancer Center) from Agenus, IMMATICS, Novocure, OBI Pharma, Parker Institute for Cancer Immunotherapy, Tachyon, Tempus and Tvardi; fees for consulting or advisory roles for Avstera Therapeutics, Bioeclipse, BrYet, Diaccurate, Macrogenics, NEX-I, and VinceRx; and travel expenses from ASCO, Cancer Care Crossroads, GenomeWeb conference, and Precision Medicine World Conference. The remaining authors declare no relevant conflict of interest.
FUNDING
This work was supported by funds given generously to A.M.T. from Mr. and Mrs. Steven McKenzie’s Endowment and donor funds from Jamie’s Hope and Mrs. and Mr. Zane W. Arrott for Dr. Tsimberidou’s Personalized Medicine Program at The University of Texas MD Anderson Cancer Center. The laboratory work was also supported by Dr. Andersson’s Fund from Steven and Lavinia Boyd for Leukemia Research. The work of the authors is also supported in part by the NIH National Cancer Institute award number P30 CA016672 (to The University of Texas MD Anderson Cancer Center).
References
1. Kuo MH, Allis CD. Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays. 1998; 20:615–26. https://doi.org/10.1002/(SICI)1521-1878(199808)20:8<615::AID-BIES4>3.0.CO;2-H. [PubMed].
2. Glozak MA, Seto E. Histone deacetylases and cancer. Oncogene. 2007; 26:5420–32. https://doi.org/10.1038/sj.onc.1210610. [PubMed].
3. Kim HJ, Bae SC. Histone deacetylase inhibitors: molecular mechanisms of action and clinical trials as anti-cancer drugs. Am J Transl Res. 2011; 3:166–79. [PubMed].
4. Nieto Y, Valdez BC, Thall PF, Jones RB, Wei W, Myers A, Hosing C, Ahmed S, Popat U, Shpall EJ, Qazilbash M, Gulbis A, Anderlini P, et al. Double epigenetic modulation of high-dose chemotherapy with azacitidine and vorinostat for patients with refractory or poor-risk relapsed lymphoma. Cancer. 2016; 122:2680–88. https://doi.org/10.1002/cncr.30100. [PubMed].
5. Valdez BC, Li Y, Murray D, Brammer JE, Liu Y, Hosing C, Nieto Y, Champlin RE, Andersson BS. Differential effects of histone deacetylase inhibitors on cellular drug transporters and their implications for using epigenetic modifiers in combination chemotherapy. Oncotarget. 2016; 7:63829–38. https://doi.org/10.18632/oncotarget.11561. [PubMed].
6. Bachy E, Camus V, Thieblemont C, Sibon D, Casasnovas RO, Ysebaert L, Damaj G, Guidez S, Pica GM, Kim WS, Lim ST, André M, García-Sancho AM, et al. Romidepsin Plus CHOP Versus CHOP in Patients With Previously Untreated Peripheral T-Cell Lymphoma: Results of the Ro-CHOP Phase III Study (Conducted by LYSA). J Clin Oncol. 2022; 40:242–51. https://doi.org/10.1200/JCO.21.01815. [PubMed].
7. Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003; 421:499–506. https://doi.org/10.1038/nature01368. [PubMed].
8. Ha K, Fiskus W, Choi DS, Bhaskara S, Cerchietti L, Devaraj SG, Shah B, Sharma S, Chang JC, Melnick AM, Hiebert S, Bhalla KN. Histone deacetylase inhibitor treatment induces ‘BRCAness’ and synergistic lethality with PARP inhibitor and cisplatin against human triple negative breast cancer cells. Oncotarget. 2014; 5:5637–50. https://doi.org/10.18632/oncotarget.2154. [PubMed].
9. Baldan F, Mio C, Allegri L, Puppin C, Russo D, Filetti S, Damante G. Synergy between HDAC and PARP Inhibitors on Proliferation of a Human Anaplastic Thyroid Cancer-Derived Cell Line. Int J Endocrinol. 2015; 2015:978371. https://doi.org/10.1155/2015/978371. [PubMed].
10. Min A, Im SA, Kim DK, Song SH, Kim HJ, Lee KH, Kim TY, Han SW, Oh DY, Kim TY, O’Connor MJ, Bang YJ. Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (SAHA), enhances anti-tumor effects of the poly (ADP-ribose) polymerase (PARP) inhibitor olaparib in triple-negative breast cancer cells. Breast Cancer Res. 2015; 17:33. https://doi.org/10.1186/s13058-015-0534-y. [PubMed].
11. Yin L, Liu Y, Peng Y, Peng Y, Yu X, Gao Y, Yuan B, Zhu Q, Cao T, He L, Gong Z, Sun L, Fan X, Li X. Correction to: PARP inhibitor veliparib and HDAC inhibitor SAHA synergistically co-target the UHRF1/. BRCA1 DNA damage repair complex in prostate cancer cells. J Exp Clin Cancer Res. 2022; 41:72. https://doi.org/10.1186/s13046-022-02290-9. [PubMed].
12. Liszczak G, Diehl KL, Dann GP, Muir TW. Acetylation blocks DNA damage-induced chromatin ADP-ribosylation. Nat Chem Biol. 2018; 14:837–40. https://doi.org/10.1038/s41589-018-0097-1. [PubMed].
13. Valdez BC, Nieto Y, Yuan B, Murray D, Andersson BS. HDAC inhibitors suppress protein poly(ADP-ribosyl)ation and DNA repair protein levels and phosphorylation status in hematologic cancer cells: implications for their use in combination with PARP inhibitors and chemotherapeutic drugs. Oncotarget. 2022; 13:1122–35. https://doi.org/10.18632/oncotarget.28278. [PubMed].
14. Holter S, Borgida A, Dodd A, Grant R, Semotiuk K, Hedley D, Dhani N, Narod S, Akbari M, Moore M, Gallinger S. Germline BRCA Mutations in a Large Clinic-Based Cohort of Patients With Pancreatic Adenocarcinoma. J Clin Oncol. 2015; 33:3124–29. https://doi.org/10.1200/JCO.2014.59.7401. [PubMed].
15. Brand R, Borazanci E, Speare V, Dudley B, Karloski E, Peters MLB, Stobie L, Bahary N, Zeh H, Zureikat A, Hogg M, Lee K, Tsung A, et al. Prospective study of germline genetic testing in incident cases of pancreatic adenocarcinoma. Cancer. 2018; 124:3520–27. https://doi.org/10.1002/cncr.31628. [PubMed].
16. Lowery MA, Wong W, Jordan EJ, Lee JW, Kemel Y, Vijai J, Mandelker D, Zehir A, Capanu M, Salo-Mullen E, Arnold AG, Yu KH, Varghese AM, et al. Prospective Evaluation of Germline Alterations in Patients With Exocrine Pancreatic Neoplasms. J Natl Cancer Inst. 2018; 110:1067–74. https://doi.org/10.1093/jnci/djy024. [PubMed].
17. Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, Park JO, Hochhauser D, Arnold D, Oh DY, Reinacher-Schick A, Tortora G, Algül H, et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N Engl J Med. 2019; 381:317–27. https://doi.org/10.1056/NEJMoa1903387. [PubMed].
18. Kindler HL, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, Park JO, Hochhauser D, Arnold D, Oh DY, Reinacher-Schick A, Tortora G, Algül H, et al. Overall Survival Results From the POLO Trial: A Phase III Study of Active Maintenance Olaparib Versus Placebo for Germline BRCA-Mutated Metastatic Pancreatic Cancer. J Clin Oncol. 2022; 40:3929–39. https://doi.org/10.1200/JCO.21.01604. [PubMed].
19. Stresemann C, Lyko F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int J Cancer. 2008; 123:8–13. https://doi.org/10.1002/ijc.23607. [PubMed].
20. Estey EH. Epigenetics in clinical practice: the examples of azacitidine and decitabine in myelodysplasia and acute myeloid leukemia. Leukemia. 2013; 27:1803–12. https://doi.org/10.1038/leu.2013.173. [PubMed].
21. Jüttermann R, Li E, Jaenisch R. Toxicity of 5-aza-2’-deoxycytidine to mammalian cells is mediated primarily by covalent trapping of DNA methyltransferase rather than DNA demethylation. Proc Natl Acad Sci U S A. 1994; 91:11797–801. https://doi.org/10.1073/pnas.91.25.11797. [PubMed].
22. Mottini C, Tomihara H, Carrella D, Lamolinara A, Iezzi M, Huang JK, Amoreo CA, Buglioni S, Manni I, Robinson FS, Minelli R, Kang Y, Fleming JB, et al. Predictive Signatures Inform the Effective Repurposing of Decitabine to Treat KRAS-Dependent Pancreatic Ductal Adenocarcinoma. Cancer Res. 2019; 79:5612–25. https://doi.org/10.1158/0008-5472.CAN-19-0187. [PubMed].
23. Goggins M, Schutte M, Lu J, Moskaluk CA, Weinstein CL, Petersen GM, Yeo CJ, Jackson CE, Lynch HT, Hruban RH, Kern SE. Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Cancer Res. 1996; 56:5360–64. [PubMed].
24. Liu X, Zou H, Slaughter C, Wang X. DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell. 1997; 89:175–84. https://doi.org/10.1016/s0092-8674(00)80197-x. [PubMed].
25. Gursoy-Yuzugullu O, House N, Price BD. Patching Broken DNA: Nucleosome Dynamics and the Repair of DNA Breaks. J Mol Biol. 2016; 428:1846–60. https://doi.org/10.1016/j.jmb.2015.11.021. [PubMed].
26. Smeenk G, Wiegant WW, Vrolijk H, Solari AP, Pastink A, van Attikum H. The NuRD chromatin-remodeling complex regulates signaling and repair of DNA damage. J Cell Biol. 2010; 190:741–49. https://doi.org/10.1083/jcb.201001048. [PubMed].
27. Ryu B, Jones J, Blades NJ, Parmigiani G, Hollingsworth MA, Hruban RH, Kern SE. Relationships and differentially expressed genes among pancreatic cancers examined by large-scale serial analysis of gene expression. Cancer Res. 2002; 62:819–26. [PubMed].
28. Kwon Y, Rösner H, Zhao W, Selemenakis P, He Z, Kawale AS, Katz JN, Rogers CM, Neal FE, Badamchi Shabestari A, Petrosius V, Singh AK, Joel MZ, et al. DNA binding and RAD51 engagement by the BRCA2 C-terminus orchestrate DNA repair and replication fork preservation. Nat Commun. 2023; 14:432. https://doi.org/10.1038/s41467-023-36211-x. [PubMed].
29. Knoche SM, Brumfield GL, Goetz BT, Sliker BH, Larson AC, Olson MT, Poelaert BJ, Bavari A, Yan Y, Black JD, Solheim JC. The histone deacetylase inhibitor M344 as a multifaceted therapy for pancreatic cancer. PLoS One. 2022; 17:e0273518. https://doi.org/10.1371/journal.pone.0273518. [PubMed].
30. Donadelli M, Costanzo C, Faggioli L, Scupoli MT, Moore PS, Bassi C, Scarpa A, Palmieri M. Trichostatin A, an inhibitor of histone deacetylases, strongly suppresses growth of pancreatic adenocarcinoma cells. Mol Carcinog. 2003; 38:59–69. https://doi.org/10.1002/mc.10145. [PubMed].
31. Arnold NB, Arkus N, Gunn J, Korc M. The histone deacetylase inhibitor suberoylanilide hydroxamic acid induces growth inhibition and enhances gemcitabine-induced cell death in pancreatic cancer. Clin Cancer Res. 2007; 13:18–26. https://doi.org/10.1158/1078-0432.CCR-06-0914. [PubMed].
32. Shinke G, Yamada D, Eguchi H, Iwagami Y, Asaoka T, Noda T, Wada H, Kawamoto K, Gotoh K, Kobayashi S, Takeda Y, Tanemura M, Mori M, Doki Y. Role of histone deacetylase 1 in distant metastasis of pancreatic ductal cancer. Cancer Sci. 2018; 109:2520–31. https://doi.org/10.1111/cas.13700. [PubMed].
33. Lee HS, Park SB, Kim SA, Kwon SK, Cha H, Lee DY, Ro S, Cho JM, Song SY. A novel HDAC inhibitor, CG200745, inhibits pancreatic cancer cell growth and overcomes gemcitabine resistance. Sci Rep. 2017; 7:41615. https://doi.org/10.1038/srep41615. [PubMed].
34. Edderkaoui M, Chheda C, Soufi B, Zayou F, Hu RW, Ramanujan VK, Pan X, Boros LG, Tajbakhsh J, Madhav A, Bhowmick NA, Wang Q, Lewis M, et al. An Inhibitor of GSK3B and HDACs Kills Pancreatic Cancer Cells and Slows Pancreatic Tumor Growth and Metastasis in Mice. Gastroenterology. 2018; 155:1985–98.e5. https://doi.org/10.1053/j.gastro.2018.08.028. [PubMed].
35. Liu S, Zhao S, Dong Y, Wang T, Niu X, Zhao L, Wang G. Antitumor activity and mechanism of resistance of the novel HDAC and PI3K dual inhibitor CUDC-907 in pancreatic cancer. Cancer Chemother Pharmacol. 2021; 87:415–23. https://doi.org/10.1007/s00280-020-04210-0. [PubMed].
36. Shouksmith AE, Shah F, Grimard ML, Gawel JM, Raouf YS, Geletu M, Berger-Becvar A, de Araujo ED, Luchman HA, Heaton WL, Bakhshinyan D, Adile AA, Venugopal C, et al. Identification and Characterization of AES-135, a Hydroxamic Acid-Based HDAC Inhibitor That Prolongs Survival in an Orthotopic Mouse Model of Pancreatic Cancer. J Med Chem. 2019; 62:2651–65. https://doi.org/10.1021/acs.jmedchem.8b01957. [PubMed].
37. Chan E, Arlinghaus LR, Cardin DB, Goff L, Berlin JD, Parikh A, Abramson RG, Yankeelov TE, Hiebert S, Merchant N, Bhaskara S, Chakravarthy AB. Phase I trial of vorinostat added to chemoradiation with capecitabine in pancreatic cancer. Radiother Oncol. 2016; 119:312–18. https://doi.org/10.1016/j.radonc.2016.04.013. [PubMed].
38. Wilson AJ, Gupta VG, Liu Q, Yull F, Crispens MA, Khabele D. Panobinostat enhances olaparib efficacy by modifying expression of homologous recombination repair and immune transcripts in ovarian cancer. Neoplasia. 2022; 24:63–75. https://doi.org/10.1016/j.neo.2021.12.002. [PubMed].
39. Podhorecka M, Skladanowski A, Bozko P. H2AX Phosphorylation: Its Role in DNA Damage Response and Cancer Therapy. J Nucleic Acids. 2010; 2010:920161. https://doi.org/10.4061/2010/920161. [PubMed].
40. Basta J, Rauchman M. The nucleosome remodeling and deacetylase complex in development and disease. Transl Res. 2015; 165:36–47. https://doi.org/10.1016/j.trsl.2014.05.003. [PubMed].
41. Lai AY, Wade PA. Cancer biology and NuRD: a multifaceted chromatin remodelling complex. Nat Rev Cancer. 2011; 11:588–96. https://doi.org/10.1038/nrc3091. [PubMed].
42. Liu B, Yip RKh, Zhou Z. Chromatin remodeling, DNA damage repair and aging. Curr Genomics. 2012; 13:533–47. https://doi.org/10.2174/138920212803251373. [PubMed].
43. Konstantinopoulos PA, Wilson AJ, Saskowski J, Wass E, Khabele D. Suberoylanilide hydroxamic acid (SAHA) enhances olaparib activity by targeting homologous recombination DNA repair in ovarian cancer. Gynecol Oncol. 2014; 133:599–606. https://doi.org/10.1016/j.ygyno.2014.03.007. [PubMed].
44. Rasheed WK, Johnstone RW, Prince HM. Histone deacetylase inhibitors in cancer therapy. Expert Opin Investig Drugs. 2007; 16:659–78. https://doi.org/10.1517/13543784.16.5.659. [PubMed].
45. Shroff RT, Hendifar A, McWilliams RR, Geva R, Epelbaum R, Rolfe L, Goble S, Lin KK, Biankin AV, Giordano H, Vonderheide RH, Domchek SM. Rucaparib Monotherapy in Patients With Pancreatic Cancer and a Known Deleterious BRCA Mutation. JCO Precis Oncol. 2018; 2:1–15. https://doi.org/10.1200/PO.17.00316. [PubMed].
46. Tsimberidou AM, Said R, Culotta K, Wistuba I, Jelinek J, Fu S, Falchook G, Naing A, Piha-Paul S, Zinner R, Siddik ZH, He G, Hess K, et al. Phase I study of azacitidine and oxaliplatin in patients with advanced cancers that have relapsed or are refractory to any platinum therapy. Clin Epigenetics. 2015; 7:29. https://doi.org/10.1186/s13148-015-0065-5. [PubMed].
47. Wildner O, Blaese RM, Morris JC. Synergy between the herpes simplex virus tk/ganciclovir prodrug suicide system and the topoisomerase I inhibitor topotecan. Hum Gene Ther. 1999; 10:2679–87. https://doi.org/10.1089/10430349950016726. [PubMed].
48. Lenth R. Estimated Marginal Means, aka Least-Squares Means. 2023.
49. Pinheiro JC, Bates DM. Mixed-Effects Models in S and S-PLUS. 2000.
50. Pinheiro JC, Bates DM, and R Core Team. Linear and Nonlinear Mixed Effects Models. 2023.