
Introduction
Colorectal cancer (CoCa) ranks on the 3rd position of cancer incidence worldwide and accounts for the 4th most common cancer death [1]. Though prognosis is improving with a 65% cure rate in high income countries, death rate still ranges between 20.1 for men and 12.2 for women / 100.000 persons in western countries [1-3]. Like for most cancer types, it is suggested that a small population of cancer initiating cells (CIC) [4] accounts for primary tumor growth as well as metastatic spread [5].
CIC are slowly progressing through the cell cycle [6], are highly radiation and drug resistant [7] and may use signaling pathways guiding the fate of embryonic and adult stem cells [8]. CIC can also be defined by a set of markers, which for CoCIC include CD44/CD44v6, EpCAM (EpC), Lgr5, CD133, CD166, Msi-1, CD29, CD24 [9], Tspan8 and claudin7 (cld7) [10]. The contribution of these markers to oncogenesis and/or tumor progression was, so far, unequivocally demonstrated only for CD44 [11,12]. But great efforts are undertaken to elaborate, whereby CIC account for tumor progression and therapy resistance, as it is hoped that attacking CIC offers more efficient therapeutics [13].
From the described CoCIC markers we are particularly interested in EpC, which interferes with E-cadherin mediated cell-cell adhesion via disrupting the link between β-catenin and F-actin [14]. EpC is also engaged in Wnt/β-catenin signaling [15], down-regulation of PKC [16] and regulation of MMP7 expression [17,18]. Furthermore, EpC cross-linking triggers TACE (TNFα converting enzyme) and PS2/NTF (presenilin 2 N-terminal fragment). The latter cleaves the intracellular domain of EpC, EpIC, which together with β-catenin, FHL2 (four-and-half-LIM-only) and Lef-1, relocates to the nucleus and initiates, besides others, c-myc, cyclinA and E [19,20], Oct4 and Nanog transcription [21]. EpIC also contributes to the process of epithelial-mesenchymal transition (EMT) by upregulation of vimentin, Snail, Slug and downregulation of E-cadherin [22]. We experienced that in colon and pancreatic cancer, EpC associates with cld7 [23], also described for hepatocyte progenitors [24].
Claudins, tight junction proteins [25], are partitioned into glycolipid-enriched membrane microdomains (GEM) upon palmitoylation, where they interact with scaffold proteins creating a platform for signal transduction and a linkage to the cytoskeleton [26]. Claudins are targets for several phosphokinases [27,28], cld phosphorylation prohibiting integration into tight junctions [29]. In line with this, a cld7 knockout (cld7ko) is lethal within 10 days after birth due to destruction of the intestine [30]. The authors speculate on the importance of a missing association with integrins and a striking upregulation of MMP3 contributing to gut destruction [30]. Notably, an EpCko also is associated with intestine destruction-promoted death within one week after birth, due to the missing association of EpC with cld7 [31].
Based on our experience in a rat pancreatic adenocarcinoma model, where the metastatic capacity was strongly impaired by an EpC knockdown (EpCkd) or a cld7kd [10] we suggested that cld7 and EpC act as a tandem in tumor progression. To substantiate our hypothesis, cld7 was knocked down in 2 CoCa lines highly expressing EpC and cld7. We show that the EpC-cld7 association particularly contributes to EMT and that exosomes derived from EpC-cld7 competent CoCa suffice for EMT induction.
Results
EpC is known as a CoCIC marker [9], where we recently provided evidence in a rat tumor model that a concerted action of cld7 and EpC is required for tumor progression / metastasis formation [10]. Thus, we now asked, using human CoCa lines, for the underlying mechanism.
The impact of cld7 on EpC and other potential CoCIC markers
To support the hypothesis that cld7 contributes to tumor progression-related activities of EpC, which frequently is associated with cld7 in CoCa, cld7 was knocked-down by shRNA transfection in the human CoCa lines HT29 and SW948, controlling common CIC features. The cld7kd did not significantly affect EpC expression (Fig.1A). However, HT29-cld7kd cells form less filipodia and lamellae than HT29wt cells. The cell-cell contact of SW948-cld7kd cells is loosened compared to SW948wt cells and cells grow in multiple layers (Fig.1B). Besides a slight downregulation of CD184, which is weakly expressed by both lines, expression of additional CIC markers remained unimpaired (Fig.1C). Instead, the cld7kd affected EpC recruitment in glycolipid-enriched, light sucrose density fractions (GEM) (Fig.1D). This is important, as GEM-located EpC does not form tetramers [23], tetramers being required for homophilic binding [14]. Co-immunoprecipitation of EpC with GEM-located proteins like the tetraspanin Tspan8 was strongly reduced (Fig.1E,1F). This suggested that in cld7kd cells not only the membrane subdomain localization of EpC, but also the associations of EpC with additional membrane molecules are distorted (Fig.1E,1F).
Taken together, reduced cld7 expression hardly affects expression of additional CoCIC markers. However, the GEM-recruitment of EpC is strongly affected by reduced cld7 expression.
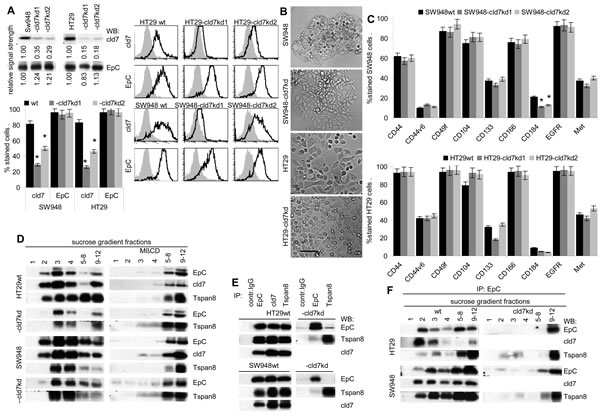
Figure 1: Characterization of cld7kd colorectal cancer cells. (A) WB and flow cytometry of SW948 and HT29 cells that were stably transfected with cld7 shRNA. Two clones of each line were selected for further experiments. WB and flow cytometry (mean % stained cells±SD, 3 assays and examples) of parental cells and two cld7kd clones are shown. (B) Morphology of SW948wt and HT29wt and -cld7kd cells (bar size: 25µm). (C) Expression of additional CoCIC markers in SW948wt and HT29wt and -cld7kd clones (mean % stained cells±SD). (A,C): significant differences between wt and cld7kd cells: *. (D) Sucrose density gradient fractionation of SW948wt and HT29wt and -cld7kd lysates and WB of light density fractions 1-4 and pools of fractions 5-8 and 9-12 with anti-EpC, anti-cld7 and as control for a GEM-located molecule with anti-Tspan8. (E) Lysate of SW948wt and HT29wt and -cld7kd cells were immunoprecipitated with control IgG, anti-EpCAM, anti-cld7 and anti-Tspan8. After SDS-PAGE separation, immunoprecipitates were blotted with anti-EpC, anti-cld7 and anti-Tspan8. (F) Immunoprecipitates as in (E) were separated by sucrose density gradient centrifugation. Light and pooled heavy fractions were separated by SDS-PAGE and blotted with anti-EpC, anti-cld7 and anti-Tspan8. A knockdown of cld7 in CoCa lines is not accompanied by downregulated expression of directly cld7-associated EpC and expression of additional CoCIC markers is not severely affected. However, only a minor part of EpC remains located in light density fractions (GEM).
Cld7 expression and CIC growth features of CoCa lines
CIC are characterized by anchorage-independent growth that was reduced by 50% in HT29-cld7kd clones and by ~30% in SW948-cld7kd clones (Fig.2A). Furthermore, the capacity of SW948-cld7kd cells to grow as spheres was reduced over 10-fold compared to SW948wt cells. When selecting first passage spheres and seeding them again under spheroid growth conditions, the percentage of spheres derived from SW948wt 1st passage spheres increased significantly. Instead, SW948-cld7kd 1st passage spheres did not gain in sphere forming capacity upon repassage. Similarly, the capacity of HT29wt cells to grow as holoclones was reduced in HT29-cld7kd clones and was only slightly increased upon repassage, whereas the number of repassaged HT29wt 1st passage holoclones was more than doubled upon repassage (Fig.2B). Notably, cld7 and EpC expression as well as expression of the CIC markers CD44v6 and Tspan8 was significantly increased in SW948 spheres and HT29 holoclones (Fig.2C).
The proliferation rate of HT29-cld7kd and SW948-cld7kd cells was slightly, but at least in HT29-cld7kd cells significantly enhanced, whereas the proliferation rate of holoclones and spheres was slightly reduced (Fig.2D). Cell cycle progression was accelerated in HT29-cld7kd and SW948-cld7kd compared to wt cells and was reduced in HT29 holoclones (Fig.2E).
Thus, suggested CIC growth features are affected in cld7kd CoCa lines. This accounts for sphere and holoclone formation and anchorage independent growth. As in spheres and holoclones high cld7 expression was seen in >90% of cells, spheres / holoclones of these two lines are well suited as internal controls for the cld7kd.
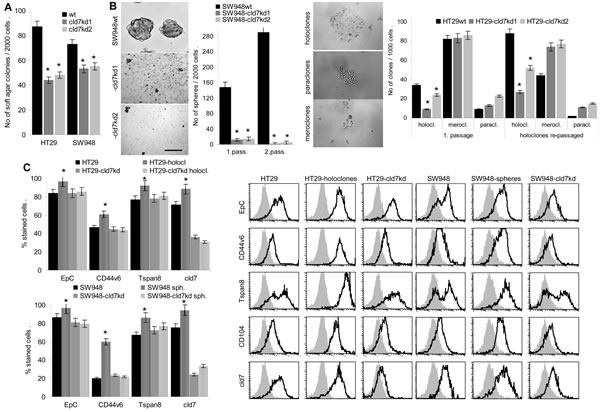
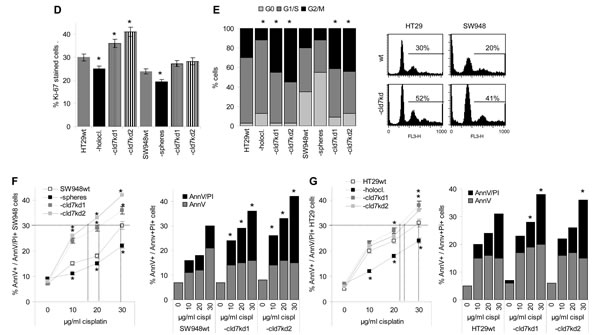
Figure 2: CIC growth features of CoCa-cld7kd cells. (A) No (mean±SD, 3 assays) of soft agar colonies of wt and cld7kd cells; (B) SW948 cells were grown under spheroid growth condition and HT29 cells under condition for holoclone formation. Examples and mean numbers±SD (3 assays) of spheres (scale bar: 100µm) and holoclones, meroclones and paraclones (scale bar: 30µm) in a first round of cloning as well as after re-seeding 1st passage spheres and holoclones are shown; (C) Expression of EpCAM, cld7, CD44v6 and Tspan8 in 1st and 2nd passage spheres and holoclones was evaluated by flow cytometry; % stained cells (±SD, 3 assays) and representative examples; (D) Ki-67 expression was evaluated by flow cytometry in wt, cld7kd and holoclone- / sphere-derived cells; the mean percent±SD of stained cells is shown; (E) Cell cycle progression was evaluated by PI staining after starved cells had recovered for 2h in medium with FCS. The % of cells in G0, G1/S and G2/M and a representative example are shown. (F,G) SW948wt and HT29wt, -cld7kd and holoclone- / sphere-derived cells were cultured for 48h in the presence of titrated amounts of cisplatin. Apoptosis was determined after AnnV/PI staining by flow cytometry; % of cells stained for AnnV and AnnV plus PI (mean±SD, triplicates) and % of cells stained for AnnV or AnnV plus PI (mean of triplicates); (A-G) significant differences between wt versus cld7kd and sphere- / holoclone-derived cells: *. Anchorage-independent growth and sphere / holoclone formation are impaired in cld7kd cells, which is accompanied by a slightly increased proliferation rate, more rapid cycling and a distinct loss in apoptosis resistance.
Cld7 expression and CIC apoptosis-resistance of CoCa lines
Cisplatin resistance of SW948 spheres and HT29 holoclones was reduced as compared to wt cells. But cisplatin resistance of SW948-cld7kd and, less pronounced, HT29-cld7kd cells was impaired compared to wt cells. This is demonstrated for mitochondrial integrity, 3H-thymidine uptake (Suppl.Fig.1A-1D) and AnnV binding / PI uptake. While 25-30µg / ml cisplatin were required for a 30% death rate of SW948 and HT29, 15-20µg / ml cisplatin sufficed for a 30% death rate of SW948-cld7kd and 20-22µg / ml for HT29-cld7kd clones. Instead, only 22% of SW948 spheres and 24% of HT29 holoclones became apoptotic in the presence of 30µg/ml cisplatin (Fig.2F,2G). Evaluation of caspase activity in cisplatin-treated HT29wt and SW948wt and cld7kd cells showed unaltered Casp8 expression; cleaved Casp9 and activated Casp3 were slightly upregulated in cld7kd clones. Furthermore, PI3K, Akt and BAD phosphorylation was reduced. Accordingly, Bcl2 and BclXl (SW948-cld7kd) expression was low, but BAX expression remained largely unaltered in SW948-cld7kd and HT29-cld7kd cells. With respect to MDR expression, HT29 differed from SW948 cells, MDR was downregulated in HT29-cld7kd cells, but low expression in SW948-cld7kd cells remained unaltered (Suppl.Fig.1E,1F).
Cld7 upregulation is accompanied by slightly increased drug resistance and downregulation is accompanied by a minor loss in drug resistance that is promoted by effector caspase activation and reduced activation of the anti-apoptotic PI3K/Akt pathway.
The impact of cld7 on motility and invasiveness
Metastasizing CIC are supposed to have high migratory potential. As demonstrated in an in vitro wound healing assay, a cld7kd is accompanied by strongly reduced migratory activity, whereas motility of HT29 holoclone-derived cells is increased (Fig.3A,Suppl.Fig.2). Transwell migration confirmed the contribution of cld7 to tumor cell motility. Compared to wt cells, migration of cld7kd cells was reduced, but holoclone-derived cells displayed significantly enhanced migratory activity (Fig.3B). The contribution of cld7 to transwell migration relies on its association with integrins rather than EpC, as migration was weakly or not inhibited by anti-EpC, but strongly inhibited by anti-CD49c. Reduced migration of cld7kd cells was not affected by anti-CD49c (Fig.3C). Cld7-promoted motility was accompanied by strong colocalization and co-immunoprecipitation of EpC and cld7 with CD49c, CD29 and CD104 that was enhanced in holoclones and reduced in cld7kd cells (Fig.3D,3F). Confocal microscopy and co-immunoprecipitation revealed that integrin-associated cld7 is phosphorylated (Fig.3E,3F).
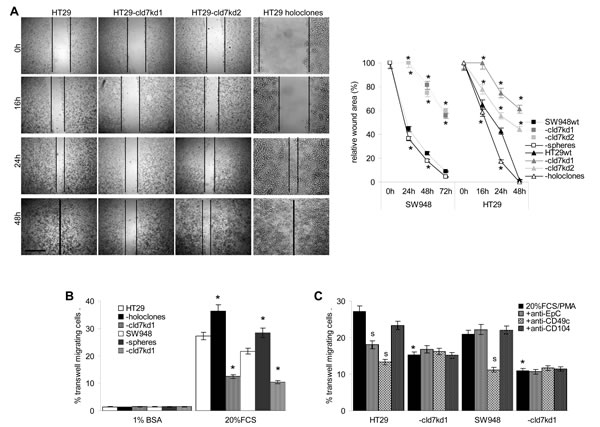
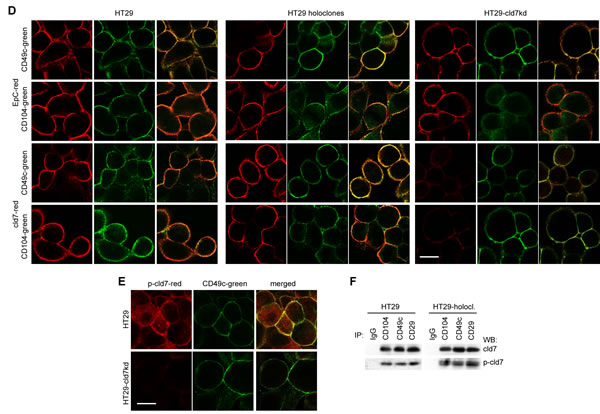
Figure 3: The impact of cld7 on CoCa cell migration. (A) SW948wt and HT29wt, -cld7kd and holoclone- / sphere-derived cells were seeded in 24-well plates. When reaching subconfluence, the monolayer was scratched with a yellow pipette tip; wound healing was followed for 48h-72h; representative examples (HT29, scale bar: 250µm) and the wound area (mean±SD, triplicates) after 16h-72h; (B) transwell migration of SW948wt and HT29wt, -cld7kd and holoclone- / sphere-derived cells was evaluated in Boyden chambers after overnight incubation using RPMI with 20% FCS and 10-8M PMA in the lower chamber as stimulus; in (C) cells were preincubated with anti-EpC, anti-CD49c or anti-CD104. Cells at the lower membrane site were stained with crystal violet and lysed. The % migrating cells (mean±SD, triplicates) is shown; (A-C) significant differences between wt and cld7kd cells: *; (C) significant antibody inhibition: s. (D) HT29wt, -cld7kd and holoclone-derived cells were seeded on coverslides and were stained after overnight incubation in the presence of PMA with anti-cld7 or anti-EpC and anti-CD49c or anti-CD104. Single fluorescence and overlays of double staining are shown; scale bar: 10µm; (E) HT29wt and –cld7kd cells were stained with anti-CD49c and counterstained with anti-p-cld7; representative single fluorescence and overlays (scale bar: 10µm) are shown; (F) HT29wt and holoclone-derived cells were PMA-treated and lysed. Lysates were precipitated with control IgG, anti-α3 or anti-β1. Precipitates were separated by SDS-PAGE and blotted with anti-cld7 and anti-p-cld7. Cld7 expression promotes motility by preferentially associating with CD49c / CD29. Integrin-associated cld7 is phosphorylated.
Finally, cld7 contributed to HT29 and SW948 invasiveness, which was enhanced in HT29 holoclone- and SW948 sphere-derived cells and was strongly decreased in the kd lines (Fig.4A). Invasiveness might be supported by the co-localization of cld7 with MMP14 and MMP7 and, weaker, but distinct colocalization with MMP2 and MMP9 (Fig.4B). TACE, MMP14 and CD26 are recovered in GEM (Fig.4C), suggesting a possible association with cld7. Indeed, TACE, MMP14 and CD26 co-immunoprecipitated with cld7, but not with EpC. Weak coimmunoprecipitation of MMP2 and MMP9 with cld7 under mild lysis conditions might be a sequel of MMP14 focalizing these MMPs at the plasma membrane (Fig.4D). Notably, MMP14 is not downregulated in cld7kd cells. This also accounts for additional MMPs, the dipeptidases CD13 and CD26 as well as uPAR. Instead, MMP9 and MMP3 expression is slightly increased in the cld7kd cells (Suppl.Fig.3).
Taken together, cld7 promotes tumor cell motility, which is accompanied by phosphorylated cld7 associating with CD49c and CD104, and supports invasiveness, which could be provoked by the colocalization of cld7 with MMP14 in GEM. MMP14 focalizes MMP2 and MMP9 close to the cell membrane and supports their activation [35,36], which contributes to invasiveness.
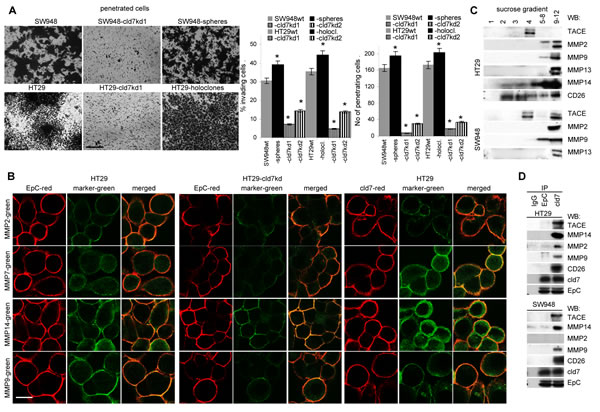
Figure 4: Cld7 and CoCa cell invasiveness. (A) SW948wt and HT29wt, -cld7kd and sphere- / holoclone-derived cells were seeded on matrigel-coated inserts of a Boyden chamber; the lower chamber contained RPMI supplemented with 20% FCS. After overnight incubation, the matrigel block was stained with crystal violet and the cells that had invaded or penetrated the matrigel were counted; representative examples (scale bar: 250µm) and the number of invading and penetrating cells (mean±SD, triplicates) are shown; significant differences between wt versus cld7kd or sphere- / holoclone-derived cells: *; (B) confocal microscopy of HT29 cells stained with anti-EpC or anti-cld7 and anti-MMP2, -MMP7, -MMP14 or –MMP9; single fluorescence and overlays of representative examples are shown (scale bar: 10µm). (C) Sucrose gradient fractions of lysates of HT29wt and Sw948wt and -cld7kd cells were separated by SDS-PAGE and blotted with anti-TACE, -MMP2, -MMP9, -MMP13, -MMP14 and -CD26; (D) Lysates of HT29wt and Sw948wt and -cld7kd cells were precipitated with control IgG, anti-EpC or anti-cld7. After SDS-PAGE separation, precipitates were blotted with anti-TACE, -MMP2, -MMP9, -MMP14 or -CD26. Cld7 contributes to invasiveness that might be promoted by proximity to MMP14 in GEM.
Cld7 supports metastasis formation
Cld7-promoted motility and invasiveness supports metastasis formation. SCID mice received an s.c. or i.v. application of HT29 cells. Subcutaneous growth of HT29-cld7kd cells started with delay and the survival time was significantly prolonged. Instead, the survival time of mice receiving holoclones was significantly shortened as compared to that of mice receiving wt cells (Fig.5A). Macroscopic metastases were seen in the draining LN of all 5 mice receiving holoclones and in 3 of 5 mice receiving HT29wt, but not in the draining LN of mice receiving cld7kd cells. Nonetheless, as revealed by flow cytometry of dispersed organs after double staining with anti-EpC and anti-Tspan8, draining LN of HT29-cld7kd-bearing mice contained few tumor cells. Few HT29-cld7kd cells were also recovered in the peripheral blood and very few in lung, liver, BM and spleen. All these organs contained a significantly higher number of HT29wt cells. With exception of the lung, recovery was further increased in mice receiving HT29 holoclone-derived cells. In ex vivo cultures HT29wt cells grew in draining LN, the peripheral blood and the lung of all 5 mice; HT29-cld7kd cells grew only in draining LN, spleen, BM and peripheral blood of 1 or 2 mice. In lung cultures, HT29-cld7kd cells were recovered in 3 of 5 mice. With exception of the liver (3 of 5 mice), HT29 holoclone-derived cells were recovered in the organs of all 5 mice (Fig.5B). After i.v. tumor cell application, the survival time of HT29-cld7kd bearing mice was significantly prolonged and 5 of the 10 mice receiving HT29-cld7kd cells were still healthy 210d after tumor cell application. Mice that were sacrificed as they started to loose weight, showed lung metastases with the exception of one HT29-cld7kd bearing mouse. HT29wt bearing mice showed around 80 metastatic nodules, the 5 HT29-cld7kd bearing mice that became sick showed 0-25 metastatic nodules (Fig.5C,5D). The tumor-load in the BM did not significantly differ between mice bearing wt or cld7kd tumors, but was increased in mice receiving HT29 holoclone-derived cells. The tumor load in the peripheral blood, the spleen and the lung was reduced in cld7kd bearing animals. Ex vivo outgrowth of tumor cells from dispersed organs confirmed that HT29-cld7kd cells hardly settled and/or survived in liver and lung (Fig.5E).
Thus, cld7 promotes settlement in metastatic organs after s.c. and i.v. tumor cell application. In line with the strong impact on motility, these findings point towards cld7 being engaged in the transition from the sessile towards the motile state (EMT).
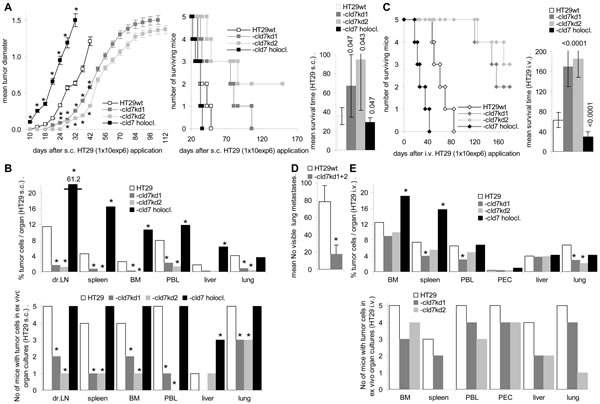
Figure 5: Metastatic settlement of HT29 cells in dependence on cld7 expression. SCID mice (5/group) were treated with anti-asialoGM1, 24h in advance of receiving 1x106 HT29 cells s.c. (A,B) or i.v. (C,D,E); (A) mean tumor diameter after s.c. injection of HT29wt, -cld7kd or holoclone-derived cells, survival time and rate and mean survival time; significant differences in the tumor growth rate and the mean survival time depending on cld7 expression: * and p-values. (B) mice were sacrificed, when the subcutaneous tumor reached a mean diameter of 1.5cm, but latest after 210d. All mice were bled, and single cell suspensions were prepared from bone marrow, spleen, lymph nodes, lung and liver; the presence of tumor cells was evaluated by flow cytometry after staining with anti-EpC and anti-Tspan8; dispersed organs also were cultured for up to 4wk to observe tumor cell outgrowth. The mean percent of tumor cells in dispersed organs and the number of mice with tumor cell outgrowth in ex vivo organ cultures is shown; significant differences between mice bearing HT29wt, -cld7kd or holoclone-derived cells: *. (C) Survival time, survival rate and mean survival time after i.v. tumor cell application; significant differences in the mean survival time depending on cld7 expression are indicated; (D) number of visible lung metastases (mean±SD) of mice that became sick after i.v. HT29 cell application; significant differences in the number of visible lung metastases: *; (E) All mice were bled, and single cell suspensions were prepared for flow cytometry and ex vivo cultures; the percent of tumor cells / organ and the number of mice with tumor cell outgrowth in ex vivo cultured organ suspension; significant differences between HT29wt, -cld7kd and holoclone-derived tumor bearing mice: *. HT29-cld7kd cell growth in vivo starts with delay and metastatic spread is impaired after s.c. and, less pronounced, i.v. application. HT29 holoclone-derived cells show a significantly accelerated growth rate and most efficiently settle and grow in draining lymph nodes after s.c. application.
EMT gene expression in cld7kd cells
Searching for EMT-related protein expression in holoclone-derived, wt and kd cells showed reduction of FN, N-cadherin and vimentin and upregulation of E-cadherin expression in cld7kd compared to wt cells and an opposite regulation in HT29 holoclone-derived cells, which was confirmed for E-cadherin and N-cadherin by WB. Similar findings accounted for SW948wt versus SW498-cld7kd and -sphere-derived cells (Fig.6A,6B). As demonstrated for HT29wt and HT29 holoclone-derived cells, FN and N-cadherin colocalize with cld7 in the plasma membrane, whereas vimentin is organized in the submembrane region. The reduction in vimentin, fibronectin and N-cadherin expression in cld7kd cells is accompanied by redistribution with predominantly cytoplasmic localization. E-cadherin localization remained unaltered (Fig.6C).
The suggested engagement of cld7 in EMT prompted us to search for expression of factors known to contribute to EMT. Flow cytometry and WB showed upregulated expression of the transcription factors Snail, Slug, Twist, ZEB1, TCF4 and Notch in holoclones, but downregulation, though mostly to a minor degree in cld7kd cells. FGF and TGFβ that support EMT protein expression [37,38] were downregulated in cld7kd cells with a stronger impact of cld7 on FGF expression in HT29 than SW948 cells (Fig.6D,6E). Confocal microscopy confirmed downregulated expression of Slug, Snail and Twist in HT29-cld7kd cells and upregulation in HT29 holoclones (Fig.6F).
Thus, a cld7kd affected EMT gene expression. A possible explanation could rely on cld7 guiding EpC into GEM, where it becomes susceptible to digestion by TACE and subsequently by presenilin2, EpIC acting as a cotranscription factor besides others in cooperation with β-catenin [19-22].
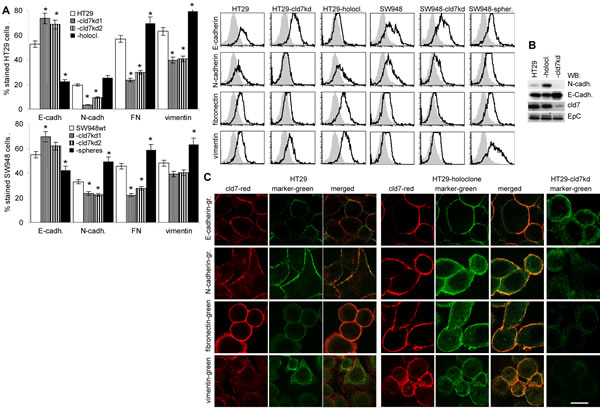
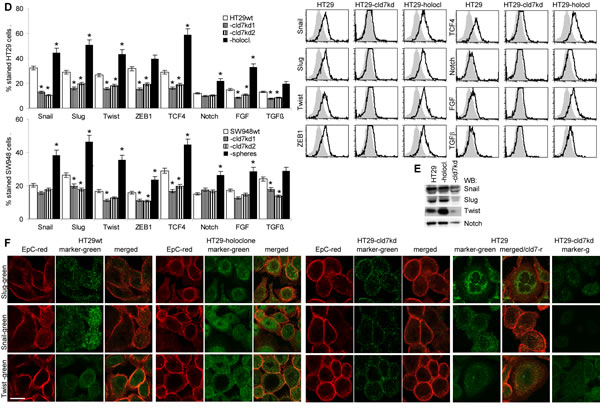
Figure 6: Expression of mesenchymal markers and EMT transcription factors in dependence on cld7 expression. Expression of E-cadherin, N-cadherin, FN and vimentin was evaluated by (A) flow cytometry, the percent stained cells (mean±SD, 3 assays) and representative examples are shown; significant differences between wt, cld7kd and holoclone- / sphere-derived cells: *; (B) WB and (C) confocal microscopy of HT29wt, -cld7kd and holoclone-derived cells stained with anti- E-cadherin, -N-cadherin, -FN and -vimentin and counterstained with anti-cld7. Where indicated, single fluorescence staining and overlays of red and green fluorescence or green fluorescence are shown (scale bar: 10µm); (D) Flow cytometry analysis of transcription factors and cytokines supporting mesenchymal gene expression; the % stained cells (mean±SD, three assays) and representative examples are shown, significant differences between wt versus cld7kd or holoclone- / sphere-derived cells: *; (E) WB of lysates as above, blotted with anti-Snail, -Slug, -Twist and -Notch; (F) Confocal microscopy of HT29wt, -cld7kd and holoclone-derived cells stained with anti-EpC or anti-cld7 and/or anti-Slug, -Snail and -Twist; single fluorescence and overlays of red and green fluorescence or green fluorescence are shown (scale bar: 10µm). Cld7 expression supports the expression of mesenchymal proteins, which is accompanied by higher expression of transcription factors and cytokines supporting EMT protein expression.
Cld7, EpIC and EMT
TACE, presenilin and β-catenin expression is not significantly altered in HT29 holoclone- and SW948 sphere-derived cells compared to -cld7kd and wt cells (Fig.7A,7B). However, in cld7kd cells the intracellular distribution of β-catenin appears affected with a preferential submembrane localization (Fig.7C). Importantly, EpIC recovery is strikingly reduced (Fig7D). To confirm that the transitional state of CoCa cells requires EpIC and that cld7 provides one of the initial triggers for EpIC generation, we searched for mesenchymal gene expression in PMA-stimulated cld7wt and cld7kd cells with a transient EpCkd (Fig.7E). Indeed, a transient EpCkd was accompanied by downregulation of FN, vimentin, N-cadherin, Snail, Slug, Twist and ZEB1 expression (Fig.7F,7G).
These findings indicating a major contribution of cld7 to EMT via supporting the generation of EpIC, we finally asked, whether EMT can be transferred from metastasizing to poorly metastasizing cells via exosomes.
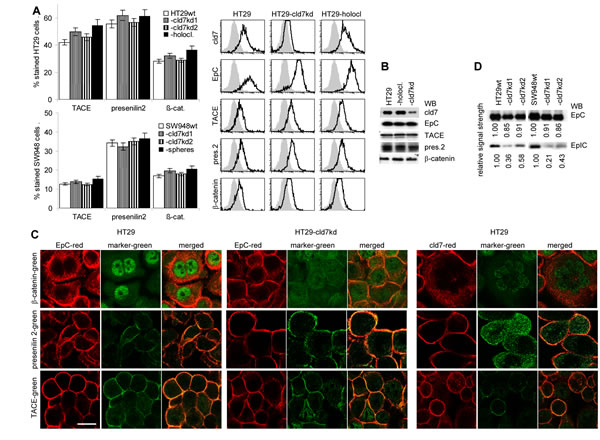
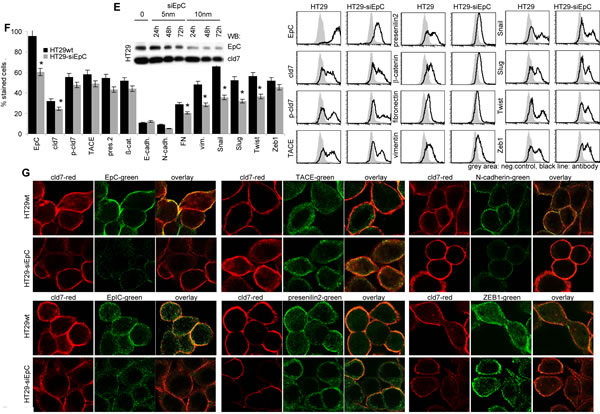
Figure 7: Cld7 expression and EpIC generation. (A) Flow cytometry analysis of TACE, presenilin2 and β-catenin expression in HT29wt and SW948wt, -cld7kd and holoclone- / sphere-derived cells; the percent stained cells (mean±SD, three assays) and representative examples are shown; (B) WB of HT29wt, -cld7kd and holoclone-derived lysates with the indicated antibodies; (C) confocal microscopy of β-catenin, presenilin2 and TACE staining and counterstaining with anti-EpC or -cld7 (single fluorescence and overlays of red and green fluorescence); scale bar: 10µm; (D) WB for EpC and EpIC in HT29wt, SW948wt and -cld7kd cells; (E) HT29 were transfected with titrated doses of EpC siRNA (Suppl.Table1). Downregulation of EpC expression was evaluated by WB after 24h, 48h and 72h. According to the expression profile, 10nm EpC siRNA were used and cells were tested after 48h. (F) EpC was transiently kd in HT29 and SW948 cells; EpC, TACE, presenilin, β-catenin and EMT gene expression was evaluated by flow cytometry (mean±SD, triplicates and representative examples); (G) confocal microscopy of single fluorescence staining and overlays of EpC, EpIC, TACE, presenilin2, N-cadherin and Zeb1 with cld7 (scale bar: 10µm). TACE, presenilin and β-catenin expression is not affected by a cld7kd. However, EpIC generation is severely impaired and Snail, Slug and Twist expression is reduced. This also accounts for a transient EpCkd.
Exosomes and EMT
EpC and cld7 are recovered at a high level in exosomes. EpC expression was slightly reduced in exosomes from cld7kd cells (Fig.8A), which fits the requirement of cld7 for the recruitment of EpC towards internalization prone GEM [10]. When HT29-cld7kd and SW948-cld7kd cells were cultured with exosomes derived from HT29wt and SW948wt cells, expression of N-cadherin, vimentin and FN (only SW948-cld7kd cells) as well as of Snail, Twist, ZEB1 and TCF4 became upregulated, though more pronounced in HT29-cld7kd than SW948-cld7kd cells. Upregulated expression became more pronounced after coculture of cld7kd cells with sphere- or holoclone-derived exosomes. This accounted particularly for vimentin and Notch. Instead, none of these effects where seen, when cld7kd cells were cocultured with cld7kd exosomes (Fig.8B), which was confirmed by confocal microscopy (Fig.8C). Furthermore, upon coculture with exosomes derived from holoclones or spheres the round / epitheloid shape of HT29-cld7kd cells changed towards fibroid and SW948-cld7kd cells again formed most tightly packed clusters (Fig.8D). Changes in cell shape were accompanied by reorganization of the actin cytoskeleton with actin bundle formation (Fig8E). Finally, HT29- and SW498-cld7kd cells treated with wt exosomes regained full motility. Wound closure was further accelerated by coculture with exosomes from holoclones or spheres. Coculture with exosomes from cld7kd cells exerted no effect (Fig.8F,Suppl.Fig.4). Treatment with wt or holoclone- / sphere-derived exosome, but not with cld7kd-derived exosomes also restored transwell migration (Fig.8G) and matrigel invasion, the efficacy of holoclone- / sphere-derived exosomes exceeding that of wt exosomes (Fig.8H). Thus, cld7-competent exosomes from metastasizing tumor cells suffice to initiate EMT in poorly metastasizing cld7kd cells.
spheres or holoclones from
In brief, cld7 downregulation in CoCa is accompanied by reduced anchorage-independent growth, sphere or holoclone formation, slightly reduced apoptosis resistance and a striking loss in motility. This corresponds to poor metastasis formation associated with reduced EMT. These phenomena are due to cld7 promoting motility by associating with integrins and by cld7 initiating the generation of EpIC, which contributes to EMT gene expression.
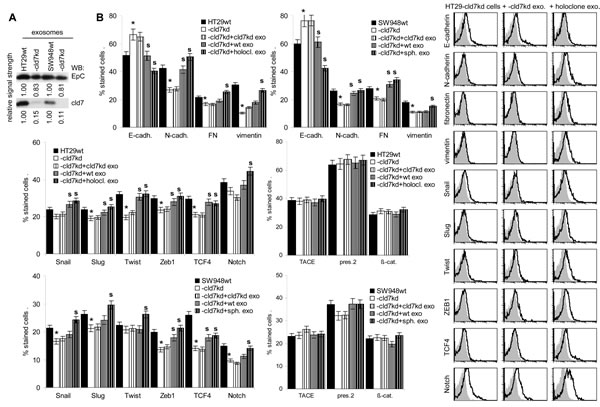
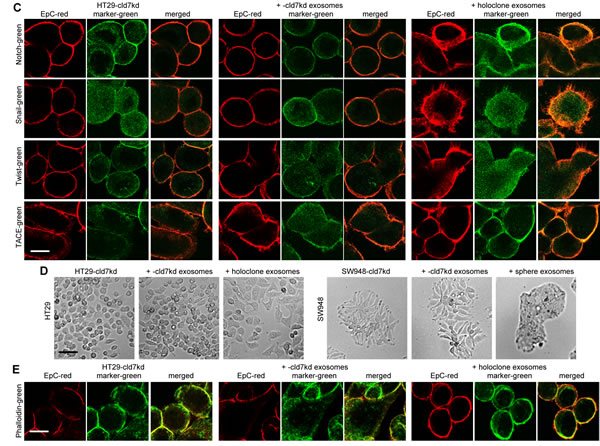
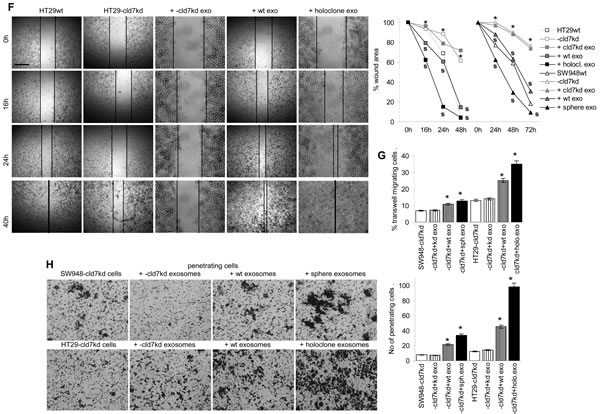
Figure 8: EMT induction by exosomes. (A) WB of EpC and cld7 expression in HT29wt and SW948wt and -cld7kd exosomes. (B-F) HT29-cld7kd and SW948-cld7kd cells were cultured for 48h with 10µg/ml exosomes derived from HT29wt and SW948wt, -cld7kd or sphere- / holoclone-derived cells. (B) Flow cytometry analysis of cld7, EpC and EMT gene expression; the % stained cells (mean±SD, triplicates) and representative examples are shown; significant differences between wt, cld7kd and holoclone- / sphere-derived cells: *; (C) confocal microscopy of EMT gene expression in HT29-cld7kd cells cocultured with cld7kd or holoclone-derived exosomes; single fluorescence and overlays with anti-EpC are shown (scale bar: 10µm); (D) light microscopy of HT29-cld7kd and SW948-cld7kd cells after coculture with cld7kd or holoclone- / sphere-derived exosomes (scale bar: 25µm); (E) confocal microscopy of HT29-cld7kd cells cultured with/without cld7kd or holoclone-derived exosomes; EpC (red), phalloidin-FITC and overlays are shown (scale bar: 10µm); (F) HT29-cld7kd and SW948-cld7kd cells were seeded in 24w plates; subconfluent cultures were scratched with a pipette tip. Wound healing was observed for 40h or 72h in the presence of medium with exosome-depleted FCS, or with exosomes from wt, cld7kd or holoclone / sphere-derived cells; representative examples (scale bar: 250µm) and the wound area (mean±SD, triplicates) are shown; significant differences between wt versus cld7kd cells: *, significant differences by coculture with wt, cld7kd and holoclone- / sphere-derived exosomes: s. (G) HT29-cld7kd and SW948-cld7kd cells were seeded in the upper part of a Boyden chamber. The lower chamber contained 20% exosome-depleted FCS with/without 10µg/ml cld7kd, wt or holoclone- / sphere-derived exosomes; the percent migrating cells is shown; significant differences in the presence of exosomes: *; (H) SW948-cld7kd and HT29-cld7kd cells were seeded on matrigel-coated inserts of a Boyden chamber; the lower chamber contained RPMI supplemented with 20% exosome-depleted FCS with/without 10µg/ml cld7kd, wt or holoclone- / sphere-derived exosomes. After overnight incubation, the matrigel block was stained with crystal violet and the cells that had invaded or penetrated the matrigel were counted; representative examples (scale bar: 250µm) and the number of invading and penetrating cells (mean±SD, triplicates) are shown; significant differences in the presence of cld7kd, wt, or sphere- / holoclone-derived exosomes: *. Exosomes from cld7 competent cells suffice for upregulated expression of mesenchymal markers and genes in cld7kd cells, which is accompanied by changes in cell shape as well as increased motility and invasiveness.
Discussion
EpC is known as a CIC marker in colorectal cancer, where we suggested that a contribution of EpC to tumor progression essentially depends on cooperativity with cld7 [9,23]. In line with our hypothesis, a cld7kd as well as an EpCkd of a rat pancreatic cancer line showed a striking loss in metastasis formation, where cld7 appeared to be the driving force [10]. Building on these findings, we here confirmed by a cld7kd in two human colorectal cancer lines the general validity for the cld7-EpC cooperation being required for tumor progression. We suggest that cld7-promoted EpIC generation strongly supports the shift towards EMT.
Impact of cld7 on CoCIC markers
The cld7kd affected the cell shape, most pronounced of SW948 cells, where the extremely tight cell-cell contacts became weakened. HT29 cells shifted from a fibroid towards a more round shape. However, neither EpC nor expression of additional CoCIC markers was seriously affected. Instead, the recruitment of EpC in GEM was distorted and co-immunoprecipitation with the GEM-located CIC marker Tspan8 [39] was strikingly reduced. From there we conclude that cld7 does not significantly influence transcription / translation of EpC or additional CoCIC markers, but affects the distribution of EpC in the cell membrane.
Cld7 influences stem cell features of CoCIC
Cld7kd cells displayed a strongly reduced capacity for anchorage-independent growth, sphere (SW948) or holoclone (HT29) formation, whereas proliferation and cell cycle progression were slightly accelerated. It is tempting to speculate that the reduced activation of β-catenin (see below) accounts, at least in part, for the loss of CIC growth characteristics. The finding is in line with the Wnt/β-catenin pathway regulating maintenance of CoCa spheres [40]. Cld7 also supported apoptosis resistance, which became stronger in SW948 spheres than wt cells, whereas it was weakened in SW948-cld7kd cells, which showed impaired activation of the PI3K/Akt pathway. Instead, high apoptosis resistance of HT29 cells was only weakly affected by the cld7kd. One possible explanation could be the distinct MDR gene expression, which is regulated by the CoCIC marker CD44v6 [41]. As CD44v6 also associates with cld7 in GEM [42,43], we suggest that the impact of a cld7kd on apoptosis resistance, including the reduced activation of the PI3K/Akt pathway in cld7kd lines is indirect and mediated by cld7-associated CD44v6 [44] such that a higher level of CD44v6 and MDR may compensate for reduced cld7 expression.
In brief, a cld7kd hardly affected proliferation and had no strong impact on drug resistance. Instead, anchorage-independent growth was strongly impaired, which points towards cld7 being engaged in selective CIC activities.
Cld7, motility, invasion and metastasis
Both HT29-cld7kd and SW948-cld7kd cells display strongly reduced motility. In line with this finding, recovery of migrating tumor cells in the peripheral blood was impaired after s.c. HT29-cld7kd cell application. In a gastric cancer line and ovarian cancer, too, overexpression of cld7 was found to be associated with increased motility and invasiveness [45,46], although the opposite was described for lung cancer [47]. Cell migration in vitro was inhibited by anti-CD49c, but not in cld7kd cells. Fittingly, the strong co-localization of EpC, cld7 and CD49c in the cell membrane is reduced in cld7kd cells. Also, CD49c colocalization and co-immunoprecipitation with cld7 requires cld7 phosphorylation. In cld7ko mice, expression of CD49b is significantly impaired and CD49b localization is distorted [30]. The finding suggests a major contribution of the cld7-integrin cooperation in promoting motility, which assumption is supported by cld7 associating with phosphorylated ezrin [9].
Cld7kd cells are also characterized by strongly reduced matrigel invasion and after s.c. HT29-cld7kd cell application only very few tumor cells are recovered from the lung; after i.v. application only 50% of HT29-cld7kd-bearing mice developed visible lung metastasis compared to 100% in HT29wt-bearing mice. A cld7ko was reported to be accompanied by a strong upregulation of MMP3 [30]. However, in the cld7kd tumor lines the effect was comparably weak and expression of other MMPs, two dipeptidases and uPAR was not consistently affected by either cld7 overexpression in spheres / holoclones or by low level expression in cld7kd lines. Instead, MMP14 colocalizes and associates with cld7 in GEM fractions. MMP14 focalizes and supports activation of non-membrane-bound MMPs close to the cell membrane [35,36] and this could well explain higher protease activity in cld7wt compared to cld7kd cells.
Taken together, the association of phosphorylated cld7 with integrins, which is accompanied by FAK activation and ezrin association, contributes to tumor cell motility and the association with MMP14 in GEM supports invasiveness. Notably, there was no evidence for a contribution of EpC to these cld7 activities, i.e. motility and invasiveness promotion likely are genuine cld7 activities.
There remained the question, why the impact of cld7 and EpC on tumor progression appears to be linked. We speculated that cld7 supports EpIC generation, which may contribute to EMT.
Cld7, EpC and EMT
A cld7kd was accompanied by a minor upregulation in E-cadherin expression and reduced expression of N-cadherin, FN and vimentin. Furthermore, EMT-related transcription factors Snail, Slug, Twist, ZEB1 and TCF4 were downregulated. β-catenin was not enriched in the nucleus and EpIC generation was severely impaired, which features were also seen in HT29 cells after a transient EpCkd. On the other hand, EMT gene expression was more pronounced in spheres / holoclones, which express cld7 at a higher level than wt cells. High EpIC acts as a cotranscription factor of β-catenin, FHL2 and Lef-1, the complex initiating, besides others, c-myc, Oct4, Sox2 and Nanog transcription, which was abolished by a γ-secretase inhibitor [19,22]. We observed, as also described before [48-50], that downregulation of EpIC sufficed for a reduction in EMT-related transcription factors Snail, Slug, Twist and TCF4. The same effects being seen in cld7kd and EpCkd cells suggests that the cld7-mediated recruitment of EpC and presenilin2 into GEM facilitates the shift of EpC from a cell-cell adhesion molecule towards a component of EMT.
These data are interpreted in the sense that the metastasis-promoting activity of EpC essentially depends on the association with cld7, which supports EpIC generation.
Exosome transfer of EMT
CIC are supposed to present a minority of cells that accounts for primary tumor growth as well as for metastasis formation [51]. Furthermore, there is increasing evidence that CIC may fulfill their tasks, at least in part, via exosomes [52]. This was amply demonstrated for preparing a premetastatic niche [53] and was also described for the transfer into non-CIC [54]. As CIC might require a niche [55], but migrating tumor cells are suggested to derive from the rim of the primary tumor [56], we hypothesized that CIC could initiate EMT in non-CIC via exosomes. To obtain a hint towards the likeliness of our hypothesis, holoclones or spheres were taken as CIC-enriched exosome donors and cld7kd cells as poorly metastatic recipients. After coculture, the cld7kd cells changed shape, showed downregulation of E-cadherin, upregulation of N-cadherin and vimentin and gained in motility. The high efficacy of exosomes from spheres or holoclones to induce EMT in poorly metastatic cells requires further elucidation. Nonetheless, taking into account that EMT is transient [38], induction via exosomes could explain the rapid reversion towards MET by dilution / degradation of the initiating signal delivered via exosomes [13,57,58]. This would imply that tumor cells may not require to repeatedly changing their phenotype, but that CIC-derived exosomes suffice for transient EMT.
Conclusion
Cld7 is a CIC marker in CoCa, mostly contributing to tumor progression. This is partly due to the association of p-cld7 with integrins converting them into the activated state that promotes tumor cell motility. More important is the co-operation of cld7 with EpC that allows for the generation of EpIC, a cotranscription factor for β-catenin, which supports EMT transcription factor upregulation. Most notably, spheres and holoclones from cld7-competent cells release exosomes that suffice to transiently induce the EMT phenotype in poorly metastatic cells that exhibit low cld7, but high EpC expression. As the finding implies that the EMT phenotype can be passively and transiently required by the uptake of “CIC”-derived exosomes, it deserves an in depth analysis to decipher whether by a blockade of CIC-derived exosomes the initial step in metastasis can be prevented.
Material and methods
Tumor lines
The CoCa lines Sw948 [32] and HT29 [33] were transfected with cld7 shRNA (Suppl.Tab.1A) using the pSuper/GFP/neo vector. Stable kd lines were established by cloning. For a transient kd of EpC, FlexiTube siRNA Hs_EPCAM_1 was used (Suppl.Tab.1B) (Qiagen, Hilden, Germany). Cells were maintained in RPMI 1640/10%FCS w/wo 0.5µg/ml G418.
Antibodies and chemicals
see Suppl.Tab.2.
Tissue preparation and cell isolation
Mice were anesthesized with CO2 or were sacrificed by cervical dislocation. Draining lymph nodes (LN), spleen, femora and tibiae, peripheral blood leukocytes (PBL), peritoneal exudate cells (PEC), liver and lung were collected. PEC and bone marrow cells (BMC) were collected by flushing the peritoneal cavity and bones with PBS. PBL were collected by heart puncture. Single cell suspensions were prepared by pressing through fine gauze. Viability (trypan blue exclusion) ranged between 95%-98%.
Sphere and holoclone selection
Cells (103/ml) were seeded in serum-free RPMI1640 on 0.5% agar precoated 6-well plates. After 1wk half of the medium was exchanged every third day. Spheres were counted after 3wk. Single spheres were picked, dispersed and further passaged. For holoclone selection, 50 cells / cm2 were seeded in 3ml RPMI1640/5% FCS in 6 well plates. After 2wk, cells were stained with crystal violet to count the number of holoclones. Alternatively, holoclones were picked and recultured.
Exosome preparation
Cells were cultured (48h) in serum-free medium. Cleared supernatants (2x10min, 500g, 1x20min, 2000g, 1x30min, 10000g) were centrifuged (90min, 100000g) and washed (PBS, 90min, 100000g). The exosome-containing pellet was resuspended and exosomes were further enriched by sucrose gradient centrifugation. In brief, 1ml exosomes were suspended in 1ml 80% sucrose in HEPES buffer, which was overlaid with 2ml 30% sucrose, 1ml 5% sucrose. After 16h of centrifugation (100000g), 0.4ml fractions were collected. According to the lipid composition of the exosome membrane, exosomes are recovered in the 3rd-6th fraction, which corresponds to a density of 1.122-1.171 [34]. Exosomes were resuspended in 50ml PBS and pelleted by centrifugation (90min, 100000g). The pellet was resuspended in a small volume of PBS, filter-sterilized (0.22µm) and stored at -80oC.
Sucrose density gradient centrifugation
Cell lysates in 2.5M sucrose were overlaid by a continuous sucrose gradient (0.25M-2M) and centrifuged (15h, 150000g), collecting twelve 1ml fractions. Proteins located in glycolipid-enriched membrane subdomains (GEM) are recovered in fraction 2-5.
Immunoprecipitation (IP), Western blot (WB)
Lysates (30min, 4°C, HEPES buffer, 1% Lubrol or 1% TritonX-100, 1mM PMSF, 1mM NaVO4, 10mM NaF, protease inhibitor mix) were centrifuged (13000g, 10min, 4°C), mixed with antibody (1h, 4°C) and incubated with ProteinG-Sepharose (1h). Washed complexes/lysates, dissolved in Laemmli buffer, were resolved on 10%-12% SDS-PAGE. For the recovery of EpIC, Tris-Tricine and 16% SDS-PAGE was used under non-reducing condition. After protein transfer, blocking, blotting with antibodies, blots were developed with ECL.
Flow-cytometry followed routine procedures
In brief, 1-2.5x105 cells were seeded in 96-well plates. After washing with PBS/1%BSA, cells were incubated with the primary antibody (2-10µg/ml, 40µl, 30min, 4oC). Cells were washed 3-times with PBS/1%BSA and incubated with a secondary dye-labeled antibody at predetermined concentration. Negative controls were only incubated with the secondary dye-labeled antibody (40µl, 30min, 4oC). For intracellular staining, cells/exosomes were fixed (PBS/1% formaldehyde, 30min, 4oC) and permeabilized (PBS/0.5%Tween-20, 20min, 4oC). Samples were processed in a FACS-Calibur and evaluated with the CellQuest program.
Confocal microscopy
Cells on glass-slides were fixed (4% paraformaldehyde, 20min on ice), permeabilized (1% Triton-X100, 4min, on ice), blocked (PBS/1% gelatine, 30min, on ice), incubated with primary antibody (60min, on ice), washed, incubated with fluorochrome-conjugated secondary antibody (60min, on ice), blocked (IgG with irrelevant specificity of the same species as the primary antibody), incubated with a second, dye-labeled primary antibody and washed. Slides were mounted in Elvanol. Digitized images were generated using a Leica LMS780 microscope and the Carl Zeiss Vision software for evaluation. The Z-stack offers 30 positions through the depth of the cell. All pictures were taken at Z-stack 14-16. Depending on the quality of the antibody and the density of marker expression, the intensity for the green channel varied between 700-900 master gain values and for the red channel between 500-750 master gain values. The photosystem automatically generates the single fluorescence and overlay pictures.
Migration
Cells, in the upper part of a Boyden chamber (RPMI/0.1%BSA), were separated from the lower part (RPMI/20%FCS) by 8µm pore size polycarbonate-membranes. After 24h the lower membrane side was stained (crystal-violet), measuring OD595 after lysis. Migration is presented as % input cells. In an in vitro wound healing assay, a subconfluent monolayer was scratched with a pipette tip. Wound closure was controlled by light microscopy.
Apoptosis
Cells (1x105) were grown for 48h in RPMI/10%FCS containing titrated amounts of cisplatin. Survival was monitored by annexinV/PI staining. In addition, mitochondrial integrity was evaluated by the MTT assay and proliferative activity by 3H-thymidine uptake.
Soft agar assay
Tumor cells in 0.3% agar were seeded on a preformed 1% agar layer counting colonies after 3wk.
In vivo assays
SCID mice received 1x106 tumor cells subcutaneously (sc) or intravenously (iv). Mice were controlled weekly for local tumor growth, short breathing or weight loss. Animals were sacrificed when the local tumor reached 1.5cm diameter, mice lost >10% weight or latest after 210d. Animals were bled by heart puncture, all hematopoietic organs, as well as liver and lung were excised and single cell suspensions were obtained by meshing through fine gauze. Cell suspensions were subjected to flow cytometry evaluating the percent of EpChigh and Tspan8+ cells. Remaining cells were cultured in RPMI1640/10%FCS for several weeks. The non-transformed cells die, but mostly dormant tumor cells start to form colonies after several weeks. Animal experiments were Government-approved (Baden-Wuerttemberg, Germany).
Statistics
P values <0.05 (two-tailed Student’s t-test, Kruskal-Wallis test) were considered significant.
Authorship
RP, SH, WM, FT and MZ performed and analyzed experiments, MWB helped with writing the manuscript, FT and MZ planned and analyzed experiments and wrote the manuscript.
Acknowledgements
This investigation was supported by the Deutsche Krebshilfe with a MD student grant (RP) and a research grant (MZ). We cordially thank Angela Frank for help with animal experiments and flow-cytometry.
Conflict of Interest Disclosure
The authors declare that they have no competing interests.
Abbreviations
BM: bone marrow, BMC: BM cells, CIC: cancer initiating cells, cld7: claudin-7, CoCa: colon cancer, EMT: epithelial mesenchymal transition, EpC: EpCAM, EpIC: EpCAM intracellular domain, GEM: glycolipid-enriched membrane domains, IP: immunoprecipitation, iv: intravenous, kd: knockdown, ko: knockout, LN: lymph node, LNC: LN cells, PBL: peripheral blood leukocytes, PEC: peritoneal exudate cells, SC: spleen cells, sc: subcutaneous, TB: tumor-bearer, WB: Western blot, wt: wild type,
References
1. Brenner H, Kloor M, Pox CP. Colorectal cancer. Lancet. 2014; 383:1490-1502.
2. Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010; 127: 2893-2917.
3. Siegel R, Desantis C, Jemal A. Colorectal cancer statistics, 2014. CA Cancer J Clin. 2014; 64: 104-117.
4. Chen SY, Huang YC, Liu SP, Tsai FJ, Shyu WC, Lin SZ. An overview of concepts for cancer stem cells. Cell Transplant. 2011; 20: 113-120.
5. Scopelliti A, Cammareri P, Catalano V, Saladino V, Todaro M, Stassi G. Therapeutic implications of Cancer Initiating Cells. Expert Opin Biol Ther. 2009; 9: 1005-1016.
6. Zaidi SK, Grandy RA, Lopez-Camacho C, Montecino M, van Wijnen AJ, Lian JB, Stein JL, Stein GS. Bookmarking target genes in mitosis: a shared epigenetic trait of phenotypic transcription factors and oncogenes? Cancer Res. 2014; 74: 420-425.
7. Malik B, Nie D. Cancer stem cells and resistance to chemo and radio therapy. Front Biosci (Elite Ed). 2012; 4: 2142-2149.
8. Bigas A, Espinosa L. Hematopoietic stem cells: to be or Notch to be. Blood. 2012; 119: 3226-3235.
9. Fanali C, Lucchetti D, Farina M, Corbi M, Cufino V, Cittadini A, Sgambato A. Cancer stem cells in colorectal cancer from pathogenesis to therapy: controversies and perspectives. World J Gastroenterol. 2014; 20: 923-942.
10. Thuma F, Zöller M. EpCAM-associated claudin-7 supports lymphatic spread and drug resistance in rat pancreatic cancer. Int J Cancer. 2013; 133: 855-866.
11. Vaiopoulos AG, Kostakis ID, Koutsilieris M, Papavassiliou AG. Colorectal cancer stem cells. Stem Cells. 2012; 30: 363-371.
12. Zöller M. CD44: can a cancer-initiating cell profit from an abundantly expressed molecule? Nat Rev Cancer. 2011; 11: 254-267.
13. Roma-Rodrigues C, Fernandes AR, Baptista PV. Exosome in Tumour Microenvironment: Overview of the Crosstalk between Normal and Cancer Cells. Biomed Res Int. 2014; 2014: 179486.
14. Winter MJ, Nagelkerken B, Mertens AE, Rees-Bakker HA, Brtiaire-deBruijn IH, Litvinov SV. Expression of EpCAM shifts the state of cadherin-mediated adhesions from strong to weak. Exp Cell Res. 2003; 285: 50-58.
15. Yamashita T, Budhu A, Forgues M, Wang XW. Activation of hepatic stem cell marker EpCAM by Wnt-beta-catenin signaling in hepatocellular carcinoma. Cancer Res. 2007;.67: 10831-10839.
16. Maghzal N, Vogt E, Reintsch W, Fraser JS, Fagotto F. The tumor-associated EpCAM regulates morphogenetic movements through intracellular signaling. J Cell Biol. 2010; 191: 645-659.
17. Denzel S, Mack B, Eggert C, Massoner P, Stöcklein N, Kemming D, Harréus U, Gires O. MMP7 is a target of the tumour-associated antigen EpCAM. Int J Exp Pathol. 2012; 93: 341-353.
18. Sankpal NV, Willman MW, Fleming TP, Mayfield JD, Gillanders WE. Transcriptional repression of epithelial cell adhesion molecule contributes to p53 control of breast cancer invasion. Cancer Res. 2009; 69: 753-757.
19. Maetzel D, Denzel S, Mack B, Eggert C, Bärr G, Gires O. Nuclear signalling by tumour-associated antigen EpCAM. Nat Cell Biol. 2009; 11: 162-171.
20. Münz M, Kieu C, Mack B, Schmitt B, Zeidler R, Gires O. The carcinoma-associated antigen EpCAM upregulates c-myc and induces cell proliferation. Oncogene. 2004; 23: 5748-5758.
21. Chen HF, Chuang CY, Lee WC, Huang HP, Wu HC, Ho HN, Chen YJ, Kuo HC. Surface marker epithelial cell adhesion molecule and E-cadherin facilitate the identification and selection of induced pluripotent stem cells. Stem Cell Rev. 2011; 7: 722-735.
22. Lin CW, Liao MY, Lin WW, Wang YP, Lu TY, Wu HC. Epithelial Cell Adhesion Molecule Regulates Tumor Initiation and Tumorigenesis via Activating Reprogramming Factors and Epithelial-Mesenchymal Transition Genes Expression in Colon Cancer. J Biol Chem. 2012; 287: 39449-39459.
23. Ladwein M, Schmidt DS, Schnölzer M, Kempf T, Pape UF, Langbein L, Franke WW, Moldenhauer G, Zöller M. The cell-cell adhesion molecule EpCAM associates with the tight junction protein claudin 7. Exp Cell Res. 2005; 309: 345-357.
24. Yovchev MI, Grozdanov PN, Zhou H, Racherla H, Guha C, Dabeva MD. Identification of adult hepatic progenitor cells capable of repopulating injured rat liver. Hepatology. 2008; 47: 636-647.
25. Tsukita S, Yamazaki Y, Katsuno T, Tamura A, Tsukita S. Tight junction-based epithelial microenvironment and cell proliferation. Oncogene. 2008; 27: 6930-6938.
26. Yeaman C, Grindstaff KK, Hansen MD, Nelson WJ. Cell polarity: Versatile scaffolds keep things in place. Curr Biol. 1999; 9: 515-517.
27. Ikari A, Ito M, Okude C, Sawada H, Harada H, Degawa M, Sakai H, Takahashi T, Sugatani J, Miwa M. Claudin-16 is directly phosphorylated by protein kinase A independently of a vasodilator-stimulated phosphoprotein-mediated pathway. J Cell Physiol. 2008; 214: 221-229.
28. French AD, Fiori JL, Camilli TC, Leotlela PD, O’Connell MP, Frank BP, Subaran S, Indig FE, Taub DD, Weeraratna AT. PKC and PKA phosphorylation affect the subcellular localization of claudin-1 in melanoma cells. Int J Med Sci. 2009; 6: 93-101.
29. Sjö A, Magnusson KE, Peterson KH. Protein kinase C activation has distinct effects on the localization, phosphorylation and detergent solubility of the claudin protein family in tight and leaky epithelial cells. J Membr Biol. 2010; 236: 181-189.
30. Ding L, Lu Z, Foreman O, Tatum R, Lu Q, Renegar R, Cao J, Chen YH. Inflammation and disruption of the mucosal architecture in claudin-7-deficient mice. Gastroenterology. 2012; 142: 305-315.
31. Lei Z, Maeda T, Tamura A, Nakamura T, Yamazaki Y, Shiratori H, Yashiro K, Tsukita S, Hamada H. EpCAM contributes to formation of functional tight junction in the intestinal epithelium by recruiting claudin proteins. Dev Biol. 2012; 371: 136-145.
32. Leibovitz A, Stinson JC, McCombs WB 3rd, McCoy CE, Mazur KC, Mabry ND. Classification of human colorectaladenocarcinoma cell lines. Cancer Res. 1976; 36: 4562-4569.
33. von Kleist S, Chany E, Burtin P, King M, Fogh J. Immunohistology of the antigenic pattern of a continuous cell line from a human colon tumor. J Natl Cancer Inst. 1975; 55: 555-560.
34. Rana S, Yue S, Stadel D, Zöller M. Toward tailored exosomes: the exosomal tetraspanin web contributes to target cell selection. Int J Biochem Cell Biol. 2012; 44: 1574-1584.
35. Holmbeck K, Bianco P, Yamada S, Birkedal-Hansen H. MT1-MMP: a tethered collagenase. J Cell Physiol. 2004; 200: 11-19.
36. Murphy DA, Courtneidge SA. The ‘ins’ and ‘outs’ of podosomes and invadopodia: characteristics, formation and function. Nat Rev Mol Cell Biol. 2011; 12: 413-426.
37. Katoh M. Network of WNT and other regulatory signaling cascades in pluripotent stem cells and cancer stem cells. Curr Pharm Biotechnol. 2011; 12: 160-170.
38. Katsuno Y, Lamouille S, Derynck R. TGF-β signaling and epithelial-mesenchymal transition in cancer progression. Curr Opin Oncol. 2013; 25: 76-84.
39. Wang H, Rana S, Giese N, Büchler MW, Zöller M. Tspan8, CD44v6 and alpha6beta4 are biomarkers of migrating pancreatic cancer-initiating cells. Int J Cancer. 2013; 133: 416-426.
40. Kanwar SS, Yu Y, Nautiyal J, Patel BB, Majumdar AP. The Wnt/beta-catenin pathway regulates growth and maintenance of colonospheres. Mol Cancer. 2010; 9: 212.
41. Soslow RA, Slomovitz BM, Saqi A, Baergen RN, Caputo TA. Tumor suppressor gene, cell surface adhesion molecule, and multidrug resistance in Müllerian serous carcinomas: clinical divergence without immunophenotypic differences. Gynecol Oncol. 2000; 79: 430-437.
42. Okada T, Nakamura T, Watanabe T, Onoda N, Ashida A, Okuyama R, Ito K. Coexpression of EpCAM, CD44 variant isoforms and claudin-7 in anaplastic thyroid carcinoma. PLoS One. 2014; 9: e94487.
43. Kuhn S, Koch M, Nübel T, Ladwein M, Antolovic D, Klingbeil P, Hildebrand D, Moldenhauer G, Langbein L, Franke WW, Weitz J, Zöller M. A complex of EpCAM, claudin-7, CD44 variant isoforms, and tetraspanins promotes colorectal cancer progression. Mol Cancer Res. 2007; 5: 553-567.
44. Jung T, Gross W, Zöller M. CD44v6 coordinates tumor matrix-triggered motility and apoptosis resistance. J Biol Chem. 2011; 286: 15862-15874.
45. Zavala-Zendejas VE, Torres-Martinez AC, Salas-Morales B, Fortoul TI, Montaño LF, Rendon-Huerta EP. Claudin-6, 7, or 9 overexpression in the human gastric adenocarcinoma cell line AGS increases its invasiveness, migration, and proliferation rate. Cancer Invest. 2011; 29: 1-11.
46. Dahiya N, Becker KG, Wood WH 3rd, Zhang Y, Morin PJ. Claudin-7 is frequently overexpressed in ovarian cancer and promotes invasion. PLoS One. 2011; 6: e22119.
47. Lu Z, Ding L, Hong H, Hoggard J, Lu Q, Chen YH. Claudin-7 inhibits human lung cancer cell migration and invasion through ERK/MAPK signaling pathway. Exp Cell Res. 2011; 317: 1935-1946.
48. Forrest MP, Hill MJ, Quantock AJ, Martin-Rendon E, Blake DJ. The emerging roles of TCF4 in disease and development. Trends Mol Med. 2014; 20: 322-331.
49. Muqbil I, Wu J, Aboukameel A, Mohammad RM, Azmi AS. Snail Nuclear Transport: the Gateways Regulating Epithelial-to-Mesenchymal Transition? Semin Cancer Biol. 2014; 27C: 39-45.
50. Tania M, Khan MA, Fu J. Epithelial to mesenchymal transition inducing transcription factors and metastatic cancer. Tumour Biol. 2014 Jun 2. [Epub ahead of print]
51. Elshamy WM, Duhé RJ. Overview: cellular plasticity, cancer stem cells and metastasis. Cancer Lett. 2013; 341: 2-8.
52. Bussolati B, Dekel B, Azzarone B, Camussi G. Human renal cancer stem cells. Cancer Lett. 2013; 338: 141-146.
53. Alderton GK. Metastasis. Exosomes drive premetastatic niche formation. Nat Rev Cancer. 2012; 12: 447.
54. Atay S, Godwin AK. Tumor-derived exosomes: A message delivery system for tumor progression. Commun Integr Biol. 2014; 7: e28231.
55. Thuma F, Zöller M. Outsmart tumor exosomes to steal the cancer initiating cell its niche. Semin Cancer Biol. 2014 Mar 12. pii: S1044-579X(14)00035-2.
56. Nabeshima K, Inoue T, Shimao Y, Kataoka H, Koono M. Cohort migration of carcinoma cells: differentiated colorectal carcinoma cells move as coherent cell clusters or sheets. Histol Histopathol. 1999; 14: 1183-1197.
57. Azmi AS, Bao B, Sarkar FH. Exosomes in cancer development, metastasis, and drug resistance: a comprehensive review. Cancer Metastasis Rev. 2013; 32: 623-642.
58. Ye J, Wu D, Wu P, Chen Z, Huang J. The cancer stem cell niche: cross talk between cancer stem cells and their microenvironment. Tumour Biol. 2014; 35: 3945-3951.