
Introduction
In EGFR-mutant-dependent non-small cell lung cancer (NSCLC), first- or second-generation EGFR-TKI (e.g., gefitinib, erlotinib, afatinib, and dacomitinib) select for resistance due to the T790M point mutation (EGFR T790M) in 50% of patients [1–4]. In other words, before therapy, 50% of patients have pre-existing resistant T790M mutations. Only one or a few cells contain the T790M mutation, because this resistant mutation confers no selective advantage prior to therapy. As soon as treatment starts, these rare cells selectively proliferate and eventually produce billions of cells [5], rendering the tumor resistant to the 1st/2nd generation of tyrosine kinase inhibitors (TKI). The cell with T790M is sensitive to the 3rd generation TKI osimertinib.
In 2015, the FDA approved osimertinib for NSCLC with a T790M mutation as a second-line therapy. This approval as a second-line therapy was based on the understanding that an untreated tumor cannot be T790M-positive; the mutation may initially occur in only one cell. Furthermore, 50% of patients do not harbor this mutation. It might seem logical to administer osimertinib after a tumor develops resistance to first- or second-generation TKIs, given the uncertainty regarding which patients will acquire the T790M mutation.
Consider a hypothetical scenario: If osimertinib were a combination of two medications (the first targets oncogenic EGFR without secondary T790M and the second targets only oncogenic EGFR with secondary T790M), then an oncologist would not prescribe the second drug to a patient sensitive to the first drug. And why would an oncologist prescribe the second (anti-resistant) drug? The tumor is not resistant to the first drug. Even if a patient has a single cell with T790M, its killing will be unnoticed at first, even in the months. The response rate will not be affected too. But after a year, even a transient addition of the anti-T790M drug at the beginning of treatment would prevent acquiring the resistance by the entire tumor and dramatically extend progression-free survival (PFS) and overall survival (OS). Osimertinib should be administered without waiting for the tumor to develop resistance, as it is feasible to eliminate a T790M-positive cell, but impossible to eradicate all million cells once the mutation is widespread. The latter scenario fails because of tumor heterogeneity, bad luck, and a mere probability. If the probability to kill one cell out of one is 0.99, then the probability to kill 2 cells out of 2 is 0.99 × 0.99 = 0.98, and the probability of killing all million cells is practically zero.
Preemptive two-drug combinations
Here I will re-introduce the notion of preemptive combinations (PC) of targeted drugs. Such combinations include the therapeutic response activities and anti-resistant activities that eliminate a few resistant cells. In other words, preemptive combinations induce both a therapeutic response and eliminate a few resistant cells with pre-existing mutations [6]. For example, if we started treatment with osimertinib and the tumor may have one cell with the C797S mutation (resistant to osimertinib but sensitive to gefitinib), then the addition of gefitinib to osimertinib renders this combination preemptive. The goal is to eliminate this one resistant cell. And vice versa, if we treat the tumor with gefitinib and this tumor may have a single cell with the T790M mutation (resistant to gefitinib but sensitive to osimertinib), then the addition of osimertinib to gefitinib creates the preemptive combination. Figuratively, osimertinib is equivalent to a preemptive combination of two activities: one activity causes tumor shrinkage (by targeting billions of cells without T790M and the second activity just to kill one cell with T790M. One cell is not noticeable. At first, its presence or absence does not affect the response rate and, if a response occurs, the degree of therapeutic response. Killing one resistant cell cannot increase the response rate and initial degree of response. But killing of this one cell (if a patient has it) extends PFS and OS. This must be done to prevent future resistance in 50% of patients and extend PFS. We should not wait for radiological progression or detection of T790M by biopsy to kill a resistant cell. One cell can be killed and all cells out of a billion cells cannot. The view on osimertinib as an analog of preemptive combination predicts the outcome of first-line treatment, when a tumor is not yet T790M-positive. The response rate will not be increased. In responders, the degree of initial response will not be changed. But PFS and OS will be extended in responders. That is exactly what was observed in a famous clinical trial published in 2018 [7]. The median progression-free survival (PFS) was significantly longer with osimertinib than with the 1st generation of EGFR inhibitors (18.9 months vs. 10.2 months; P < 0.001). The median duration of response was 17.2 months with osimertinib versus 8.5 months with 1st generation inhibitors. Yet, the objective response rate was similar in the two groups: 80% with osimertinib and 76% with 1st generation EGFR-TKIs [7]. Compared with the 1st generation of TKI, osimertinib prolongs PFS almost twofold (Figure 1A). This is especially dramatic given that not all patients have T790M. But osimertinib must be given to all patients because we do not know which patient has this mutation. If T790M were the only resistance mechanism, then PFS would be eternal, so patients would have a normal lifespan on chronic treatment with osimertinib. But roughly half of the resistance mechanisms are on-target such as secondary L718, G724, L792, G796, C797 in EGFR, and all off-target mechanisms such as MET and HER2 amplifications. Osimertinib selects for on-target resistance due to secondary mutations such as L718, G724, L792, G796, C797 [1, 8, 9] and the cells with these mutations can be targeted by the 1st or/and 2nd generation of EGFR inhibitors such as gefitinib and afatinib. So, addition of these EGFR inhibitors to osimertinib would prevent on-target resistance.
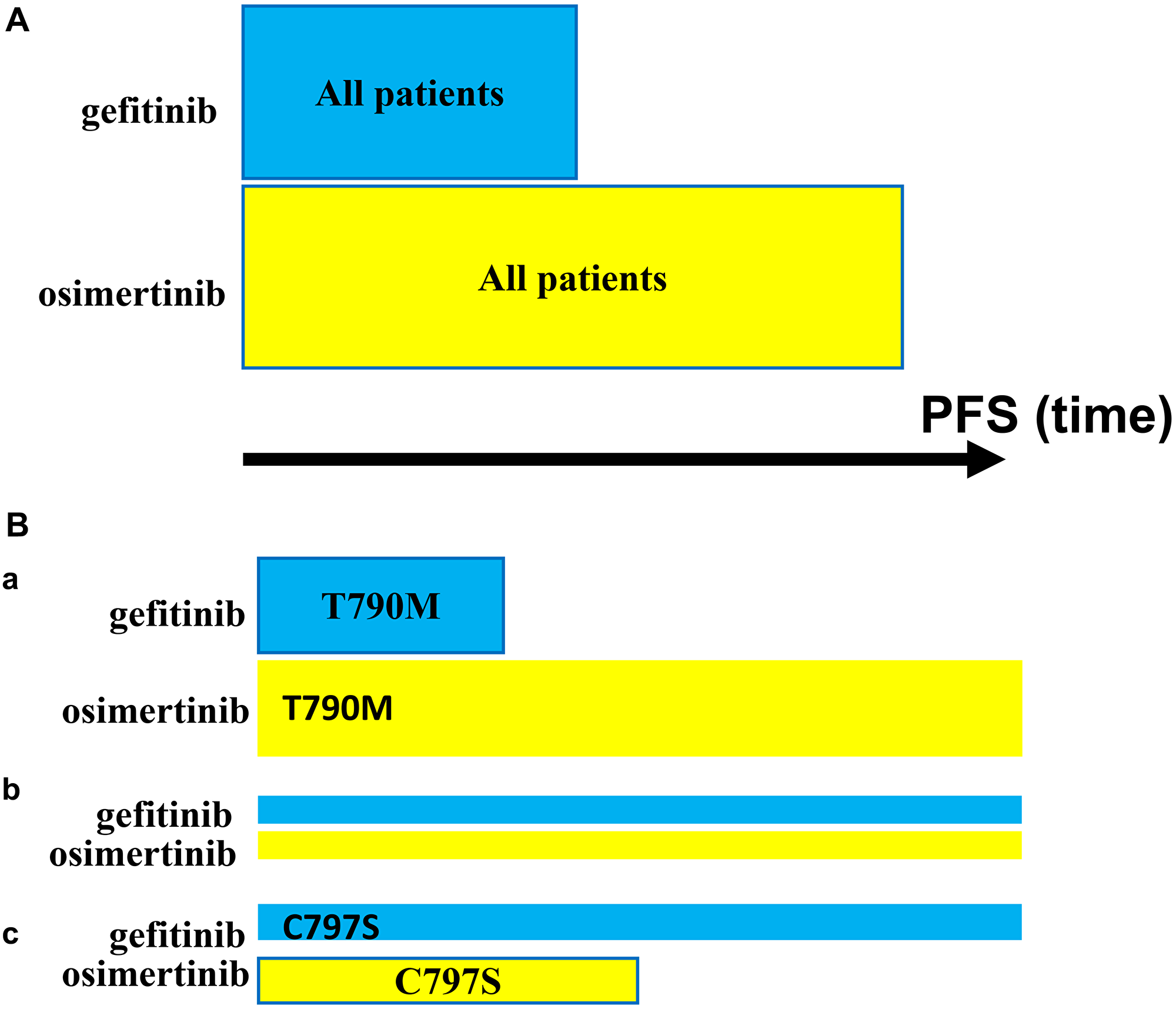
Figure 1: Progression-free survival (PFS): Gefitinib vs. Osimertinib
(A) PFS in a Total Cohort with EGFR-Mutant-Driven NSCLC: Patients Treated with Gefitinib vs. Osimertinib. (B) PFS in Cohorts Depending on Pre-existing Secondary Mutation within EGFR-Mutant: T790M vs. C797S. (a) T790M. (b) Neither T790M nor C797S. (c) C797S. To simplify, other pre-existing alterations are not shown.
Preemptive multi-drug combinations
The most common off-target resistances acquired after treatment with EGFR inhibitors are amplification of MET and HER2/3/4, and the latter is sensitive to afatinib. Adding capmatinib (the most effective MET inhibitor) would prevent MET-dependent resistance.
What percentage of patients with EGFR-driven cancer would benefit from a preemptive combination like Osimertinib, afatinib, and capmatinib?
Monotherapy with 1st or 2nd-generation EGFR TKIs causes resistance in 75% of patients due to either secondary T790M EGFR mutation plus MET or HER2 amplification [2–4, 10].
Monotherapy with 3rd-generation EGFR-TKI (osimertinib) causes at least 50% of resistance cases due to secondary mutations such as L718, G724, L792, G796, C797 plus MET and HER2 amplification [1, 9]. Specifically, resistance mechanisms to second-line osimertinib include MET 5–50%; HER2 5%; on-target 10–26% (mostly C797X); KRAS 2–8% [9]. Resistance mechanisms to first-line osimertinib include MET 7–15%; HER2 1–2%; on-target 6–10% (mostly C797X); KRAS 3–4% [9].
Thus, we can calculate that at least 75% of resistance mechanisms are preventable by a combination of osimertinib, afatinib, and capmatinib (OAC). Whether the combination should include afatinib or a 1st-generation TKI may depend on the primary activating mutation in the EGFR: del19 or L858R or uncommon. Osimertinib, afatinib, capmatinib (OAC) is most preferable because afatinib (i) inhibits HER2-4 [11] (ii) shows improved PFS compared with 1st generation [12, 13], especially when followed by osimertinib [14], and is effective in targeting brain metastases [11].
I estimate that by preventing 75% of resistance mechanisms, a combination of osimertinib, afatinib, and capmatinib (OAC) would extend median PFS from 18 months (osimertinib alone) to approximately 40 months. Such a remarkable extension takes into account that 25% of patients cannot benefit from OAC because they do not have pre-existing mechanisms of relevant resistance. However, all EGFR-dependent patients must be treated because it is not clear who will benefit. It is a mistake to wait for tumor progression or for resistance detection by biopsy.
Even superior monotherapy hurts 20% patients
Osimertinib prolongs median progression-free survival (PFS) and overall survival (OS) compared with first-generation tyrosine kinase inhibitors (TKIs), such as gefitinib. Osimertinib is superior because the osimertinib-sensitive mutation (T790M) is more common than osimertinib-resistant mutations, such as C797S. We may suggest that osimertinib extends median PFS by prolonging PFS only in patients who have the T790M mutation (Figure 1B (a)). In patients lacking the T790M mutation, osimertinib should not extend PFS beyond the extension afforded by first-generation TKIs such as gefitinib (Figure 1B (b)). Furthermore, in a patient lacking T790M and having a preexisting C797S mutation, osimertinib selects for C797S (and L718, G724, L792, G796), making the tumor resistant and thus shortening PFS compared to first-generation TKIs such as gefinitib (Figure 1B (c)). Therefore, although most patients (approximately 55%) benefit from osimertinib, some patients (approximately 20%) are harmed. It is impossible to detect one (or a few) cells with a resistant mutation in untreated patients, so we must choose osimertinib for all patients. If we are obligated to use monotherapy, many patients (approximately 20%) will be adversely affected, even though the median PFS for the entire cohort is improved. Neither the unfortunate patient nor patient’s doctor can know that superior TKI shortened PFS in this particular patient. The solution is remarkably simple: to use preemptive combinations of osimertinib + gefitinib (O+G) and osimertinib + gefitinib + afatinib (O+G+A) from the start. Current therapy drives resistance and even finds a way to achieve the infamous cis-T790M/C797S triple mutations that are resistant to all existing EGFR inhibitors and any of their combinations [15]. This cis mutation will not appear when the O+G combination is used preemptively. What is more, there will be no need for the development of a fourth generation of EGFR inhibitors to target this cis mutation. In addition, preemptive combinations should include drugs that target the most common off-target mechanisms, such as MET inhibitors (capmatinib). Preemptive combinations can be used in a sequence of transient two-drug combinations (Figure 2A) if an oncologist is uncomfortable with three- and four-drug combinations. (Ironically, an oncologist is comfortable with multi-kinase inhibitors such as lenvatinib and cabozantinib against one intended target, which is akin to a multi-drug combo with one intended target and all other random drugs. If so, why then an oncologist is uncomfortable with preemptive combinations of selective inhibitors). Preemptive combinations to prevent both on-target and off-target resistance have been discussed [6]. In general, combinations are necessary to abrogate resistance [10, 16–20].
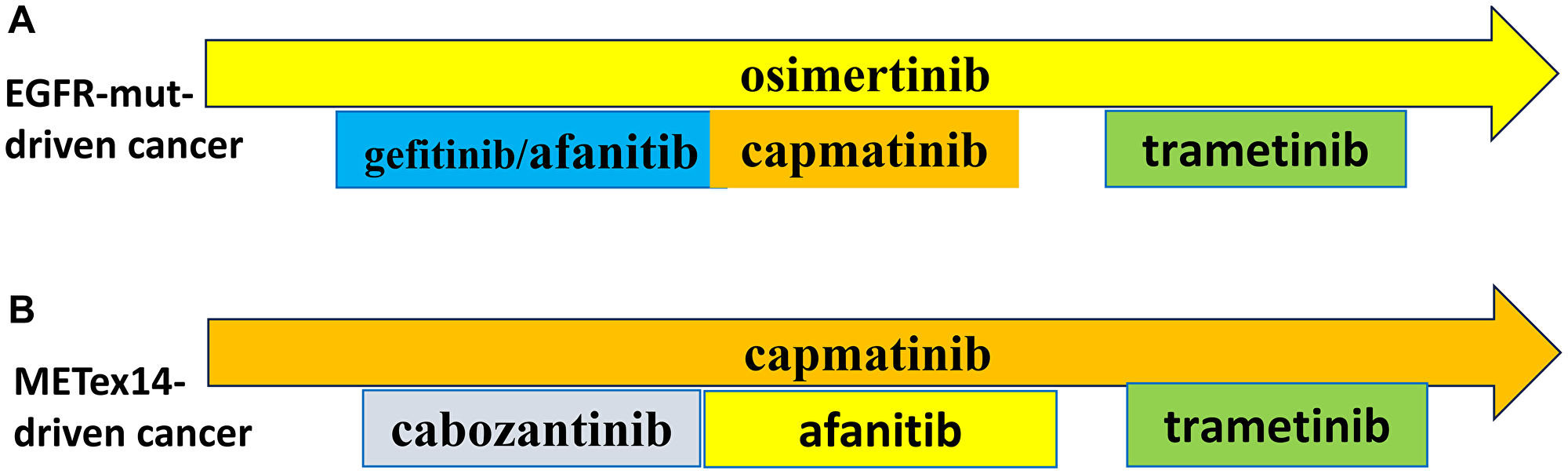
Figure 2: Preemptive combinations.
(A) EGFR-Mutant-Driven NSCLC. See text. (B) METex14-Driven NSCLC. See text.
If a patient has EGFR-driven NSCLC, they are in some ways fortunate because this is the most common type of lung cancer, and it has been studied extensively. There are three generations of inhibitors available, and drug combinations, especially with MET inhibitors, are occasionally used. The treatment for MET-driven lung cancer is less developed, but the insights gained from EGFR-driven cancer can be beneficial. The approaches to treatment can be similar.
Preemptive combination for MET-driven NSCLC
A year ago, I was hospitalized at Massachusetts General Hospital (Boston) with multiple brain metastases from lung cancer, driven by a MET exon 14 skipping mutation (METex14) [6]. The response to capmatinib (a selective MET inhibitor, type I) was outstanding. However, resistance tends to develop within a year of treatment [21]. The most common mechanism of on-target resistance involves secondary mutations in METex14, such as D1228 and Y1230 [22]. Resistance mutations against type I MET inhibitors are sensitive to type II inhibitors, and vice versa [22–26]. In patients with METex14 NSCLC, cabozantinib can overcome resistance selected by type I MET inhibitors [27–29]. Furthermore, cabozantinib is effective in METex14-positive NSCLC with brain metastases [30].
Cabozantinib is the only FDA-approved type II MET inhibitor, so our choices are limited to this multi-kinase inhibitor, which has unpleasant side effects if used long-term. Yet, cabozantinib can be used transiently for a few weeks, for example, to eliminate cells with pre-existing mutations such as D1228 or Y1230 before they expand due to capmatinib. Simultaneous treatment with type I and type II MET inhibitors may delay the emergence of on-target MET resistance mutations [31].
The most common off-target mechanisms of resistance include alterations in EGFR and HER2 [26, 32, 33]. These can be targeted by afatinib (Figure 2B). Combinations including a MEK inhibitor (trametinib) will be discussed in the forthcoming article “My Battle with Cancer: Part III.”
Preemptive treatment with capmatinib, afatinib, and cabozantinib may prevent 50% of all potential resistance, and half of all patients with METex14-driven lung cancer may experience prolonged progression-free survival (PFS), leading to a longer and happier life.
CONFLICTS OF INTEREST
Author has no conflicts of interest to declare.
FUNDING
No funding was used for this paper.
References
1. Rotow JK, Lee JK, Madison RW, Oxnard GR, Jänne PA, Schrock AB. Real-World Genomic Profile of EGFR Second-Site Mutations and Other Osimertinib Resistance Mechanisms and Clinical Landscape of NSCLC Post-Osimertinib. J Thorac Oncol. 2024; 19:227–39. https://doi.org/10.1016/j.jtho.2023.09.1453. [PubMed].
2. Wu L, Ke L, Zhang Z, Yu J, Meng X. Development of EGFR TKIs and Options to Manage Resistance of Third-Generation EGFR TKI Osimertinib: Conventional Ways and Immune Checkpoint Inhibitors. Front Oncol. 2020; 10:602762. https://doi.org/10.3389/fonc.2020.602762. [PubMed].
3. Fu K, Xie F, Wang F, Fu L. Therapeutic strategies for EGFR-mutated non-small cell lung cancer patients with osimertinib resistance. J Hematol Oncol. 2022; 15:173. https://doi.org/10.1186/s13045-022-01391-4. [PubMed].
4. Liao BC, Griesing S, Yang JC. Second-line treatment of EGFR T790M-negative non-small cell lung cancer patients. Ther Adv Med Oncol. 2019; 11:1758835919890286. https://doi.org/10.1177/1758835919890286. [PubMed].
5. Blagoev KB, Iordanov R, Zhou M, Fojo T, Bates SE. Drug resistant cells with very large proliferative potential grow exponentially in metastatic prostate cancer. Oncotarget. 2021; 12:15–21. https://doi.org/10.18632/oncotarget.27855. [PubMed].
6. Blagosklonny MV. My battle with cancer. Part 1. Oncoscience. 2024; 11:1–14. https://doi.org/10.18632/oncoscience.593. [PubMed].
7. Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, Dechaphunkul A, Imamura F, Nogami N, Kurata T, Okamoto I, Zhou C, Cho BC, et al, and FLAURA Investigators. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N Engl J Med. 2018; 378:113–25. https://doi.org/10.1056/NEJMoa1713137. [PubMed].
8. Nishino M, Suda K, Kobayashi Y, Ohara S, Fujino T, Koga T, Chiba M, Shimoji M, Tomizawa K, Takemoto T, Mitsudomi T. Effects of secondary EGFR mutations on resistance against upfront osimertinib in cells with EGFR-activating mutations in vitro. Lung Cancer. 2018; 126:149–55. https://doi.org/10.1016/j.lungcan.2018.10.026. [PubMed].
9. Leonetti A, Sharma S, Minari R, Perego P, Giovannetti E, Tiseo M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br J Cancer. 2019; 121:725–37. https://doi.org/10.1038/s41416-019-0573-8. [PubMed].
10. Husain H, Scur M, Murtuza A, Bui N, Woodward B, Kurzrock R. Strategies to Overcome Bypass Mechanisms Mediating Clinical Resistance to EGFR Tyrosine Kinase Inhibition in Lung Cancer. Mol Cancer Ther. 2017; 16:265–72. https://doi.org/10.1158/1535-7163.MCT-16-0105. [PubMed].
11. Harvey RD, Adams VR, Beardslee T, Medina P. Afatinib for the treatment of EGFR mutation-positive NSCLC: A review of clinical findings. J Oncol Pharm Pract. 2020; 26:1461–74. https://doi.org/10.1177/1078155220931926. [PubMed].
12. Park K, Tan EH, O’Byrne K, Zhang L, Boyer M, Mok T, Hirsh V, Yang JC, Lee KH, Lu S, Shi Y, Kim SW, Laskin J, et al. Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non-small-cell lung cancer (LUX-Lung 7): a phase 2B, open-label, randomised controlled trial. Lancet Oncol. 2016; 17:577–89. https://doi.org/10.1016/S1470-2045(16)30033-X. [PubMed].
13. Soria JC, Felip E, Cobo M, Lu S, Syrigos K, Lee KH, Göker E, Georgoulias V, Li W, Isla D, Guclu SZ, Morabito A, Min YJ, et al, and LUX-Lung 8 Investigators. Afatinib versus erlotinib as second-line treatment of patients with advanced squamous cell carcinoma of the lung (LUX-Lung 8): an open-label randomised controlled phase 3 trial. Lancet Oncol. 2015; 16:897–907. https://doi.org/10.1016/S1470-2045(15)00006-6. [PubMed].
14. Tamiya M, Tamiya A, Suzuki H, Moriizumi K, Nakahama K, Taniguchi Y, Kunimasa K, Kimura M, Inoue T, Kuhara H, Nishino K, Hirashima T, Atagi S, et al. Which Is Better EGFR-TKI Followed by Osimertinib: Afatinib or Gefitinib/Erlotinib? Anticancer Res. 2019; 39:3923–29. https://doi.org/10.21873/anticanres.13544. [PubMed].
15. Niederst MJ, Hu H, Mulvey HE, Lockerman EL, Garcia AR, Piotrowska Z, Sequist LV, Engelman JA. The Allelic Context of the C797S Mutation Acquired upon Treatment with Third-Generation EGFR Inhibitors Impacts Sensitivity to Subsequent Treatment Strategies. Clin Cancer Res. 2015; 21:3924–33. https://doi.org/10.1158/1078-0432.CCR-15-0560. [PubMed].
16. Adashek JJ, Subbiah V, Westphalen CB, Naing A, Kato S, Kurzrock R. Cancer: slaying the nine-headed Hydra. Ann Oncol. 2023; 34:61–69. https://doi.org/10.1016/j.annonc.2022.07.010. [PubMed].
17. Fountzilas E, Tsimberidou AM, Vo HH, Kurzrock R. Clinical trial design in the era of precision medicine. Genome Med. 2022; 14:101. https://doi.org/10.1186/s13073-022-01102-1. [PubMed].
18. Nikanjam M, Tinajero J, McGann M, Li J, Yang J, Shen F, Sicklick JK, Kato S, Capparelli E, Kurzrock R. Dosing of 3 Targeted Agents in Novel Drug Combinations Used at the Precision Medicine Clinic of the University of California San Diego. J Hematol Oncol Pharm. 2023; 13:19–25. [PubMed].
19. Wahida A, Buschhorn L, Fröhling S, Jost PJ, Schneeweiss A, Lichter P, Kurzrock R. The coming decade in precision oncology: six riddles. Nat Rev Cancer. 2023; 23:43–54. https://doi.org/10.1038/s41568-022-00529-3. [PubMed].
20. Henary H, George GC, Wheler J, Naing A, Piha-Paul S, Fu S, Mistry R, Zinner R, Kurzrock R, Hong DS. A phase 1 study of intermittently administered pazopanib in combination with continuous daily dosing of lapatinib in patients with solid tumors. Cancer Chemother Pharmacol. 2015; 76:597–603. https://doi.org/10.1007/s00280-015-2824-6. [PubMed].
21. Wolf J, Seto T, Han JY, Reguart N, Garon EB, Groen HJM, Tan DSW, Hida T, de Jonge M, Orlov SV, Smit EF, Souquet PJ, Vansteenkiste J, et al, and GEOMETRY mono-1 Investigators. Capmatinib in MET Exon 14-Mutated or MET-Amplified Non-Small-Cell Lung Cancer. N Engl J Med. 2020; 383:944–57. https://doi.org/10.1056/NEJMoa2002787. [PubMed].
22. Fujino T, Kobayashi Y, Suda K, Koga T, Nishino M, Ohara S, Chiba M, Shimoji M, Tomizawa K, Takemoto T, Mitsudomi T. Sensitivity and Resistance of MET Exon 14 Mutations in Lung Cancer to Eight MET Tyrosine Kinase Inhibitors In Vitro. J Thorac Oncol. 2019; 14:1753–65. https://doi.org/10.1016/j.jtho.2019.06.023. [PubMed].
23. Bahcall M, Sim T, Paweletz CP, Patel JD, Alden RS, Kuang Y, Sacher AG, Kim ND, Lydon CA, Awad MM, Jaklitsch MT, Sholl LM, Jänne PA, Oxnard GR. Acquired METD1228V Mutation and Resistance to MET Inhibition in Lung Cancer. Cancer Discov. 2016; 6:1334–41. https://doi.org/10.1158/2159-8290.CD-16-0686. [PubMed].
24. Engstrom LD, Aranda R, Lee M, Tovar EA, Essenburg CJ, Madaj Z, Chiang H, Briere D, Hallin J, Lopez-Casas PP, Baños N, Menendez C, Hidalgo M, et al. Glesatinib Exhibits Antitumor Activity in Lung Cancer Models and Patients Harboring MET Exon 14 Mutations and Overcomes Mutation-mediated Resistance to Type I MET Inhibitors in Nonclinical Models. Clin Cancer Res. 2017; 23:6661–72. https://doi.org/10.1158/1078-0432.CCR-17-1192. [PubMed].
25. Li A, Yang JJ, Zhang XC, Zhang Z, Su J, Gou LY, Bai Y, Zhou Q, Yang Z, Han-Zhang H, Zhong WZ, Chuai S, Zhang Q, et al. Acquired MET Y1248H and D1246N Mutations Mediate Resistance to MET Inhibitors in Non-Small Cell Lung Cancer. Clin Cancer Res. 2017; 23:4929–37. https://doi.org/10.1158/1078-0432.CCR-16-3273. [PubMed].
26. Recondo G, Bahcall M, Spurr LF, Che J, Ricciuti B, Leonardi GC, Lo YC, Li YY, Lamberti G, Nguyen T, Milan MSD, Venkatraman D, Umeton R, et al. Molecular Mechanisms of Acquired Resistance to MET Tyrosine Kinase Inhibitors in Patients with MET Exon 14-Mutant NSCLC. Clin Cancer Res. 2020; 26:2615–25. https://doi.org/10.1158/1078-0432.CCR-19-3608. [PubMed].
27. Wang SXY, Zhang BM, Wakelee HA, Koontz MZ, Pan M, Diehn M, Kunder CA, Neal JW. Case series of MET exon 14 skipping mutation-positive non-small-cell lung cancers with response to crizotinib and cabozantinib. Anticancer Drugs. 2019; 30:537–41. https://doi.org/10.1097/CAD.0000000000000765. [PubMed].
28. Pruis MA, Paats MS, Geurts WRR, Dubbink HJ, Dingemans AC. Overcoming Acquired Resistance Mutation MET D1228N to Crizotinib With Cabozantinib in NSCLC With MET Exon 14 Skipping Mutation. JCO Precis Oncol. 2021; 5:849–53. https://doi.org/10.1200/PO.21.00076. [PubMed].
29. Cai B, Li X, Huang X, Ma T, Qu B, Yu W, Yang W, Zhang P, Chen J, Liu F. Case Report: Sequential Combination Targeted Therapy With Type I and II MET Inhibitors in a Metastatic EGFR-Mutated, MET-Amplified NSCLC Patient With Acquired MET Y1230H Mutation. Front Oncol. 2021; 11:738832. https://doi.org/10.3389/fonc.2021.738832. [PubMed].
30. Klempner SJ, Borghei A, Hakimian B, Ali SM, Ou SI. Intracranial Activity of Cabozantinib in MET Exon 14-Positive NSCLC with Brain Metastases. J Thorac Oncol. 2017; 12:152–56. https://doi.org/10.1016/j.jtho.2016.09.127. [PubMed].
31. Bahcall M, Paweletz CP, Kuang Y, Taus LJ, Sim T, Kim ND, Dholakia KH, Lau CJ, Gokhale PC, Chopade PR, Hong F, Wei Z, Köhler J, et al. Combination of Type I and Type II MET Tyrosine Kinase Inhibitors as Therapeutic Approach to Prevent Resistance. Mol Cancer Ther. 2022; 21:322–35. https://doi.org/10.1158/1535-7163.MCT-21-0344. [PubMed].
32. Rotow JK, Gui P, Wu W, Raymond VM, Lanman RB, Kaye FJ, Peled N, Fece de la Cruz F, Nadres B, Corcoran RB, Yeh I, Bastian BC, Starostik P, et al. Co-occurring Alterations in the RAS-MAPK Pathway Limit Response to MET Inhibitor Treatment in MET Exon 14 Skipping Mutation-Positive Lung Cancer. Clin Cancer Res. 2020; 26:439–49. https://doi.org/10.1158/1078-0432.CCR-19-1667. [PubMed].
33. Awad MM, Lee JK, Madison R, Classon A, Kmak J, Frampton GM, Alexander BM, Venstrom J, Schrock AB. Characterization of 1,387 NSCLCs with MET exon 14 (METex14) skipping alterations (SA) and potential acquired resistance (AR) mechanisms. J Clin Oncol. 2020; 38:9511. https://doi.org/10.1200/JCO.2020.38.15_suppl.9511.