
Introduction
Rituximab is a chimeric monoclonal antibody that targets the cluster of differentiation (CD) 20 and is highly effective in treating several B-cell malignancies and inflammatory diseases [1–3]. Combination therapy with chemotherapeutic agents such as R-CHOP has become the standard treatment for non-Hodgkin’s lymphoma (NHL) and chronic lymphocytic leukemia (CLL). However, 30–40% of patients do not respond to rituximab-based therapy initially or the disease progresses after an initial response to rituximab-based therapy [1, 4, 5]. Moreover, rituximab resistance is acquired through various mechanisms, such as the loss of CD20 expression on the B-cell surface [6, 7] or overexpression of anti-apoptotic factors [8, 9]. Recently, several therapeutic antibodies and chimeric antigen receptor T cell (CAR-T) therapies have been approved for relapse and refractory B-cell malignancies after rituximab-based therapy; however, their efficacy is limited. For example, tafasitamab, an anti-CD19 antibody that has enhanced antibody-dependent cell-mediated cytotoxicity (ADCC) and polatuzumab vedotin, a drug-conjugated anti-CD79b antibody, have demonstrated a 43% and 45% overall objective response, respectively, in relapse and refractory diffuse large B-cell lymphoma (DLBCL) [10, 11]. In addition, axicabtagene ciloleucel and lisocabtagene maraleucel (anti-CD19 CAR-T) have become applicable for treating relapse and refractory large B-cell lymphoma [12]. However, treatments in 49% of patients with these types of CAR-T therapy fail [13]. Although these new therapies provide favorable therapeutic effects, some patients still experience refractory disease and disease relapse. Therefore, alternative clinical options are required to treat B-cell malignancies.
IgM is a component of the B-cell receptor (BCR), which functions as a regulator of B-cell proliferation, differentiation, and apoptosis, depending on the cellular state and extracellular environment [14, 15]. Constitutive activation of BCR signaling is associated with the pathogenesis of B-cell malignancies, including mantle cell lymphoma (MCL), DLBCL and CLL [16–20]. MCL is an aggressive NHL subtype, accounting for 3% to 6% of NHL cases. The phenotype of MCL is characterized by the intense expression of surface immunoglobulins, including IgM and IgD [21, 22]. DLBCL is the most common subtype of NHL, accounting for 30–40% of NHL cases, and exhibits complex and heterogeneous characteristics in biological and clinical presentations [23]. Gene expression profiling has been employed to divide DLBCL into two subtypes based on its cell-of-origin: germinal center B cell-like (GCB)-DLBCL and activated B cell-like (ABC)-DLBCL [24]. Patients with ABC-DLBCL have shown shorter survival and a higher frequency of treatment failure than those with GCB-DLBCL [24]. Interestingly, approximately 88.5% of patients with ABC-DLBCL express a primary isotype of immunoglobulin, mainly IgM, whereas 88.2% of patients with GCB-DLBCL express secondary types of immunoglobulins, IgG and IgA [25]. Moreover, regardless of the cell-of-origin, high cell-surface expression of IgM is associated with a poor prognosis in DLBCL [25]. Ruminy et al. [25] reported that defective isotype class-switching has contributed to the high expression of IgM and may be involved in the development of the pathogenesis of ABC-DLBCL. CLL is the most common type of leukemia in adults and is characterized by the accumulation of monoclonal B cells in peripheral blood, bone marrow, and lymphoid tissues. CLL is divided into two major subtypes, mutated-CLL (M-CLL) and unmutated-CLL (U-CLL), based on the presence and absence of somatic hypermutations in the immunoglobulin heavy chain variable region (IGHV) gene, respectively [26]. U-CLL shows more rapid progression and higher expression of IgM than M-CLL [26, 27]. Moreover, similar to DLBCL, high IgM expression is associated with a poor CLL prognosis [27]. Because IgM is highly expressed and related to a poor prognosis in these aggressive B-cell malignancies, we considered that IgM is a promising target of therapeutic antibodies for refractory and/or relapsed B-cell malignancies.
IgM is exclusively expressed in B cells; therefore, anti-IgM antibodies specifically bind to these cells. Furthermore, anti-IgM antibodies trigger cell arrest and apoptosis in B-cell lymphoma cells in vitro [28–30], suggesting that anti-IgM antibodies are therapeutic candidates for B-cell malignancies. However, in addition to its membrane form, IgM is also present in a soluble form at 0.24–7.91 mg/mL in the blood [31]. The soluble form of the antigen may capture therapeutic antibodies, resulting in loss of their activity in the blood. For instance, programmed death-ligand 1 (PD-L1) vInt4 is one of the splicing variants of PD-L1 which produces the secreted PD-L1 and is expressed in 10–20% of patients with squamous cell carcinoma [32]. The expression of PD-L1 vInt4 leads to loss of anti-tumor efficacy of anti-PD-L1 antibodies by acting as a decoy for antibodies injected in mice [32]. In addition, elevated levels of soluble PD-L1 are associated with unfavorable progression-free survival and overall survival in non-small-cell lung cancer patients treated with anti-PD-L1 antibody monotherapy [33]. Thus, the presence of soluble antigen types is one of the problems associated with antibody therapy. To overcome this problem, Welt et al. [34] produced anti-IgM antibodies targeting the proximal domain of membrane IgM, which is absent in soluble IgM. Alternatively, we used a bispecific antibody modality to improve the selectivity of anti-IgM antibodies for B cells.
IgG-like bispecific antibodies exert several advantages, including favorable pharmacokinetic properties via the neonatal Fc receptor (FcRn), and cytotoxic effects such as ADCC, complement-dependent cell cytotoxicity (CDC), and antibody-dependent cellular phagocytosis (ADCP) via the Fc region [35]. However, the efficient production of suitable bispecific antibodies is challenging because two distinct heavy chains need to bind together, and the respective light chain needs to bind to the correct heavy chain [36]. Therefore, advanced technologies are necessary to produce the correct pair of bispecific antibodies efficiently. The heterodimerization of two distinct heavy chains is enabled by the knobs-into-holes (KIH) technology [36, 37]. Recently, technologies have been developed to prevent the mispairing of light and heavy chains. One approach involves using a common light chain against two heavy chains, and this approach has been applied to emicizumab, a therapeutic antibody used in the treatment of hemophilia A [38]. Another approach involves producing two parental antibodies separately and then exchanging one pair of heavy and light chains under an oxidation-reduction reaction, as in DuoBody [39]. In addition, technologies to restrict the combination of heavy and light chains, such as CrossMab and DuetMab, have also been developed [40, 41]. In contrast, the strategy that we developed for producing IgG-like bispecific antibodies involves inserting an artificial disulfide bond between the heavy chain constant domain 1 (CH1) and light chain constant domain (CL) interface, and deleting the native disulfide bond on one of the parental antibodies. We termed this technology the Cys1m technology [42].
In this study, we aimed to produce IgM-dependent bispecific antibodies targeting IgM and the other B-cell antigens such as CD20, CD32b (FcγRIIB), CD79b, and human leukocyte antigen (HLA)-DR using the Cys1m technology [10, 43–45]. Additionally, the correct IgG-like bispecific antibody structures were confirmed and their efficacies in the presence of soluble IgM were analyzed.
Results
Production of full-length IgG-like bispecific antibodies using Cys1m technology
Because the presence of soluble-type antigens in the blood reduces antibody activity, conventional anti-IgM antibodies may lose their activity owing to soluble IgM. To improve the selectivity of anti-IgM antibodies for IgM on the B-cell surface, we produced IgM-dependent bispecific antibodies targeting IgM and other B-cell surface antigens. Conventional approaches that produce all light and heavy chains simultaneously often result in the inefficient production of IgG-like bispecific antibodies owing to the incorrect pairing of light and heavy chains. Approximately 40% of incorrect pairings between light and heavy chains occurred using the KIH technology [36]. In contrast, our developed technique, Cys1m, involves inserting an artificial disulfide bond between the CH1 and CL interface and deleting the native disulfide bond on one of the parental antibodies [42]. As shown in Figure 1A, to engineer artificial disulfide bonds, we introduced an S162C mutation in CH1 and F170C in CL, which was termed Cys1m (g), or an F126C mutation in CH1 and Q124C in CL, which was termed Cys1m (f), using a mutagenesis kit. To delete the native disulfide bond, we introduced C220S into CH1 and C214S into CL.
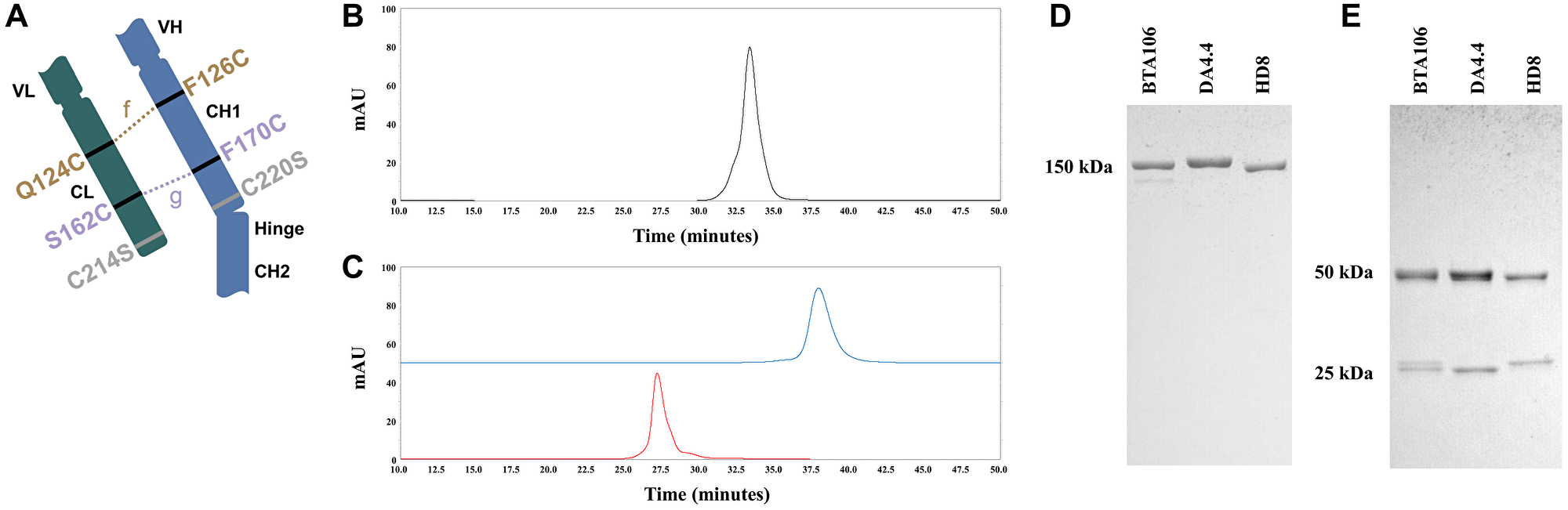
Figure 1: Production of the IgG1 bispecific antibodies using Cys1m technology.
(A) Schematic diagram of bispecific antibodies produced using Cys1m technology. Bispecific antibodies produced using Cys1m technology consisted of one wild-type fragment antigen-binding (Fab) and one mutated Fab. The mutated Fab contained artificial disulfide-bound Cys1m (g) or Cys1m (f), with the wild-type disulfide bond deleted at the interface of CH1 and CL. CEX profiles of BTA106 (B), DA4.4 (blue), and HD8 (red) of the parent antibodies (C). SDS-PAGE analysis of BTA106 and their parent antibodies under non-reducing (D) and reducing (E) conditions. The molecular weights of the whole antibody (150 kDa), heavy chain (50 kDa), and light chain (25 kDa) were calculated using the protein marker.
In this study, we produced several bispecific antibodies targeting IgM and other antigens in B-cell lymphoma cells, as presented in Table 1. These bispecific antibodies were generated using the Cys1m technology. To clarify whether differences in the core structure affected antibody activity, we produced KIH-IgM/CD20 and KIH-IgM/HLA-DR using the KIH technology (Table 1) and compared them with BTA065 and BTA106, respectively. The antibodies used in the following examinations were analyzed by cation exchange chromatography (CEX) to assess their purity and confirm the composition of the heavy and light chains. The retention time of BTA106 was localized in the middle of those of the parental antibodies, and no extra peaks were detected (Figure 1B, 1C). All antibodies listed in Table 1 showed similar CEX results (data not shown). Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis of BTA106 under non-reducing conditions confirmed that BTA106 is a 150 kDa full-length antibody (Figure 1D). In addition, 50 kDa heavy chains and 25 kDa light chains were detected under reducing conditions (Figure 1E). Notably, slightly different sizes of the two light-chain bands were observed for the bispecific antibodies under reducing conditions. BTA065 showed the same results as BTA106 did in CEX and SDS-PAGE analyses, as shown in the supplementary data (Supplementary Figure 1A–1D). These data demonstrate that the Cys1m technology is useful for the accurate production of IgG-like bispecific antibodies.
Table 1: Bispecific antibodies used in this study
| Bispecific antibody | Target 1 | Clone | Target 2 | Clone | Structure |
|---|---|---|---|---|---|
| BTA065 | IgM | DA4.4 | CD20 | Rituximab | Cys1m |
| BTA106 | IgM | DA4.4 | HLA-DR | HD8 | Cys1m |
| BTA116 | IgM | DA4.4 | CD32b | 5A6 | Cys1m |
| BTA118 | IgM | DA4.4 | CD79b | #89 | Cys1m |
| BTA124 | IgM | 4B9 | HLA-DR | HD8 | Cys1m |
| BTA125 | IgM | 6D9 | HLA-DR | HD8 | Cys1m |
| KIH-IgM/CD20 | IgM | DA4.4 | CD20 | Rituximab | KIH |
| KIH-IgM/HLA-DR | IgM | DA4.4 | HLA-DR | HD8 | KIH |
Proliferation inhibition of IgM-dependent bispecific antibodies in the presence of soluble antigens
To determine whether IgM-dependent bispecific antibodies show activity in the presence of a soluble antigen, a cell viability assay was performed in JeKo-1 cells (MCL) and B104 cells (unclassified B-cell lymphoma) at various concentrations of purified soluble IgM. Because the epithelial growth factor receptor (EGFR) is not expressed on immune cells, an anti-EGFR antibody (clone h425) was used as the control IgG. Control IgG and HD8 did not inhibit the proliferation of JeKo-1 cells, whereas DA4.4 (100 ng/mL) inhibited proliferation in the absence of purified soluble IgM. However, more than 3 μg/mL of purified soluble IgM disturbed its effects (Figure 2A). In contrast, BTA106 (100 ng/mL) and KIH-IgM/HLA-DR (100 ng/mL) inhibited the proliferation of JeKo-1 cells in the presence of 10 μg/mL purified soluble IgM. Similar to the JeKo-1 cells, BTA106 (100 ng/mL) inhibited the proliferation of B104 cells in the presence of purified soluble IgM (Figure 2B). In addition, we performed cell viability assays using the other anti-IgM antibody clones, 4B9 and 6D9. We observed that 4B9 and 6D9 (300 ng/mL) inhibited the proliferation of JeKo-1 cells in the absence of soluble IgM, whereas more than 1 μg/mL of purified soluble IgM disturbed their effects. However, BTA124 and BTA125 (300 ng/mL) inhibited the proliferation of JeKo-1 in the presence of more than 1 μg/mL of purified soluble IgM (Supplementary Figure 2A, 2B). These results indicate that the effects of IgM-dependent bispecific antibodies were independent of the anti-IgM antibody clones used.
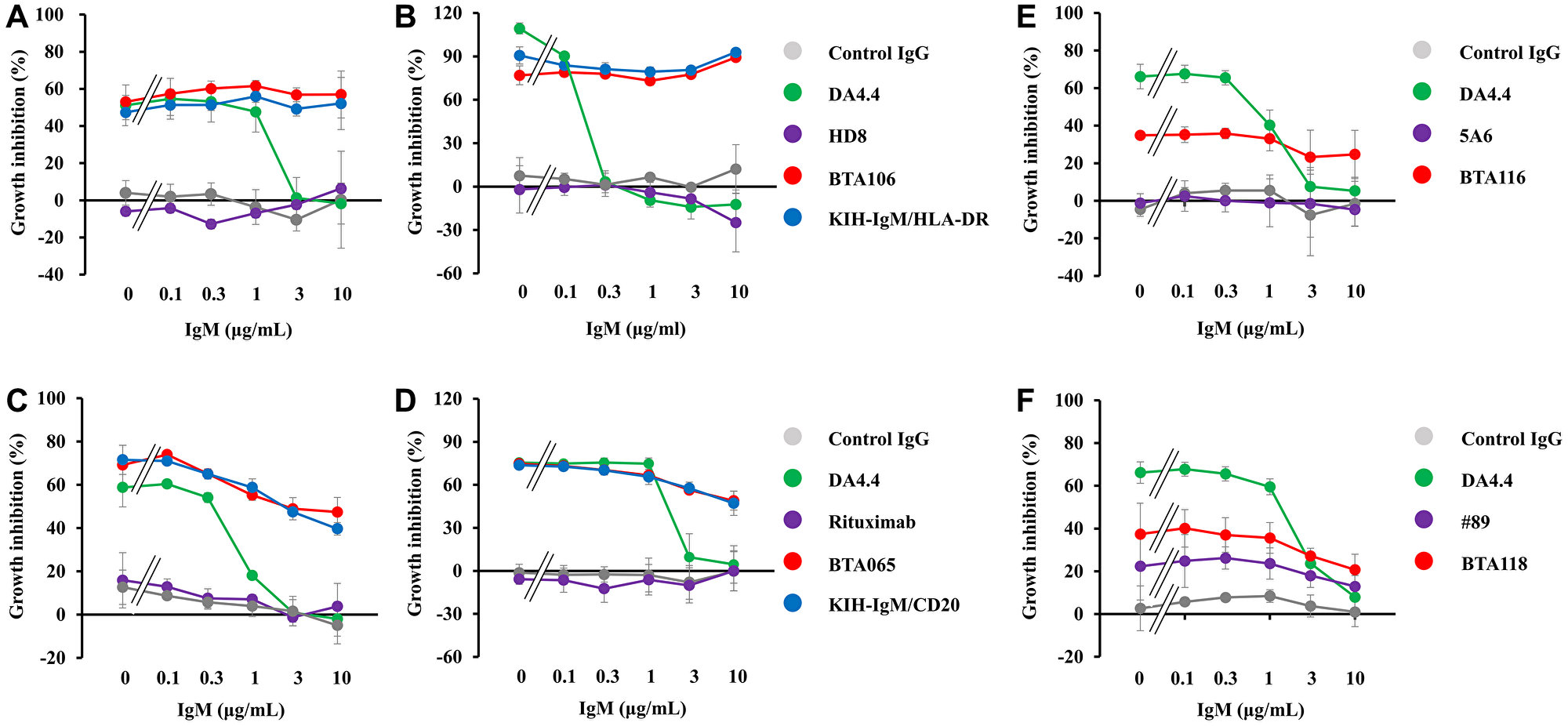
Figure 2: Effects of IgM-dependent bispecific antibodies on cell viability in the presence of soluble IgM.
BTA106 (A, B), BTA065 (C, D), BTA116 (E), BTA118 (F) and their parent antibodies were added to JeKo-1 (A, C, E, F) and B106 cells (B, D), and incubated for 72 hours in the presence of purified soluble IgM. h425 was used as the control IgG. Viable cells were measured using the Cell Counting Kit-8 (mean ± S.D. of triplicate).
Next, we investigated whether IgM-dependent bispecific antibodies targeting other B-cell antigens, such as CD20, CD32b, and CD79b, would be effective in the presence of purified soluble IgM. Treatment with control IgG and rituximab did not inhibit the proliferation of JeKo-1 or B104 cells, whereas treatment with BTA065 and KIH-IgM/CD20 inhibited the proliferation of JeKo-1 (100 ng/mL) and B106 (1,000 ng/mL) cells in the absence or presence of purified soluble IgM (Figure 2C, 2D). Furthermore, 300 ng/mL of BTA116 (Figure 2E) and 300 ng/mL of BTA118 (Figure 2F) showed activity against JeKo-1 cells in the presence of purified soluble IgM. These data indicate that IgM-dependent bispecific antibodies are less affected by soluble IgM than by conventional anti-IgM antibodies.
Proliferation inhibition of bispecific antibodies in the presence of serum
To examine the effects of IgM-dependent bispecific antibodies under conditions similar to those in the internal environment, we performed cell viability assays in the absence and presence of human serum. DA4.4 (1,000 ng/mL) inhibited the proliferation of JeKo-1 cells in the absence of human serum in contrast to the control IgG (Figure 3A). However, DA4.4 failed to inhibit the proliferation of JeKo-1 cells in the presence of human serum (Figure 3B). In contrast, BTA106 and KIH-IgM/HLA-DR retained their activities at 1,000 ng/mL in the presence of human serum. In addition, these anti-IgM/HLA-DR bispecific antibodies demonstrated efficacy against JeKo-1 cells when the donor human serum was changed (data not shown). In the case of anti-IgM/CD20 bispecific antibodies, BTA065 and KIH-IgM/CD20 (1,000 ng/mL) inhibited the proliferation of JeKo-1 cells in the absence or presence of human serum in contrast to the control IgG and DA4.4, similar to anti-IgM/HLA-DR bispecific antibodies (Figure 3C, 3D). These data suggest that IgM-dependent bispecific antibodies may be active in the body even with soluble antigens.
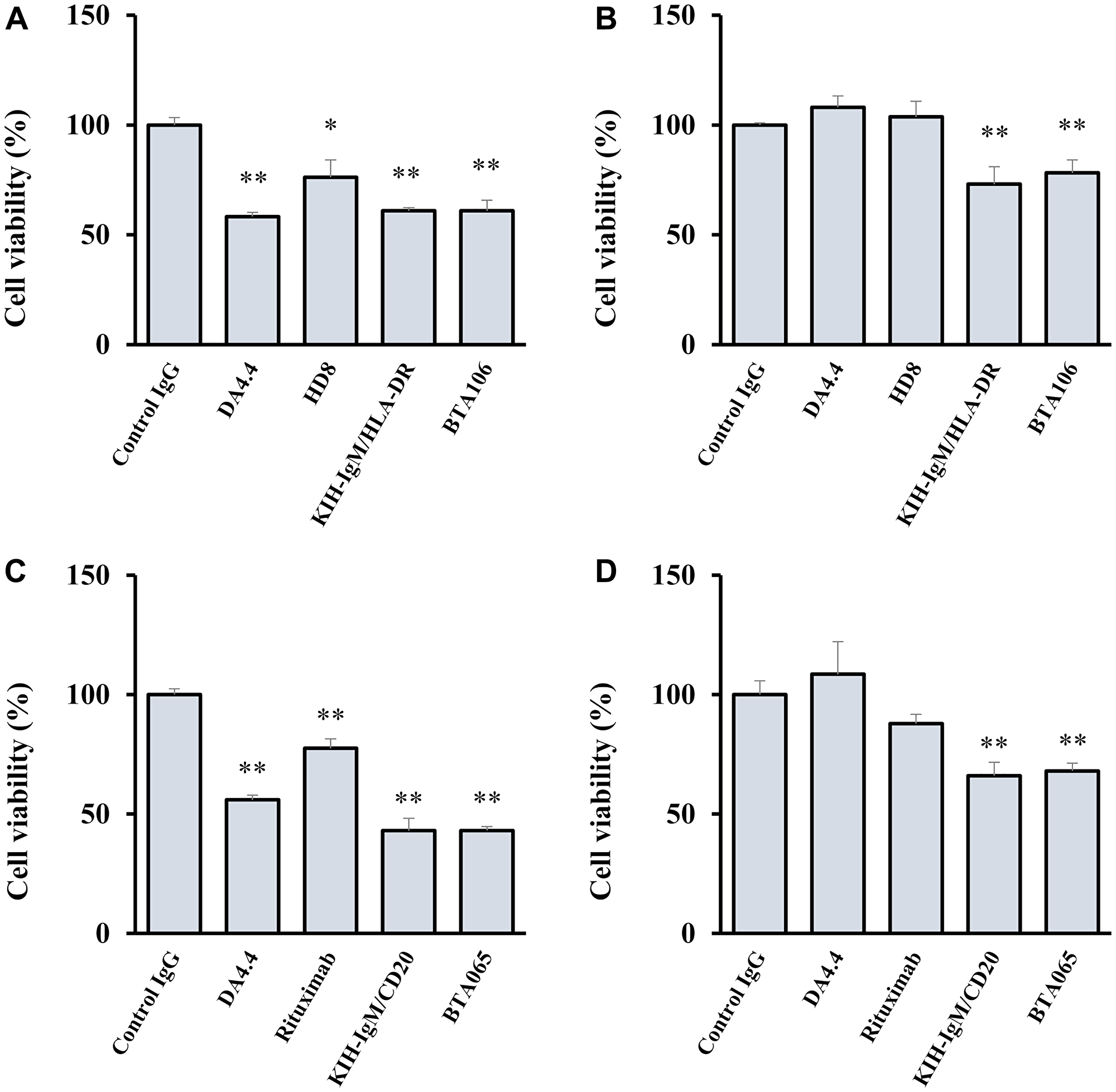
Figure 3: Effects of IgM-dependent bispecific antibodies on cell viability in the presence of human serum.
JeKo-1 cells were treated with BTA106 (A, B), BTA065 (C, D), and their parental antibodies at 1,000 ng/mL for 48 hours in the absence (A, C) or presence (B, D) of human serum (mean ± S.D. of triplicate). Viable cells were measured using the Realtime-Glo MT Cell viability assay. Human serum was obtained from a healthy donor. Statistical analysis was performed with one-way analysis of variance (ANOVA), followed by Dunnett’s test (*p < 0.05, **p < 0.01, versus control IgG).
Induction of apoptosis and cell-cycle arrest by IgM-dependent bispecific antibodies
As shown in previous reports, anti-IgM antibodies inhibit cell proliferation by inducing apoptosis and cell-cycle arrest [28–30]. First, we investigated whether IgM-dependent bispecific antibodies induce apoptosis. After treatment of JeKo-1 cells with antibodies, we analyzed the expression levels of cleaved-PARP and cleaved-caspase3 as apoptotic markers using western blotting [46]. Both cleaved-PARP and cleaved-caspase3 were detected 24 and 36 hours after treating JeKo-1 cells with DA4.4 and BTA106 at 300 ng/mL, whereas they were not detected after treatment with HD8 at 300 ng/mL (Figure 4A). As expected, the induction of cleaved-PARP and cleaved-caspase3 by DA4.4 was attenuated by the addition of 3 μg/mL purified soluble IgM. However, BTA106 retained the ability to increase the levels of these apoptotic factors at 24 and 36 hours even in the presence of 3 μg/mL purified soluble IgM. BTA065 (300 ng/mL) also induced apoptosis in the presence of 3 μg/mL purified soluble IgM (Figure 4B). Interestingly, the expression levels of apoptotic factors induced by BTA065 were higher than those induced by the parental antibodies, DA4.4, and rituximab in the absence of soluble IgM.

Figure 4: Analysis of apoptosis and cell-cycle profiles in the presence of soluble IgM.
(A, B) Expression levels of apoptotic factors in JeKo-1 cells. JeKo-1 cells were treated with antibodies (300 ng/mL) in the absence or presence of purified soluble IgM (3 μg/mL) for the indicated times. The expression levels of cleaved-caspase3 and cleaved-PARP, which are apoptotic factors, were analyzed using western blotting. The left and right sides of the dotted line represent results from different gels. (C) Cell-cycle profile of JeKo-1 cells following treatment with BTA106. JeKo-1 cells were incubated with antibodies (300 ng/mL) in the absence (left) or presence (right) of purified soluble IgM (3 μg/mL) for 24 hours. The treated JeKo-1 cells were permeabilized, stained with propidium iodide, and analyzed using flow cytometry. (D) Cell-cycle profile of JeKo-1 cells following treatment with BTA106 in the absence (left) or presence (right) of human serum. JeKo-1 cells were incubated with antibodies (1,000 ng/mL) in the presence of human serum for 24 hours. Human serum was obtained from a healthy donor.
Second, to investigate the induction of cell-cycle arrest, we performed a cell-cycle analysis 24 hours after the treatment of JeKo-1 cells with antibodies. DA4.4 (300 ng/mL) and BTA106 (300 ng/mL) treatments accumulated cells in the G0/G1 phase and decreased the number of cells in the S and G2/M phases compared with phosphate-buffered saline (PBS) treatment (Figure 4C). DA4.4 failed to induce cell-cycle arrest in the presence of 3 μg/mL purified soluble IgM, whereas BTA106 was still able to induce cell-cycle arrest under the same conditions. Next, we performed the cell-cycle analysis in the presence of human serum. Similar to the presence of purified soluble IgM, DA4.4 (1,000 ng/mL) failed to induce cell-cycle arrest (Figure 4D). However, BTA106 caused an accumulation of cells in the G0/G1 phase in the presence of human serum. Moreover, BTA106 induced cell-cycle arrest when the human serum donor was changed (data not shown).
In vitro activities of BTA106 for B-cell lymphoma cell lines
The cytotoxic activity of BTA106 was evaluated using ADCC, CDC, and cell viability assays with several B-cell lymphoma cell lines. Furthermore, its cytotoxic activities were compared with those of rituximab, which is the standard for initial treatment of NHL [1]. First, to assess the direct inhibitory activity on cell proliferation, B104, RL (GCB-DLBCL), U2932 (ABC-DLBCL), JeKo-1, Ramos (Burkitt’s lymphoma), and RRBL1 (rituximab-resistant DLBCL) cells were treated with antibodies at 1,000 ng/mL for 72 hours, and cell viability was measured using the Cell Counting Kit-8 [47]. The results demonstrated that, in comparison to the control IgG, BTA106 exhibited cytotoxicity against NHL cell lines other than RL (Figure 5A). In addition, the cytotoxic activity of BTA106 was higher than that of rituximab in B104 (72.2 ± 1.5% vs. 1.9 ± 1.5%, data are expressed as mean ± standard deviation), U2932 (44.0 ± 1.1% vs. 23.0 ± 0.6%), JeKo-1 (62.1 ± 2.1% vs. −1.9 ± 4.4%), and Ramos (35.2 ± 1.2% vs. 8.4 ± 2.3%) cells.
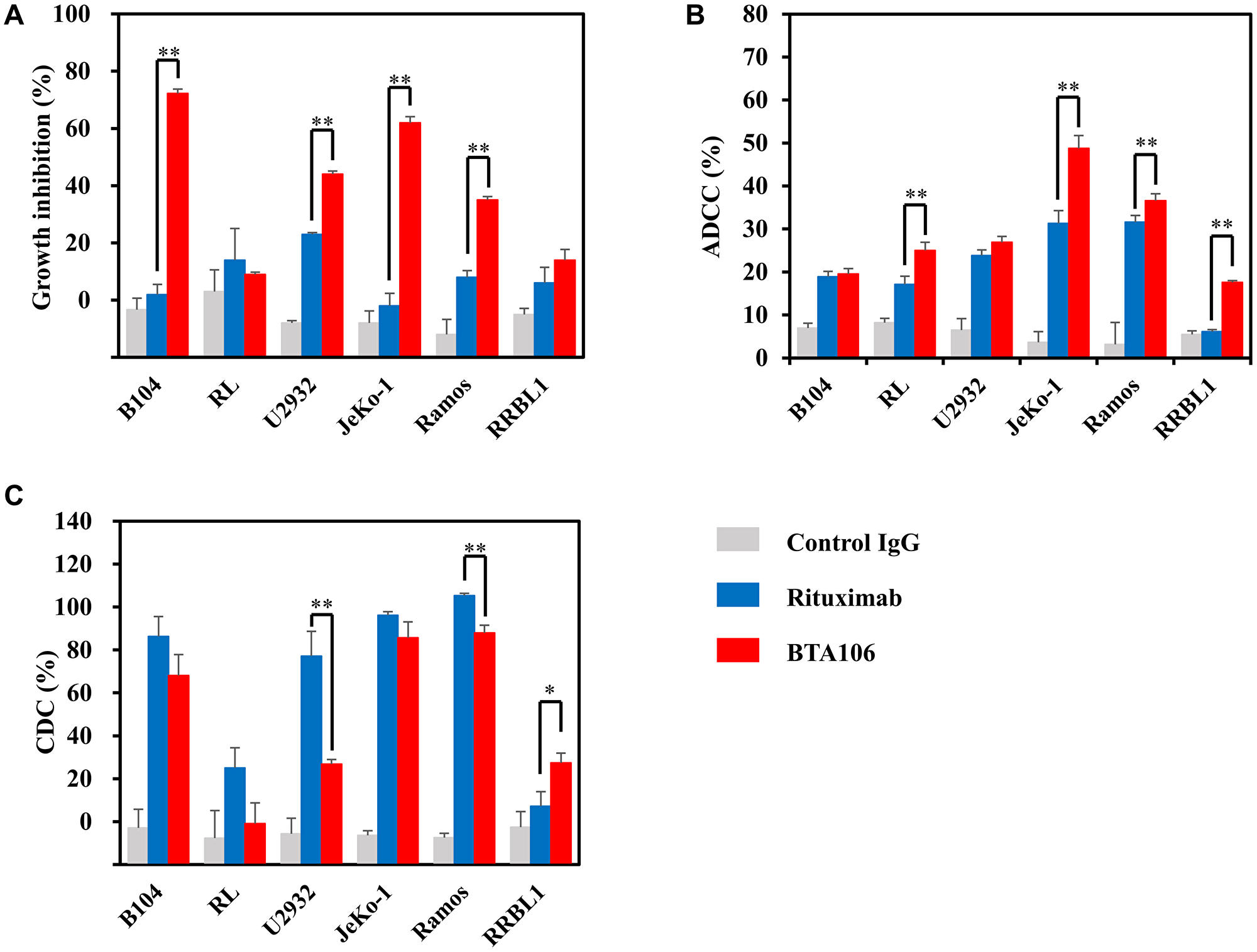
Figure 5: The in vitro activity of BTA106 compared with that of rituximab.
(A) Effects of antibodies on cell viability of several lymphoma cells (mean ± S.D. of triplicate). B104, RL, U2932, JeKo-1, Ramos, and RRBL1 cells were treated with BTA106 or rituximab at 100 ng/mL for 72 hours. Cell viability was measured using the Cell Counting Kit-8. (B) ADCC activity of antibodies against several lymphoma cells (mean ± S.D. of triplicate). B104, RL, U2932, JeKo-1, Ramos, and RRBL1 cells were fluorescently labeled and incubated with human PBMCs and antibodies at 100 ng/mL for 4 hours (mean ± S.D. of triplicate). The E:T ratio was 20:1. Flow cytometry analysis was performed to measure the percentage of the lysed tumor cells. (C) CDC activity of antibodies against several lymphoma cells (mean ± S.D. of triplicate). B104, RL, U2932, JeKo-1, Ramos, and RRBL1 cells were incubated with human complement and antibodies at 1,000 ng/mL for 4 hours. Cell viability was measured using the Cell Counting Kit-8. Statistical analysis was performed with one-way ANOVA, followed by Tukey–Kramer test (*p < 0.05, **p < 0.01).
Next, we assessed the ADCC of the antibodies at 100 ng/mL. Human peripheral blood mononuclear cells (PBMCs) were used as effector cells, and the ratio of effector to target cells was 20:1. BTA106 showed ADCC activity in all NHL cell lines (Figure 5B). Furthermore, the ADCC activity of BTA106 was higher than that of rituximab in RL (25.0 ± 1.9% vs. 17.1 ± 1.0%), JeKo-1 (48.8 ± 3.0% vs. 31.3 ± 2.5%), Ramos (36.6 ± 1.6% vs. 31.6 ± 5.1%), and RRBL1 (17.6 ± 0.4% vs. 6.1 ± 0.8%) cells, and at approximately the same levels as those of rituximab against B104 (19.6 ± 1.3% vs. 18.9 ± 1.1%) and U2932 (26.9 ± 1.3% vs. 23.8 ± 2.7%) cells.
Finally, we assessed the CDC activity of the antibodies at 1,000 ng/mL. The CDC activity of BTA106 was lower than that of rituximab against U2932 (26.8 ± 2.1% vs. 77.1 ± 11.5%) and Ramos (87.9 ± 3.7% vs. 105.3 ± 1.1%) cells (Figure 5C). Although rituximab induced CDC activity against almost all NHL cell lines used in this study, it did not show CDC activity against RRBL1 cells. These data are consistent with the fact that RRBL1 was derived from a rituximab-resistant patient [47]. In contrast, the CDC activity of BTA106 was higher than that of rituximab (27.5 ± 4.5% vs. 7.2 ± 6.8%) in RRBL1 cells. These results suggest that BTA106 is effective against various types of NHL cells, including rituximab-resistant cells.
Selective binding to B-cells by BTA106
As described previously, anti-HLA-DR antibodies are attractive partners of anti-IgM antibodies for the production of bispecific antibodies. HLA-DR is expressed not only on B cells but also on other cells [48]. A high target selectivity is desirable for a therapeutic antibody to avoid adverse events. To clarify whether BTA106 exhibited higher selectivity for B cells than HLA-DR-positive cells, a binding assay of BTA106, DA4.4, and HD8 was performed, as shown in Figure 6. JeKo-1 cells (CD19+, IgM+, HLA-DR+) and HH cells (CD19-, IgM-, HLA-DR+) were mixed at a ratio of 1:5. JeKo-1 cells were labeled with an anti-CD19 antibody conjugated to PE. The cells were then treated with each antibody conjugated to Alexa Fluor 488 (100 ng/mL) for 1 hour and analyzed using flow cytometry. The control IgG did not bind to any of the cells (Figure 6A). DA4.4 was bound only to JeKo-1 cells (Figure 6B), whereas HD8 bound equally to JeKo-1 and HH cells (Figure 6C). In contrast, BTA106 exhibited a higher selectivity for JeKo-1 cells than for HH cells (Figure 6D). These data suggest that BTA106 has a higher specificity for B cells than for other cells.
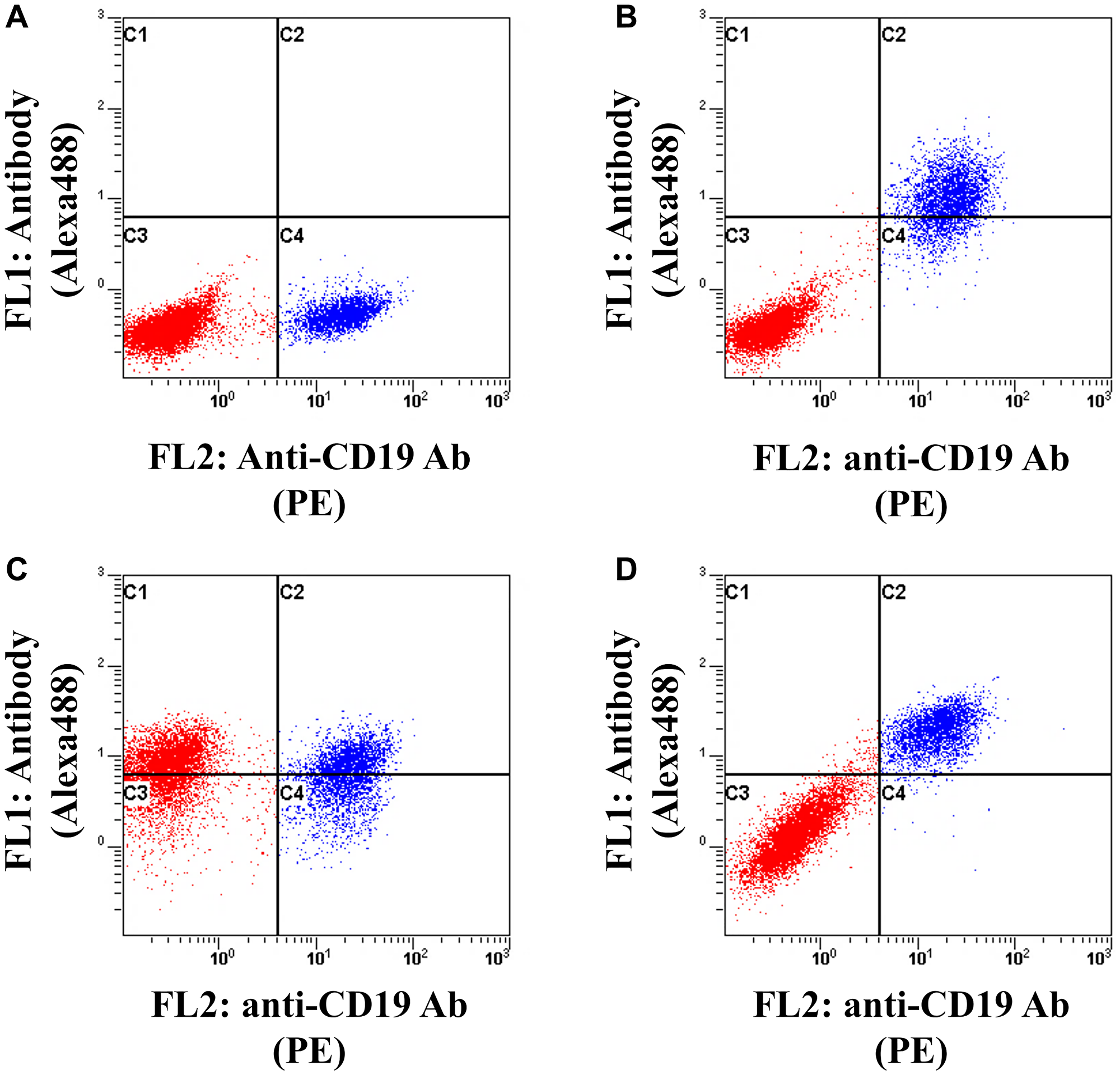
Figure 6: Analysis of selectivity of BTA106 for B cells.
JeKo-1 cells (CD19+, IgM+, HLA-DR+) were labeled with anti-CD19 antibody (HIB19) conjugated with PE and mixed with HH cells (CD19-, IgM-, HLA-DR+) derived from T cell leukemia/lymphoma at a ratio of 1:5. Subsequently, the cells were treated with control IgG (A), DA4.4 (B), HD8 (C), and BTA106 (D) conjugated with Alexa 488 (100 ng/mL) for 1 hour and detected using flow cytometry. Red and blue dots indicate HH cells and JeKo-1 cells, respectively.
B cell-depleting activity of BTA106 in cynomolgus monkeys
We evaluated the B cell-depleting activity of BTA106 in cynomolgus monkeys. At first, B cell depletion was observed in both the peripheral and lymph nodes in a dose escalation study (Supplementary Figure 3A–3C). Subsequently, in a single-administration study, a cynomolgus monkey received BTA106 at a dose of 20 mg/kg intravenously on day 0. BTA106 depleted 96.6% of the peripheral B cells, and the reduction in the number of B cells was maintained until day 14 (Figure 7A). On the other hand, the number of T cells decreased by 33.9% on day 1 and returned to their original level on day 14 (Figure 7B).
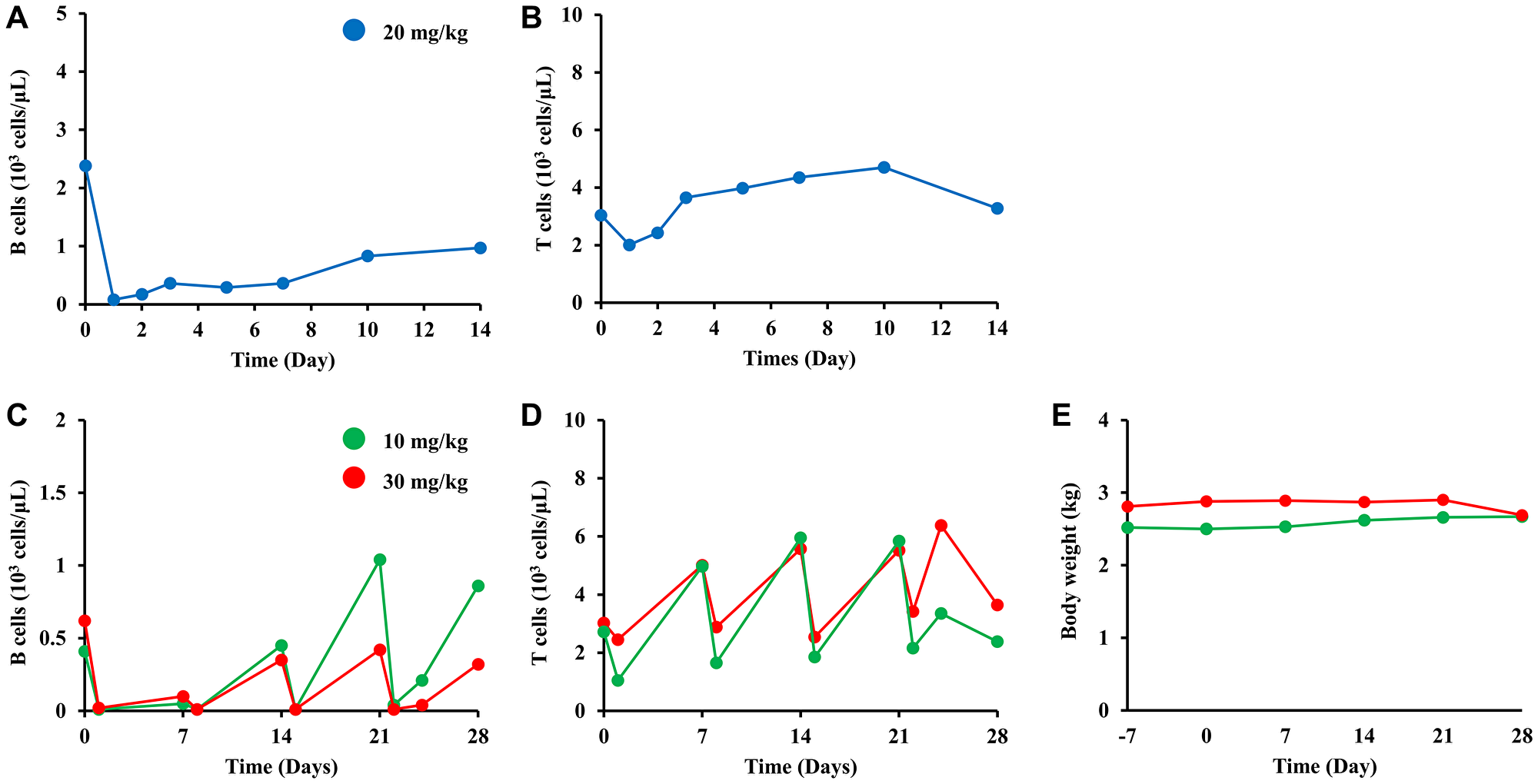
Figure 7: B-cell depletion of BTA106 in cynomolgus monkeys.
The number of B (A) and T cells (B) after single administration of BTA106 in cynomolgus monkeys. A male cynomolgus monkey was intravenously administered BTA106 at 20 mg/kg. B cells were labeled with anti-CD20 antibody (2H7) conjugated with APC, and T cells were labeled with anti-CD3 antibody (SP34-2) conjugated with Alexa 488. The numbers of peripheral B and T cells were measured on days 0, 1, 2, 3, 5, 7, 10, and 14 using flow cytometry. The numbers of B (C) and T cells (D) after repeated administrations of BTA106 in cynomolgus monkeys. Female cynomolgus monkeys were administered BTA106 at 10 or 30 mg/kg intravenously on days 0, 7, 14, and 21. The numbers of peripheral B cells and T cells were measured on days 0, 1, 7, 8, 14, 15, 21, 22, 24 and 28 using flow cytometry. Body weight was measured on days −7, 0, 7, 14, 21, and 28 (E).
Next, we evaluated the efficacy of repeated administrations of BTA106 in cynomolgus monkeys. Cynomolgus monkeys received BTA106 at doses of 10 and 30 mg/kg intravenously on days 0, 7, 14, and 21. After each administration of BTA106, 90.2–98.4% of B cells were rapidly depleted (Figure 7C). However, after the second dose, the number of B cells partially recovered until the next administration. Moreover, in the 10 mg/kg cynomolgus monkeys, recovery was accelerated by each administration. In contrast, BTA106 temporarily reduced the number of T cells, which quickly recovered (Figure 7D). We suspected the existence of anti-drug antibodies (ADAs) because the duration of B-cell depletion induced by BTA106 was shorter than those induced by rituximab and obinutuzumab (type II anti-CD20 antibody) in cynomolgus monkeys, as reported previously [49]. Enzyme-linked immunosorbent assay (ELISA) analysis of ADAs revealed that ADAs were detected from day 14 (Supplementary Figure 4A, 4B). In particular, ADA activity against DA4.4 Fab was higher than that against HD8 Fab in animals treated with BTA106 at 10 mg/kg.
In the repeated-administration study, body weight loss (Figure 7E) and abnormal findings at necropsy were not observed in the animals treated with BTA106. Complete blood counts revealed transient alterations in platelets, neutrophils, eosinophils, basophils, and monocytes after administration of BTA106; however, rapid recovery was observed (Supplementary Figure 5A–5E). Additionally, no abnormalities were observed in the red blood cell parameters (Supplementary Figure 5F–5L). These data suggested that BTA106 was well tolerated.
DISCUSSION
IgM is a notable target for lymphoma therapy; however, the presence of a soluble form disturbs the therapeutic efficacy of conventional anti-IgM antibodies. We succeeded in improving the selectivity for B cells by producing IgM-dependent bispecific antibodies and allowing the antibodies to reach IgM on the B-cell membrane. These bispecific antibodies were effective in the presence of soluble IgM in vitro. Moreover, BTA106, an IgM-dependent bispecific antibody targeting IgM and HLA-DR, depleted B cells after single or repeated administrations in cynomolgus monkeys. Additionally, BTA106 was well tolerated. Our results indicate that IgM-dependent bispecific antibodies are potent therapeutic agents for B-cell malignancies.
Mispairing of antibodies is one of the problems to be solved during the production of bispecific antibodies as it reduces the production efficiency of the desired bispecific antibodies [36]. To improve efficiency, we developed a novel solution called the Cys1m technology [42]. This method involves inserting an ectopic disulfide bond at the CH1-CL interface and deleting the native disulfide bond on one of the parent antibodies to suppress mispairing of the light and heavy chains. The Cys1m technology enables a reduction in the mispairing and the efficient production of IgG-like bispecific antibodies, similar to CrossMab, DuoBody, and DuetMab technologies [39–42]. Because IgG-like bispecific antibodies retain the entire Fc region, they exert ADCC, CDC, and ADCP activities through interactions between Fc and the Fc receptor and have an advantage regardless of pharmacokinetics, as follows [35]. Blinatumomab is an anti-CD19/CD3 bispecific antibody, which has significantly improved the prognosis of B-cell acute lymphoblastic leukemia. However, the lack of the Fc portion and its low molecular weight result in a short half-life and the need for frequent administrations [50]. In contrast, IgG-like bispecific antibodies are expected to have a longer half-life, as these antibodies are recycled through the Fc and FcRn interaction [35]. Using Cys1m technology, we successfully produced several IgM-dependent bispecific antibodies with a full-length IgG-like structure. Because the bispecific antibody consists of two pairs of light and heavy chains from parent antibodies, the CEX analysis revealed a peak positioned centrally between the parent antibodies. As expected, the peaks of purified BTA065 and BTA106 were detected at the midpoint of their respective parental antibodies (Figure 1B, 1C, and Supplementary Figure 1A, 1B). In SDS-PAGE analysis, a 150 kDa band corresponding to the full-length IgG1 antibody was observed for BTA065 and BTA106 under non-reducing conditions (Figure 1D and Supplementary Figure 1C). In contrast, under reducing conditions, a 25 kDa band corresponding to the light chain and a 50 kDa band corresponding to the heavy chain were observed (Figure 1E and Supplementary Figure 1D). These results suggest that the Cys1m technology is a reliable method for producing correct IgG-like bispecific antibodies.
To investigate whether IgM-dependent bispecific antibodies exhibit their effects even in the presence of soluble IgM, we produced bispecific antibodies targeting IgM and B-cell surface antigens including CD20, HLA-DR, CD32b, and CD79b (Table 1). As shown in Figure 2A, 2B, the conventional anti-IgM antibodies lost their efficacy as the concentration of purified soluble IgM increased, whereas BTA106 retained its cytotoxic activity even in the presence of soluble IgM (10 μg/mL). Furthermore, as shown in Figure 2C–2F, the bispecific antibodies targeting other B-cell surface antigens such as CD20, CD32b, and CD79b, instead of HLA-DR, could also exhibit their effects in the presence of purified soluble IgM. The anti-IgM antibody DA4.4 could be replaced with alternative clones, such as 6D9 and 4B9 (Supplementary Figure 2A, 2B). The concentration of the purified soluble IgM used in the cell viability assay was lower than that normally present in human serum. To address this, we supplemented our assay with human serum. When untreated JeKo-1 cells were cultured in 90% human serum for 72 hours, cell death was evident. Therefore, we shortened the incubation time and observed that 48 hours after treatment, the effects on growth inhibition were slightly weaker than those in the previous study under 72 hours, as shown in Figure 2A, 2C. BTA106 and KIH-IgM/HLA-DR retained their cytotoxic effects in human serum, whereas conventional anti-IgM antibodies did not (Figure 3A, 3B). Similar results were obtained for BTA065 and KIH-IgM/CD20 (Figure 3C, 3D), and consistent effects were observed across different donors (data not shown). These findings indicate that IgM-dependent bispecific antibodies are effective even in the presence of soluble antigens and that any molecule present on B cells can be targeted by a pair of anti-IgM antibodies. These effects did not depend on the core structures of the bispecific antibodies.
Anti-IgM antibodies exhibit direct cytotoxicity through apoptosis and cell-cycle arrest, independent of immune mechanisms in the body such as ADCC and CDC [28–30]. This is one of the advantages of anti-IgM antibodies as therapeutic antibodies. Therefore, we investigated whether IgM-dependent bispecific antibodies induce apoptosis and cell-cycle arrest and verified whether these mechanisms are activated in the presence of soluble IgM. Figure 4A, 4B indicate that BTA106 and BTA065 increased the expression of apoptotic factors, cleaved PARP and cleaved caspase 3, similarly to the parental anti-IgM antibody. BTA106 or BTA065, but not the parental anti-IgM antibody, induced the expression of these apoptotic factors even in the presence of soluble IgM. Surprisingly, BTA065 induced the expression of apoptotic factors more potently than the parental anti-IgM antibody. Additionally, the cytotoxic effects of the IgM-dependent bispecific antibodies BTA124 and BTA125 were stronger than those of their parental anti-IgM antibodies (Supplementary Figure 2A, 2B). In Ramos cells, BTA106 exhibited a remarkable enhancement of apoptosis and growth inhibition compared with the parental antibodies (Supplementary Figure 6A, 6B). Mimori et al. [51] reported that cross-linking of anti-IgM and anti-CD20 antibodies induces stronger apoptosis than the anti-IgM antibody alone. Therefore, the advantage of an IgM-dependent bispecific antibody over conventional anti-IgM antibodies is that they not only retain their efficacy in human serum but also enhance apoptosis and inhibit proliferation. Subsequently, we performed a cell-cycle analysis and found that BTA106 induced G0/1 cell-cycle arrest. This effect was also observed in the presence of purified soluble IgM (Figure 4C) and human serum (Figure 4D). These results suggest that IgM-dependent bispecific antibodies induce apoptosis and cell-cycle arrest, even in the presence of soluble IgM, which is consistent with the results of the cell viability assay shown in Figures 2 and 3.
We evaluated the cytotoxic activity of BTA106 in several B-cell lymphoma cell lines and compared its activity with that of rituximab, a standard treatment for B-cell lymphoma. Similar to anti-HLA-DR antibodies, BTA106 exhibited potent ADCC and CDC activity against B-cell lymphoma cells (data not shown). Furthermore, BTA106 showed direct cytotoxic activity similar to that of anti-IgM antibodies (data not shown). Interestingly, BTA106 showed high efficacy against cells derived from ABC-DLBCL and MCL, which have been reported to have a poor prognosis. Approximately 30–40% of patients with B-cell lymphoma develop refractory disease and relapse after rituximab-based therapy, which is a crucial problem in lymphoma treatment [1, 4, 5]. One of the causes of refractory disease and relapse after rituximab-based therapy is the acquisition of resistance due to the loss of CD20 expression. The RRBL1 cells, established by Dr. Tomita, were derived from a patient with DLBCL transformed from follicular lymphoma and showed resistance to rituximab owing to CD20 deletion [47]. BTA106 induced more potent ADCC and CDC in RRBL1 cells than rituximab (Figure 5B, 5C). These results indicate that BTA106 may be effective in patients with various types of lymphomas. Additionally, BTA106 may be effective against rituximab resistance.
HLA-DR is an attractive target for therapeutic antibodies because anti-HLA-DR antibodies have been reported to exhibit high ADCC and CDC activities [43]. However, because HLA-DR is expressed not only on B cells but also on other immune cells, the low cell specificity of anti-HLA-DR antibodies may result in toxicity. As shown in Figure 6C, the anti-HLA-DR antibody bound equally to B and T cells. In fact, Tawara et al. [45] reported that two cynomolgus monkeys treated with the anti-HLA-DR antibody HD8 at 1.5 mg/kg showed severe reactions and died after administration, although it is not clear how HD8 administration resulted in death. In contrast, BTA106 showed improved specificity for B cells compared with HD8 (Figure 6D). Moreover, BTA106 was well tolerated in single- and repeated-administration studies conducted in cynomolgus monkeys. These results suggest that BTA106 exhibits improved toxicity compared with anti-HLA-DR antibodies.
BTA106 exhibited anti-tumor activity through mechanisms such as ADCC, CDC, and direct cytotoxic activity. Among these, ADCC and CDC activities are exerted by effector cells that recognize the Fc region of the antibody [35]. As BTA106 possesses a human Fc region and is not recognized by mouse effector cells, we considered that the anti-tumor activity of BTA106 would be significantly reduced in mouse xenograft models transplanted with human lymphoma tumors. However, the human Fc region is recognized by effector cells in cynomolgus monkeys and is therefore widely used to evaluate its therapeutic efficacy of lymphoma drugs [49, 52, 53]. Therefore, we chose cynomolgus monkeys as an animal model to evaluate the efficacy in vivo. In single-administration and repeated-administration studies, BTA106 rapidly depleted B cells (Figure 7A, 7C). Moreover, the disappearance of lymph follicles was observed after BTA106 administration in the dose-escalation study, indicating that BTA106 reduces B cells in both the peripheral and lymphoid tissues (Supplementary Figure 3A–3C). These results indicate that BTA106 functions in the body, where soluble IgM is abundant and can target B-cell lymphoma cells in both peripheral and lymph tissues. Because BTA106 reduces B cells, which are crucial for both adaptive and innate immunity, it may cause infection-related adverse events similar to other therapeutic antibodies for B-cell lymphoma, such as rituximab, obinutuzumab, tafasitamab, and polatuzumab vedotin. However, these adverse events can be controlled by symptomatic treatment or discontinuation of the medication. In addition, the frequency of infection-related adverse events (Grade ≥3) was low in rituximab-based therapy (7.5–12.6%), obinutuzumab-based therapy (7.6–12.0%), polatuzumab vedotin and rituximab-based therapy (15.2%), and tafasitamab-based therapy (all infective pneumonia 9.9%, all urinary tract infection 2.5%, and upper respiratory tract infection 2.5%) [54–56]. These antibody drugs were approved because their benefits were considered to outperform these adverse events. Our bispecific antibody may cause infection-related adverse events similar to the approved antibody drugs. However, we believe that the benefits of anti-tumor effects outperform the cytotoxic effects, and our bispecific antibody will represent a major advancement in the treatment of poor-prognosis ABC-DLBCL and U-CLL.
In a study of repeated administrations, B-cell recovery was accelerated with each administration, which is thought to be due to the development of ADAs. Indeed, high levels of ADAs against DA4.4 Fab were detected in serum after repeated administrations of BTA106 (Supplementary Figure 4A). Therefore, it is necessary to select alternative anti-IgM antibody clones, such as 4B9 and 6D9, to avoid ADA generation. We evaluated the toxicity in these experiments, and no severe toxicity was observed. For example, the body weight of the cynomolgus monkeys did not change after single (data not shown) or repeated administrations (Figure 7E). In addition, unlike B cells, the disappearance of T cells was not observed (Figure 7B, 7G), indicating that BTA106 has a high selectivity for B cells. These results suggest that BTA106 is a notable therapeutic antibody for B-cell lymphoma. We assessed the therapeutic efficacy of BTA106 based on the reduction of normal B-cell levels in cynomolgus monkeys. However, its efficacy on B-cell malignancies remains unknown in vivo. Furthermore, as DA4.4 does not bind to the mouse IgM, accurate evaluation is not feasible in animal models such as patient-derived xenografts. Thus, to assess the true therapeutic efficacy of BTA106, clinical trials are necessary.
In conclusion, IgM-dependent bispecific antibodies could overcome the decrease in anti-IgM antibody activity caused by soluble IgM. Additionally, the bispecific antibody modality is one of the approaches employed to address the interference of antibody efficacy by soluble antigens. Among IgM-dependent bispecific antibodies, the anti-IgM/HLA-DR antibody showed high activity in vitro and in vivo, indicating its potential as a therapeutic antibody for B-cell lymphoma. Specifically, it is expected to be a new treatment option for ABC-DLBCL, MCL, and rituximab-resistant B-cell lymphoma, which have a poor prognosis.
Materials and Methods
Antibodies
The sequences of anti-human IgM (clone DA4.4) [57], anti-human HLA-DR (clone HD8) [58], anti-human CD20 (clone IDEC-C2B8, rituximab), anti-human CD32b (clone 5A6) [59], anti-CD79b antibody (clone #89), anti-human IgM (clones 4B9 and 6D9), and anti-human EGFR (clone h425) antibodies are described in Supplementary Table 1. The numbering of CDR was based on the Kabat numbering system [60]. h425 was used as the control IgG. Clones #89, # 4B9, and # 6D9 were obtained as follows. BALB/c mice were immunized with CD79b-expressing Sp2/0 cells (mouse myeloma cells, CRL-1581, American Type Culture Collection, ATCC) or purified human IgM (OBT1524, Bio-Rad). Subsequently, #89, #4B9, and #6D9 were cloned from the hybridomas obtained by fusing Sp2/0 cells with spleen cells from immunized mice. Other DNA sequences for the heavy and light chains of the antibodies were synthesized (Eurofins Genomics) and fused with the 5′ end of the human IgG1 heavy chain constant region and 5′ end of the kappa light chain constant region, respectively. The constant region of the human antibody IgG1 was amplified from cDNA derived from human peripheral blood leukocytes (C1234148-10, Fujifilm Wako Pure Chemical). All antibodies were cloned into the pcDNA 3.1 (+) vector (V79020, Thermo Fisher Scientific). The bispecific antibodies used in this study are listed in Table 1. Bispecific antibodies were generated using the Cys1m or KIH technology. Importantly, control bispecific antibodies were produced using KIH technology to clarify whether differences in the core structure affected antibody activity. Cys1m (g) was introduced at the CL-CH1 interface of anti-IgM antibodies, and the Fab of antibodies against other B-cell antigens was wild-type (Figure 1A). In the case of KIH, the heavy chain of DA4.4 was introduced with a knob mutation (T366W), and the heavy chains of other B-cell antigen antibodies (HD8 and rituximab) were introduced with hole mutations (T366S, L368A, and Y407V). Cys1m and KIH mutations were introduced using the PrimeSTAR Mutagenesis Basal Kit (R046A, TaKaRa Bio). The numbering of Cys1m and KIH mutations was based on the EU numbering system [61]. Antibodies were expressed using the ExpiCHO Expression System Kit (A29133, Thermo Fisher Scientific). To produce bispecific antibodies containing Cys1m (g) and KIH, vectors for the two parental antibodies were simultaneously transfected into ExpiCHO cells at a 1:1 ratio. Subsequently, the antibodies were purified from the supernatants of ExpiCHO cells cultured for 10–14 days after transfection, using a HiTrap Protein A HP Column (17040201, Cytiva). Next, the bispecific antibodies were purified using CEX (PL-SCX 1000Å 8 μm, Agilent). Mobile phase A consisted of 10 mM 2-(N-morpholino) ethanesulfonic acid (MES, pH 6.5, 349-01623, Fujifilm Wako Pure Chemical), and mobile phase B consisted of 10 mM MES and 500 mM sodium chloride (pH 6.5, 191-01665, Fujifilm Wako Pure Chemical). The flow rate was 1.0 mL/min with a linear gradient of 0.8%/min from 2% to 40% of mobile phase B. After sample loading, absorption at 280 nm was recorded, and the elution behavior of the proteins was monitored. Finally, bispecific antibodies were dialyzed against PBS (#9808S, Cell Signaling Technology). BTA106 used in the cynomolgus monkey study was purified using a process different from that used for BTA106 in the in vitro assay. BTA106 used in the cynomolgus monkey study was purified using protein A affinity chromatography (ProSep-vA, 113115827, MERCK) and CEX (Nuvia HR-S, 1560513, Bio-Rad). Additionally, BTA106 was purified using hydrophobic interaction chromatography (CHT Type1, 7324755, Bio-Rad) to remove endotoxins. The loading buffer was 10 mM sodium phosphate buffer (pH 6.5), consisting of disodium hydrogen phosphate 12-water (196-02835, Fujifilm Wako Pure Chemical) and sodium dihydrogen phosphate dihydrate (192-02815, Fujifilm Wako Pure Chemical). The elution buffer contained 5 mM sodium phosphate and 1.5 M sodium chloride (pH 6.5). Finally, BTA106 was dialyzed against citrate buffer composed of 25 mM sodium citrate (191-01785, Fujifilm Wako Pure Chemical), 150 mM sodium chloride, and 0.014% Tween 80 (P1754-500ML, Merck) and was filtered through a 0.22 μm filter (ADVANTEC).
Analysis of antibodies
Antibodies were analyzed using CEX (PL-SCX 1000Å, 8 μm) and SDS-PAGE under non-reducing or reducing (50 mM dithiothreitol, 044-29221, Fujifilm Wako Pure Chemical) conditions. CEX analysis was performed using the conditions described in the section titled “Antibodies” in the Materials and Methods. The gels were stained with Coomassie Brilliant Blue R250 (27816-25G, MERCK) and digitally imaged using LAS 4000 (Cytiva). The molecular weights of the whole antibody (150 kDa), heavy chain (50 kDa), and light chain (25 kDa) were calculated using protein markers (1610375, Bio-Rad).
Cell lines
The human B-cell lymphoma cell lines JeKo-1 (CRL-3006), RL (CRL-2261), and Ramos (CRL-1596) and human T-cell lymphoma HH cells (CRL-2105) were obtained from the ATCC. U2932 (ACC-633) and B104 (JCRB0117) were obtained from the German Collection of Microorganisms and Cell Cultures (DSMZ) and the Japanese Collection of Research Bioresources (JCRB) cell bank, respectively. The rituximab-resistant DLBCL cell line RRBL1 was kindly gifted by Dr. Akihiro Tomita (Fujita Health University School of Medicine, Aichi, Japan) [47]. JeKo-1 cells were cultured in RPMI 1640 (11875-119, Thermo Fisher Scientific) supplemented with 20% fetal bovine serum (FBS, Thermo Fisher Scientific), 100 U/mL penicillin, and 100 μg/mL streptomycin (P0781-100ML, MERCK) at 37°C in a 5% CO2 incubator. B104, RL, Ramos, HH, U2932, and RRBL1 cells were cultured in RPMI 1640 supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C in a 5% CO2 incubator. ExpiCHO cells were cultured in ExpiCHO expression medium (A2910001, Thermo Fisher Scientific) at 37°C in an 8% CO2 incubator on an orbital shaker (125 rpm). All cell lines were utilized within 6 months after resuscitation.
Cell viability assay
Tumor cells were incubated with antibodies for 72 hours in each culture medium with or without different concentrations of purified soluble IgM in a 5% CO2 incubator at 37°C. Cell viability was determined using the Cell Counting Kit-8 (343-07623, Dojindo Laboratories). The maximum cell death was determined by lysis in 1% Triton X-100 (X100-100ML, MERCK). The absorbance was measured at 450 nm using iMark (Bio-Rad) to calculate cell viability. The incubation time for the cells treated with or without 90% human serum and 10% culture medium was 48 hours. Human serum was isolated from the blood of a healthy human. Cell viability was determined using the real-time Glo MT cell viability assay (G9711, Promega). Luminescence was measured using GloMax Explorer (GM3500; Promega).
Western blotting
Tumor cells were incubated with antibodies for the indicated times in Figure 4A, 4B. The cells were harvested and homogenized in RIPA buffer containing 25 mM Tris-HCl (207-06275, Fujifilm Wako Pure Chemical), 150 mM sodium chloride, 1% Nonidet P40 substitute (492016, MERCK), 0.25% sodium deoxycholate (044-18812, Fujifilm Wako Pure Chemical), 0.1% SDS, and 1% protease inhibitor (539134, MERCK). The protein concentration in the cell lysates was measured using the Pierce BCA protein assay kit (23227, Pierce). SDS-PAGE sample buffer was added to 5 μg of the cell lysate and incubated at 95°C for 5 minutes. Subsequently, the lysates were subjected to SDS-PAGE using 4–20% gradient polyacrylamide gels and transferred to a nitrocellulose membrane (170-4158, BIO-RAD). The blots were blocked with 5% skim milk (198-10605, Fujifilm Wako Pure Chemical) in tris buffered saline with tween 20 containing 10 mM Tris-HCl, 150 mM sodium chloride, and 0.05% Tween 20 (P7949-100ML, MERCK) and incubated with anti-cleaved-caspase 3 (#9664, Cell Signaling Technology), anti-cleaved PARP (#5625, Cell Signaling Technology), and anti-actin β antibodies (#4970, Cell Signaling Technology). The blots were detected using a horseradish peroxidase (HRP)-conjugated anti-rabbit IgG antibody (#7074, Cell Signaling Technology) and an enhanced chemiluminescence reagent (RPN2232, Cytiva). The blots were imaged using the LAS 4000 system (Cytiva).
Cell-cycle analysis
Tumor cells were treated with antibodies and incubated at 37°C in a 5% CO2 incubator for 24 hours. The cells were stored in 70% ethanol at −20°C for over 24 hours for permeabilization and to fix the cells. They were then stained with 25 μg/mL propidium iodide (PI, P4170, MERCK) for 30 minutes and analyzed using flow cytometry (CYTOMICS FP 500, Beckman Coulter).
Assessment of apoptotic cells
Ramos cells were treated with antibodies (1,000 ng/mL) at 37°C in a 5% CO2 incubator for 24 hours. The cells were fixed with 1% glutaraldehyde (073-00536, Fujifilm Wako Pure Chemical) for 24 hours. The cells were stained with 4′,6-diamidino-2-phenylindole and observed under a fluorescence microscope (FluoView FV10i, Olympus). Cells with condensed and fragmented nuclei were counted. The percentage of apoptotic cells was calculated as the number of apoptotic cells per count of total cells.
Cytotoxicity assays
In the ADCC assay, tumor cells were stained with PKH67 (MINI67-1KT, MERCK) and incubated with antibodies and human PBMCs as effector cells for 3 hours under 5% CO2 at 37°C. The effector to target ratio was 20:1. PBMCs were isolated from the blood of a healthy donor using Lympholyte (CL5115, Cedarlane Laboratories). The lysed cells were stained with PI for 30 minutes, and the percentage of lysed tumor cells stained with PKH67 and PI was measured using flow cytometry. In the CDC assay, tumor cells were incubated with antibodies and human complement (S1764-5X1ML, MERCK) for 4 hours in 5% CO2 at 37°C. Cell viability assays were performed using the Cell Counting Kit-8. The absorbance was measured at 450 nm using iMark (Bio-Rad). Maximum cell death was determined by lysis in 1% Triton X-100 (X100-100ML, MERCK).
Binding assays
The antibodies were conjugated with Alexa Fluor 488 using an Alexa Fluor 488 Monoclonal Antibody Labeling Kit (A20181, Thermo Fisher Scientific). JeKo-1 cells were labeled with an anti-human CD19 antibody conjugated with phycoerythrin (clone HIB19, 555413, BD Biosciences) and mixed with HH cells at a ratio of 1:5. Subsequently, the mixed cells were treated with antibodies conjugated with Alexa Fluor 488 for 1 hour, followed by flow cytometry.
Evaluation of efficacy and toxicity in cynomolgus monkeys
A single-dose study and dose-escalation study of BTA106 were conducted at Shin Nippon Biomedical Laboratories, Ltd. (Japan). In the single-administration study, BTA106 (20 mg/kg) was injected intravenously into male cynomolgus monkeys on day 0, and blood samples were collected on days 0, 1, 2, 3, 5, 7, 10, and 14. B and T cells were labeled with an anti-CD20 antibody conjugated with allophycocyanin (APC, clone 2H7, BioLegend) and an anti-CD3 antibody conjugated with Alexa Fluor 488 (clone SP34-2, BD Biosciences), respectively. The number of B and T cells was measured using flow cytometry (FACSCalibur, BD Biosciences). A repeated-administration study of BTA106 was conducted at the BoZo Research Center, Inc. (Japan). In the repeated administration study, BTA106 (10 and 30 mg/kg) was injected intravenously into female cynomolgus monkeys on days 0, 7, 14, and 28, and blood samples were collected on days 0, 1, 7, 8, 14, 15, 21, 22, 28, and 29. B and T cells were labeled with an anti-CD20 antibody conjugated with APC (clone 2H7) and an anti-CD3 antibody conjugated with Alexa Fluor 488 (clone SP34-2), respectively, and analyzed using flow cytometry (BD FACSCanto, BD Biosciences). Body weights were measured on days -7, 0, 7, 14, 21, and 28. In the dose-escalation study, a female cynomolgus monkey was intravenously administered BTA106 at doses of 1, 3, 10, and 20 mg/kg on days 0, 2, 4, and 6, respectively. A cynomolgus monkey was sacrificed, and axillary lymph nodes were sampled on day 8. Axillary lymph nodes were stained with hematoxylin (131-09665, Fujifilm Wako Pure Chemical) and eosin (050-06041, Fujifilm Wako Pure Chemical).
Hematological analysis
Hematological analysis was performed in a repeated-administration study. Blood samples were collected from cynomolgus monkeys on days 0, 1, 7, 8, 14, 15, 21, 22, 28, and 29. Values of platelets (104/μL), neutrophils (%), eosinophils (%), basophils (%), monocytes (%), red blood cells (104/μL), mean corpuscular volume (MCV, fL), mean corpuscular hemoglobin (MCH, pg), mean corpuscular hemoglobin concentration (MCHC, g/dL), reticulocytes (%), hemoglobin (g/dL), and hematocrit (%) were measured using ADVIA2120i (Siemens Healthineers).
ELISA for detecting ADAs
Cynomolgus serum samples were collected on days 0, 1, 7, 8, 14, 15, 21, 22, 24, and 28. DA4.4 Fab or HD8 Fab digested with papain (P3125-25MG, MERCK) was coated onto a 96-well microplate. Subsequently, it was incubated for 1 hour with serum obtained from cynomolgus monkeys diluted 1/100 in PBS. After washing with PBS with tween 20, the captured ADAs were incubated with an anti-cynomolgus IgG Fc antibody conjugated with horseradish peroxidase (SAB3700766, MERCK). Peroxidase substrate (5120-0032, SeraCare Life Science) was added to the plate and incubated for 15 minutes, followed by the measurement of absorbance at 405 nm using a microplate reader (iMark, Bio-Rad).
Statistics
Data are presented as the mean ± standard deviation (S.D.). Multiple comparisons were performed using one-way analysis of variance (ANOVA) with the Tukey–Kramer or Dunnett’s multiple comparison tests. Statistical significance was determined using Excel Statistics Ver. 7 (ESUMI). Differences were considered significant when the P-value was < 0.05.
Abbreviations
ABC: Activated B cell-like; ADAs: Anti-drug antibodies; ADCC: Antibody-dependent cell-mediated cytotoxicity; ADCP: Antibody-dependent cellular phagocytosis; ANOVA: Analysis of variance; APC: allophycocyanin; ATCC: American type culture collection; BCR: B-cell receptor; CAR-T: Chimeric antigen receptor T cell; CD: Cluster of differentiation; CDC: Complement-dependent cell cytotoxicity; CDR: Complementarity determining region; CEX: Cation exchange chromatography; CH1: Constant domain 1 of heavy chain; CL: Constant domain of light chain; CLL: Chronic lymphocytic leukemia; DLBCL: Diffuse large B-cell lymphoma; DSMZ: German Collection of Microorganisms and Cell Cultures; EGFR: Epithelial growth factor receptor; ELISA: Enzyme-linked immunosorbent assay; FBS: Fetal bovine serum; GCB: Germinal center B cell-like; HRP: Horseradish peroxidase; HLA: Human leukocyte antigen; IGHV: Immunoglobulin heavy chain variable region; JCRB: Japanese collection of research bioresources; KIH: knobs-into-holes; MCL: Mantle cell lymphoma; M-CLL: Mutated-CLL; MES: 2-(N-morpholino) ethanesulfonic acid; NHL: Non-Hodgkin’s lymphoma; PBMCs: Peripheral blood mononuclear cells; PBS: Phosphate-buffered saline; PD-L1: Programmed death-ligand 1; PI: Propidium iodide; SD: Standard deviation; SDS-PAGE: Sodium dodecyl sulfate-polyacrylamide gel electrophoresis; U-CLL: Unmutated-CLL.
ACKNOWLEDGMENTS
The authors thank Yoko Tanaka, Airi Hara, Satoko Tatebe, Kazutaka Oda, Toshifumi Kobayashi, and Hayuma Otsuka for technical assistance; Dr. Junpei Enami for helpful discussions; and Dr. Yoshimasa Yamaguchi and Dr. Kazuhiko Haruta for comments on the previous version of the manuscript. The authors thank Dr. Akihiko Tomita (Department of Hematology, Fujita Health University) for kindly providing the RRBL1 cells.
CONFLICTS OF INTEREST
Takahiro Ohashi, Sayuri Terada, Shinsuke Hiramoto, Yuko Nagata, Hirokazu Suzuki, Hitoshi Miyashita, Tetsuo Sasaki, Yasukatsu Tsukada and Keiko Fukushima are employees of Zenyaku Kogyo Co., Ltd.
ETHICAL STATEMENT
Animal experiments were conducted following protocols approved by the Animal Experimental Investigations at Zenyaku Kogyo Co., Ltd., Shin Nippon Biomedical Laboratories, Ltd., and the BoZo Research Center Inc. (Approval Numbers: 1. Dose escalation study: 16-43. 2. Single-administration study: 17-6. 3. Repeated-administration study: 18-18). Experiments with human samples were conducted following the protocols approved by the ethics committee of Zenyaku Kogyo Co., Ltd., (Approval number: 2017-04).
CONSENT
Human samples were obtained after obtaining written informed consent, in accordance with the Declaration of Helsinki.
FUNDING
No funding was used for this paper.
References
1. Salles G, Barrett M, Foà R, Maurer J, O’Brien S, Valente N, Wenger M, Maloney DG. Rituximab in B-Cell Hematologic Malignancies: A Review of 20 Years of Clinical Experience. Adv Ther. 2017; 34:2232–73. https://doi.org/10.1007/s12325-017-0612-x. [PubMed].
2. Cohen MD, Keystone E. Rituximab for Rheumatoid Arthritis. Rheumatol Ther. 2015; 2:99–111. https://doi.org/10.1007/s40744-015-0016-9. [PubMed].
3. Kaegi C, Wuest B, Schreiner J, Steiner UC, Vultaggio A, Matucci A, Crowley C, Boyman O. Systematic Review of Safety and Efficacy of Rituximab in Treating Immune-Mediated Disorders. Front Immunol. 2019; 10:1990. https://doi.org/10.3389/fimmu.2019.01990. [PubMed].
4. Vitolo U, Trněný M, Belada D, Burke JM, Carella AM, Chua N, Abrisqueta P, Demeter J, Flinn I, Hong X, Kim WS, Pinto A, Shi YK, et al. Obinutuzumab or Rituximab Plus Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone in Previously Untreated Diffuse Large B-Cell Lymphoma. J Clin Oncol. 2017; 35:3529–37. https://doi.org/10.1200/JCO.2017.73.3402. [PubMed].
5. Sehn LH, Salles G. Diffuse Large B-Cell Lymphoma. N Engl J Med. 2021; 384:842–58. https://doi.org/10.1056/NEJMra2027612. [PubMed].
6. Czuczman MS, Olejniczak S, Gowda A, Kotowski A, Binder A, Kaur H, Knight J, Starostik P, Deans J, Hernandez-Ilizaliturri FJ. Acquirement of rituximab resistance in lymphoma cell lines is associated with both global CD20 gene and protein down-regulation regulated at the pretranscriptional and posttranscriptional levels. Clin Cancer Res. 2008; 14:1561–70. https://doi.org/10.1158/1078-0432.CCR-07-1254. [PubMed].
7. Hiraga J, Tomita A, Sugimoto T, Shimada K, Ito M, Nakamura S, Kiyoi H, Kinoshita T, Naoe T. Down-regulation of CD20 expression in B-cell lymphoma cells after treatment with rituximab-containing combination chemotherapies: its prevalence and clinical significance. Blood. 2009; 113:4885–93. https://doi.org/10.1182/blood-2008-08-175208. [PubMed].
8. Jazirehi AR, Vega MI, Bonavida B. Development of rituximab-resistant lymphoma clones with altered cell signaling and cross-resistance to chemotherapy. Cancer Res. 2007; 67:1270–81. https://doi.org/10.1158/0008-5472.CAN-06-2184. [PubMed].
9. Olejniczak SH, Hernandez-Ilizaliturri FJ, Clements JL, Czuczman MS. Acquired resistance to rituximab is associated with chemotherapy resistance resulting from decreased Bax and Bak expression. Clin Cancer Res. 2008; 14:1550–60. https://doi.org/10.1158/1078-0432.CCR-07-1255. [PubMed].
10. Salles G, Duell J, González Barca E, Tournilhac O, Jurczak W, Liberati AM, Nagy Z, Obr A, Gaidano G, André M, Kalakonda N, Dreyling M, Weirather J, et al. Tafasitamab plus lenalidomide in relapsed or refractory diffuse large B-cell lymphoma (L-MIND): a multicentre, prospective, single-arm, phase 2 study. Lancet Oncol. 2020; 21:978–88. https://doi.org/10.1016/S1470-2045(20)30225-4. [PubMed].
11. Sehn LH, Herrera AF, Flowers CR, Kamdar MK, McMillan A, Hertzberg M, Assouline S, Kim TM, Kim WS, Ozcan M, Hirata J, Penuel E, Paulson JN, et al. Polatuzumab Vedotin in Relapsed or Refractory Diffuse Large B-Cell Lymphoma. J Clin Oncol. 2020; 38:155–65. https://doi.org/10.1200/JCO.19.00172. [PubMed].
12. Westin JR, Kersten MJ, Salles G, Abramson JS, Schuster SJ, Locke FL, Andreadis C. Efficacy and safety of CD19-directed CAR-T cell therapies in patients with relapsed/refractory aggressive B-cell lymphomas: Observations from the JULIET, ZUMA-1, and TRANSCEND trials. Am J Hematol. 2021; 96:1295–312. https://doi.org/10.1002/ajh.26301. [PubMed].
13. Vercellino L, Di Blasi R, Kanoun S, Tessoulin B, Rossi C, D’Aveni-Piney M, Obéric L, Bodet-Milin C, Bories P, Olivier P, Lafon I, Berriolo-Riedinger A, Galli E, et al. Predictive factors of early progression after CAR T-cell therapy in relapsed/refractory diffuse large B-cell lymphoma. Blood Adv. 2020; 4:5607–15. https://doi.org/10.1182/bloodadvances.2020003001. [PubMed].
14. Seda V, Mraz M. B-cell receptor signalling and its crosstalk with other pathways in normal and malignant cells. Eur J Haematol. 2015; 94:193–205. https://doi.org/10.1111/ejh.12427. [PubMed].
15. Kraus M, Alimzhanov MB, Rajewsky N, Rajewsky K. Survival of resting mature B lymphocytes depends on BCR signaling via the Igalpha/beta heterodimer. Cell. 2004; 117:787–800. https://doi.org/10.1016/j.cell.2004.05.014. [PubMed].
16. Saba NS, Liu D, Herman SE, Underbayev C, Tian X, Behrend D, Weniger MA, Skarzynski M, Gyamfi J, Fontan L, Melnick A, Grant C, Roschewski M, et al. Pathogenic role of B-cell receptor signaling and canonical NF-κB activation in mantle cell lymphoma. Blood. 2016; 128:82–92. https://doi.org/10.1182/blood-2015-11-681460. [PubMed].
17. Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, Kohlhammer H, Lamy L, Zhao H, Yang Y, Xu W, Shaffer AL, Wright G, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010; 463:88–92. https://doi.org/10.1038/nature08638. [PubMed].
18. Profitós-Pelejà N, Santos JC, Marín-Niebla A, Roué G, Ribeiro ML. Regulation of B-Cell Receptor Signaling and Its Therapeutic Relevance in Aggressive B-Cell Lymphomas. Cancers (Basel). 2022; 14:860. https://doi.org/10.3390/cancers14040860. [PubMed].
19. Burger JA, Chiorazzi N. B cell receptor signaling in chronic lymphocytic leukemia. Trends Immunol. 2013; 34:592–601. https://doi.org/10.1016/j.it.2013.07.002. [PubMed].
20. Ferrer G, Montserrat E. Critical molecular pathways in CLL therapy. Mol Med. 2018; 24:9. https://doi.org/10.1186/s10020-018-0001-1. [PubMed].
21. Alnassfan T, Cox-Pridmore MJ, Taktak A, Till KJ. Mantle cell lymphoma treatment options for elderly/unfit patients: A systematic review. EJHaem. 2022; 3:276–90. https://doi.org/10.1002/jha2.311. [PubMed].
22. Jares P, Campo E. Advances in the understanding of mantle cell lymphoma. Br J Haematol. 2008; 142:149–65. https://doi.org/10.1111/j.1365-2141.2008.07124.x. [PubMed].
23. Susanibar-Adaniya S, Barta SK. 2021 Update on Diffuse large B cell lymphoma: A review of current data and potential applications on risk stratification and management. Am J Hematol. 2021; 96:617–29. https://doi.org/10.1002/ajh.26151. [PubMed].
24. Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, Powell JI, Yang L, Marti GE, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000; 403:503–11. https://doi.org/10.1038/35000501. [PubMed].
25. Ruminy P, Etancelin P, Couronné L, Parmentier F, Rainville V, Mareschal S, Bohers E, Burgot C, Cornic M, Bertrand P, Lenormand B, Picquenot JM, Jardin F, et al. The isotype of the BCR as a surrogate for the GCB and ABC molecular subtypes in diffuse large B-cell lymphoma. Leukemia. 2011; 25:681–88. https://doi.org/10.1038/leu.2010.302. [PubMed].
26. Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999; 94:1848–54. [PubMed].
27. D’Avola A, Drennan S, Tracy I, Henderson I, Chiecchio L, Larrayoz M, Rose-Zerilli M, Strefford J, Plass C, Johnson PW, Steele AJ, Packham G, Stevenson FK, et al. Surface IgM expression and function are associated with clinical behavior, genetic abnormalities, and DNA methylation in CLL. Blood. 2016; 128:816–26. https://doi.org/10.1182/blood-2016-03-707786. [PubMed].
28. Banerji L, Glassford J, Lea NC, Thomas NS, Klaus GG, Lam EW. BCR signals target p27(Kip1) and cyclin D2 via the PI3-K signalling pathway to mediate cell cycle arrest and apoptosis of WEHI 231 B cells. Oncogene. 2001; 20:7352–67. https://doi.org/10.1038/sj.onc.1204951. [PubMed].
29. Carey GB, Scott DW. Role of phosphatidylinositol 3-kinase in anti-IgM- and anti-IgD-induced apoptosis in B cell lymphomas. J Immunol. 2001; 166:1618–26. https://doi.org/10.4049/jimmunol.166.3.1618. [PubMed].
30. Carey GB, Semenova E, Qi X, Keegan AD. IL-4 protects the B-cell lymphoma cell line CH31 from anti-IgM-induced growth arrest and apoptosis: contribution of the PI-3 kinase/AKT pathway. Cell Res. 2007; 17:942–55. https://doi.org/10.1038/sj.cr.2007.90. [PubMed].
31. Gonzalez-Quintela A, Alende R, Gude F, Campos J, Rey J, Meijide LM, Fernandez-Merino C, Vidal C. Serum levels of immunoglobulins (IgG, IgA, IgM) in a general adult population and their relationship with alcohol consumption, smoking and common metabolic abnormalities. Clin Exp Immunol. 2008; 151:42–50. https://doi.org/10.1111/j.1365-2249.2007.03545.x. [PubMed].
32. Sagawa R, Sakata S, Gong B, Seto Y, Takemoto A, Takagi S, Ninomiya H, Yanagitani N, Nakao M, Mun M, Uchibori K, Nishio M, Miyazaki Y, et al. Soluble PD-L1 works as a decoy in lung cancer immunotherapy via alternative polyadenylation. JCI Insight. 2022; 7:e153323. https://doi.org/10.1172/jci.insight.153323. [PubMed].
33. Himuro H, Nakahara Y, Igarashi Y, Kouro T, Higashijima N, Matsuo N, Murakami S, Wei F, Horaguchi S, Tsuji K, Mano Y, Saito H, Azuma K, Sasada T. Clinical roles of soluble PD-1 and PD-L1 in plasma of NSCLC patients treated with immune checkpoint inhibitors. Cancer Immunol Immunother. 2023; 72:2829–40. https://doi.org/10.1007/s00262-023-03464-w. [PubMed].
34. Welt RS, Welt JA, Kostyal D, Gangadharan YD, Raymond V, Welt S. Specificity and biologic activities of novel anti-membrane IgM antibodies. Oncotarget. 2016; 7:74701–23. https://doi.org/10.18632/oncotarget.12506. [PubMed].
35. Saxena A, Wu D. Advances in Therapeutic Fc Engineering - Modulation of IgG-Associated Effector Functions and Serum Half-life. Front Immunol. 2016; 7:580. https://doi.org/10.3389/fimmu.2016.00580. [PubMed].
36. Schaefer W, Völger HR, Lorenz S, Imhof-Jung S, Regula JT, Klein C, Mølhøj M. Heavy and light chain pairing of bivalent quadroma and knobs-into-holes antibodies analyzed by UHR-ESI-QTOF mass spectrometry. MAbs. 2016; 8:49–55. https://doi.org/10.1080/19420862.2015.1111498. [PubMed].
37. Ridgway JB, Presta LG, Carter P. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. 1996; 9:617–21. https://doi.org/10.1093/protein/9.7.617. [PubMed].
38. Kitazawa T, Shima M. Emicizumab, a humanized bispecific antibody to coagulation factors IXa and X with a factor VIIIa-cofactor activity. Int J Hematol. 2020; 111:20–30. https://doi.org/10.1007/s12185-018-2545-9. [PubMed].
39. Labrijn AF, Meesters JI, de Goeij BE, van den Bremer ET, Neijssen J, van Kampen MD, Strumane K, Verploegen S, Kundu A, Gramer MJ, van Berkel PH, van de Winkel JG, Schuurman J, Parren PW. Efficient generation of stable bispecific IgG1 by controlled Fab-arm exchange. Proc Natl Acad Sci U S A. 2013; 110:5145–50. https://doi.org/10.1073/pnas.1220145110. [PubMed].
40. Schaefer W, Regula JT, Bähner M, Schanzer J, Croasdale R, Dürr H, Gassner C, Georges G, Kettenberger H, Imhof-Jung S, Schwaiger M, Stubenrauch KG, Sustmann C, et al. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc Natl Acad Sci U S A. 2011; 108:11187–92. https://doi.org/10.1073/pnas.1019002108. [PubMed].
41. Mazor Y, Oganesyan V, Yang C, Hansen A, Wang J, Liu H, Sachsenmeier K, Carlson M, Gadre DV, Borrok MJ, Yu XQ, Dall’Acqua W, Wu H, Chowdhury PS. Improving target cell specificity using a novel monovalent bispecific IgG design. MAbs. 2015; 7:377–89. https://doi.org/10.1080/19420862.2015.1007816. [PubMed].
42. Enami J, Sasaki T, Suzuki H. Antibody and antibody composition production method. United states patent. US 10,344,099. 2011.
43. Rankin CT, Veri MC, Gorlatov S, Tuaillon N, Burke S, Huang L, Inzunza HD, Li H, Thomas S, Johnson S, Stavenhagen J, Koenig S, Bonvini E. CD32B, the human inhibitory Fc-gamma receptor IIB, as a target for monoclonal antibody therapy of B-cell lymphoma. Blood. 2006; 108:2384–91. https://doi.org/10.1182/blood-2006-05-020602. [PubMed].
44. Tawara T, Hasegawa K, Sugiura Y, Tahara T, Ishida I, Kataoka S. Fully human antibody exhibits pan-human leukocyte antigen-DR recognition and high in vitro/vivo efficacy against human leukocyte antigen-DR-positive lymphomas. Cancer Sci. 2007; 98:921–28. https://doi.org/10.1111/j.1349-7006.2007.00469.x. [PubMed].
45. Tawara T, Hasegawa K, Sugiura Y, Harada K, Miura T, Hayashi S, Tahara T, Ishikawa M, Yoshida H, Kubo K, Ishida I, Kataoka S. Complement activation plays a key role in antibody-induced infusion toxicity in monkeys and rats. J Immunol. 2008; 180:2294–98. https://doi.org/10.4049/jimmunol.180.4.2294. [PubMed].
46. Chang CH, Wang Y, Gupta P, Goldenberg DM. Extensive crosslinking of CD22 by epratuzumab triggers BCR signaling and caspase-dependent apoptosis in human lymphoma cells. MAbs. 2015; 7:199–211. https://doi.org/10.4161/19420862.2014.979081. [PubMed].
47. Tomita A, Hiraga J, Kiyoi H, Ninomiya M, Sugimoto T, Ito M, Kinoshita T, Naoe T. Epigenetic regulation of CD20 protein expression in a novel B-cell lymphoma cell line, RRBL1, established from a patient treated repeatedly with rituximab-containing chemotherapy. Int J Hematol. 2007; 86:49–57. https://doi.org/10.1532/IJH97.07028. [PubMed].
48. Carey BS, Poulton KV, Poles A. Factors affecting HLA expression: A review. Int J Immunogenet. 2019; 46:307–20. https://doi.org/10.1111/iji.12443. [PubMed].
49. Mössner E, Brünker P, Moser S, Püntener U, Schmidt C, Herter S, Grau R, Gerdes C, Nopora A, van Puijenbroek E, Ferrara C, Sondermann P, Jäger C, et al. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell-mediated B-cell cytotoxicity. Blood. 2010; 115:4393–402. https://doi.org/10.1182/blood-2009-06-225979. [PubMed].
50. Robinson HR, Qi J, Cook EM, Nichols C, Dadashian EL, Underbayev C, Herman SEM, Saba NS, Keyvanfar K, Sun C, Ahn IE, Baskar S, Rader C, Wiestner A. A CD19/CD3 bispecific antibody for effective immunotherapy of chronic lymphocytic leukemia in the ibrutinib era. Blood. 2018; 132:521–32. https://doi.org/10.1182/blood-2018-02-830992. [PubMed].
51. Mimori K, Kiyokawa N, Taguchi T, Suzuki T, Sekino T, Nakajima H, Saito M, Katagiri YU, Isoyama K, Yamada K, Matsuo Y, Fujimoto J. Costimulatory signals distinctively affect CD20- and B-cell-antigen-receptor-mediated apoptosis in Burkitt’s lymphoma/leukemia cells. Leukemia. 2003; 17:1164–74. https://doi.org/10.1038/sj.leu.2402936. [PubMed].
52. Reff ME, Carner K, Chambers KS, Chinn PC, Leonard JE, Raab R, Newman RA, Hanna N, Anderson DR. Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood. 1994; 83:435–45. [PubMed].
53. Li D, Lee D, Dere RC, Zheng B, Yu SF, Fuh FK, Kozak KR, Chung S, Bumbaca Yadav D, Nazzal D, Danilenko D, Go MAT, Williams M, et al. Evaluation and use of an anti-cynomolgus monkey CD79b surrogate antibody-drug conjugate to enable clinical development of polatuzumab vedotin. Br J Pharmacol. 2019; 176:3805–18. https://doi.org/10.1111/bph.14784. [PubMed].
54. Townsend W, Hiddemann W, Buske C, Cartron G, Cunningham D, Dyer MJS, Gribben JG, Phillips EH, Dreyling M, Seymour JF, Grigg A, Trotman J, Lin TY, et al. Obinutuzumab Versus Rituximab Immunochemotherapy in Previously Untreated iNHL: Final Results From the GALLIUM Study. Hemasphere. 2023; 7:e919. https://doi.org/10.1097/HS9.0000000000000919. [PubMed].
55. Tilly H, Morschhauser F, Sehn LH, Friedberg JW, Trněný M, Sharman JP, Herbaux C, Burke JM, Matasar M, Rai S, Izutsu K, Mehta-Shah N, Oberic L, et al. Polatuzumab Vedotin in Previously Untreated Diffuse Large B-Cell Lymphoma. N Engl J Med. 2022; 386:351–63. https://doi.org/10.1056/NEJMoa2115304. [PubMed].
56. Duell J, Maddocks KJ, González-Barca E, Jurczak W, Liberati AM, De Vos S, Nagy Z, Obr A, Gaidano G, Abrisqueta P, Kalakonda N, André M, Dreyling M, et al. Long-term outcomes from the Phase II L-MIND study of tafasitamab (MOR208) plus lenalidomide in patients with relapsed or refractory diffuse large B-cell lymphoma. Haematologica. 2021; 106:2417–26. https://doi.org/10.3324/haematol.2020.275958. [PubMed].
57. Maruyama S, Kubagawa H, Cooper MD. Activation of human B cells and inhibition of their terminal differentiation by monoclonal anti-mu antibodies. J Immunol. 1985; 135:192–99. [PubMed].
58. Tawara T, Kataoka S. Use of anti-HLA-DR antibody. United states patent. US 72,62,278. 2002.
59. Chan A, Shields R, Wu L. Anti-Fc-gamma RIIB receptor antibody and uses therefor. United states patent. US 20,060,073,142. 2005.
60. Kabat EA, Wu TT, Perry HM, Gottesman KS, Foeller C. Sequences of proteins of immunological interest. 5th ed. MD: United States Department of Health and Human Services, Public Health Service, National Institutes of Health; 1991.
61. Edelman GM, Cunningham BA, Gall WE, Gottlieb PD, Rutishauser U, Waxdal MJ. The covalent structure of an entire gammaG immunoglobulin molecule. Proc Natl Acad Sci U S A. 1969; 63:78–85. https://doi.org/10.1073/pnas.63.1.78. [PubMed].