
Introduction
Thyroid cancer is a disease of generally indolent course with the majority of tumors being of well-differentiated histology and amenable to cure with standard therapy. However, a subset of patients present at more advanced stages or with dedifferentiated histologies that are not cured via standard therapy alone. Dedifferentiated subtypes of thyroid cancer include anaplastic thyroid cancers (ATC) and poorly differentiated thyroid cancers (PDTC), which are thought to arise from a process of gradual microevolution from papillary thyroid cancers (PTC) [1]. Once progressed to the anaplastic form, disease is often incurable with 1-year survival rates as low as 35% with chemoradiation [2, 3]. An improved understanding of the molecular basis of thyroid cancer has led to the development of new targeted agents.
The MAPK pathway and BRAF inhibition
The mitogen-activated protein kinase (MAPK) pathway regulates processes such as cell proliferation, differentiation and death through a cascade of three sequential protein kinases [4–6]. MAPK pathways are initiated by various extracellular cues such as growth factors or stress resulting in sequential phosphorylation until target substrates in the cytosol and nucleus are activated to alter protein function and gene expression. Mutations involving the cascade can lead to constitutive activation and signal transduction resulting in inappropriate cellular proliferation and tumorigenesis [7].
The mutational landscape of PTC is characterized by several molecular subtypes with mutually exclusive effectors in the MAPK pathway [8, 9]. In PTC, the most common mutations in this pathway are BRAF V600E (60%), alterations in RAS (15%), BRAF non-V600E kinase domain mutations, and mutations of other receptor tyrosine kinases such as RET, NTRK, and ALK (12%). In contrast to PTC, in PDTC, BRAF V600E mutations are relatively less common (33% of cases), with alterations in RAS and RET more common, 28% and 6%, respectively. In ATC, BRAF V600E mutations are found in 45% and RAS in 24% of cases [10]. In thyroid cancer, the BRAF V600E mutation leads to constitutive activation of the RAF/ERK pathway and consequent deactivation of thyroid-specific genes resulting in dedifferentiation and tumor progression [11].
With the limited benefit of conventional chemotherapeutics and radiation and growing evidence of clinical activity of BRAF inhibitors in other cancer types such as melanoma, these agents have been also investigated for efficacy in thyroid cancer. The first agents approved for use in thyroid cancer were sorafenib and lenvatinib. While these agents demonstrated benefits in progression-free survival, they have several limitations including no significant improvements in overall survival [12, 13] and a broad range of toxic effects [14, 15]. Additionally, these agents are multi-kinase inhibitors and not specifically targeted to BRAF [13, 16]. Newer, more targeted agents were subsequently investigated and approved for use in thyroid cancer, including vemurafenib and dabrafenib. These agents are not only targeted to BRAF, but specifically to the V600E mutant oncoprotein [17, 18]. Early trials for these agents were promising: vemurafenib with a median progression-free survival of 18.2 months and 6-month stable disease rate of 35%, [19] and dabrafenib with a median progression-free survival of 11.3 months and partial response and stable disease rates of 29% and 45%, respectively [20]. Additionally, pre-clinical data suggested that BRAF V600E inhibitors may restore radioactive iodine (RAI) uptake in previously refractory tumor cells [18], a finding which bore out in a case series of 10 patients who underwent RAI uptake scan after treatment with dabrafenib, 6 of whom had restored RAI uptake [21]. To date, no phase III clinical trials have evaluated long-term disease control with dabrafenib or vemurafenib monotherapy; however, diverse resistance mechanisms have since been described that explain clinical observations of limited sustained disease control [22].
A number of mutations and compensatory mechanisms have been observed to mediate bypass of BRAF blockade by thyroid carcinoma cells [23]. These mutations may be primary (already present in the tumor) or secondary (acquired over the course of treatment). Examples of these mechanisms include: PIK3CA mutations that paradoxically hyperactivate ERK when BRAF is inhibited [24], expansion of subclonal populations harboring KRAS mutations leading to constitutive activation of the GTPase and the PI3K/AKT pathway [25], increased autocrine signaling via NRG1 to activate the HER2/HER3 pathway [26], autocrine activation of the c-Met receptor to activate the ERK pathway [27] upregulation of IL-6 secretion with activation of the STAT3/JAK pathway [28], and stimulation of EGFR phosphorylation to reactivate ERK/AKT signaling [29]. Another mechanism of resistance is the transition to an alternative cell state due to selective pressures exerted by targeted therapy [30]. Although rare, this can coincide with dedifferentiation of tumor histology as seen with rare cases of anaplastic transformation after BRAF inhibitor therapy [25, 31].
Anaplastic transformation in thyroid cancer
In rare instances, thyroid cancer can evolve from its well-differentiated form, to PDTC—maintaining some semblance of the follicular cells from which it originated—or to ATC, losing all of follicular architecture (Figure 1). Clinical evidence of the microevolution of PTC to ATC began with observations of ATCs occurring in older adults with histories of long-standing or incompletely treated thyroid cancers [32–37]. In addition, the majority—and perhaps nearly all ATCs, accounting for incomplete sampling—have foci of differentiated thyroid cancer, as evidenced by studies utilizing whole-organ sections [38]. Molecular techniques have since supported this conclusion. A study by Wiseman et al. [39] found conserved genomic alterations in adjacent microdissected papillary and anaplastic foci, a finding reflected by data from Hunt et al. [40].
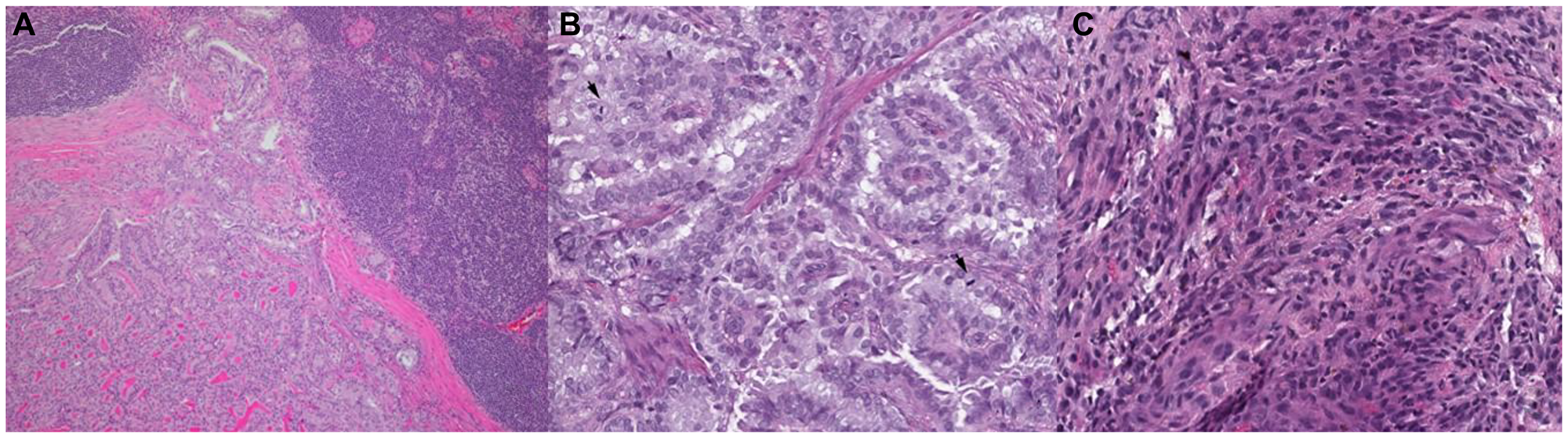
Figure 1: Representative histopathologic slides stained with hematoxylin and eosin of thyroid cancer from well-differentiated to dedifferentiated states.
(A) 55 year-old-female diagnosed with PTC, specimen taken prior to treatment with targeted therapy, (B) 52 year-old-male diagnosed with PDTC, specimen from prior to targeted therapy, (C) 66 year-old-male initially diagnosed with PDTC later transformed to ATC on subsequent biopsy (shown here) after BRAF targeted therapy and radiation.
The mechanisms underlying anaplastic transformation are not fully understood however, requiring cytologic and molecular studies for further insight. Ultrastructural analyses have shown that well-differentiated thyroid carcinomas transforming to ATCs undergo loss of tight junctions, desmosomes, and cellular polarity [41]. Molecular alterations found to be associated with ATC and thought to contribute to its pathogenesis include aneuploidy [42, 43], increased copy number alterations [44], and mutations affecting p53 [45–56], bcl-2 [57], cyclin D1 [58], β-catenin [59], Met [60], c-myc [61], Nm23 [62] and Ras [63, 64].
A number of these mutations involve or bypass the MAPK pathway, and together with observations of tumor dedifferentiation after BRAF inhibitor therapy in cancer types such as melanoma [65, 66] and thyroid cancer [25, 67], we hypothesized that mechanisms that drive BRAF inhibitor resistance may overlap with that of anaplastic evolution.
Mechanisms of BRAF inhibitor resistance and anaplastic transformation may overlap
A recent study from our group [31] observed that the genetic and transcriptomic alterations seen in BRAF inhibitor resistance were also associated with thyroid tumor dedifferentiation. In this study, we assessed mutations in patients with thyroid cancer across the spectrum of tumor dedifferentiation, and genomic and transcriptomic profiles of matched pre- and post-BRAF inhibitor treated thyroid tumors harboring BRAF V600E mutations, including a number of cases that dedifferentiated during the course of this study.
Whole exome sequencing (WES) was performed on matched samples before and after treatment with vemurafenib in a patient initially diagnosed with PTC whose tumor eventually transformed to ATC (Figure 2). Shared mutations were observed between the initial PTC sample and later ATC metastases, consistent with a process of clonal evolution during transformation. Among these shared mutations were the BRAF V600E alteration and a TERT promotor mutation. Several new mutations emerged in the ATC metastases, notably in 2 genes in the PI3K-AKT-mTOR pathway: PIK3CA and KIAA1024. Upon further evaluation with orthogonal ultra-deep next-generation sequencing, no mutations in PIK3CA were found in any of multiple regions of the initial PTC sample, while the ATC metastases contained the mutation at high cancer cell fractions (>0.90), suggesting that tumor cells harboring the PIK3CA mutation may have been present in the initial disease but only in regionally subclonal populations that later seeded the ATC metastases. In fact, we found that the clonal architecture of the ATC metastases was characterized by more subclonal populations than the initial PTC samples.
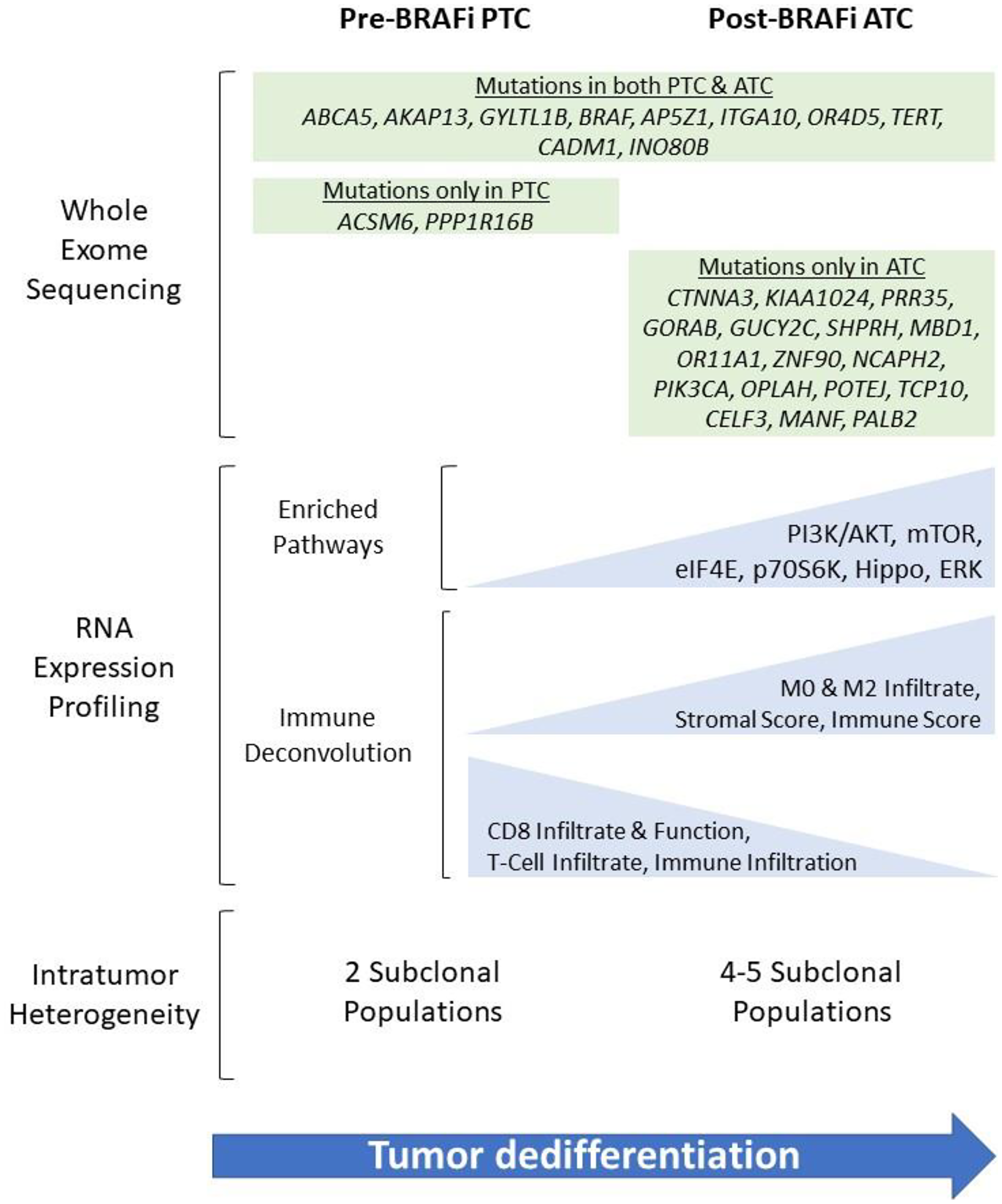
Figure 2: Summary of findings from whole exome sequencing (WES) and RNA expression profiling of tumor samples pre- and post-treatment with vemurafenib and anaplastic transformation in a 55-year-old female initially diagnosed with PTC and subsequently developed biopsy-proven ATC after targeted therapy.
In this case, because anaplastic transformation occurred after treatment with BRAF inhibition, it is possible that selective pressures exerted by BRAF inhibition contributed to this process.
Mutations in PIK3CA have been previously described as a mechanism of resistance to BRAF inhibitors and also associated with the development of anaplastic disease. Using transgenic mouse models of thyroid cancer, Roelli and colleagues [24] showed that double mutants with Braf V600E and Pik3ca H1047R mutations had paradoxically increased ERK signaling when treated with BRAF inhibitors compared to single mutant mice with Braf V600E mutation alone. This phenomenon is likely due to cross inhibition between the PI3K-AKT and MAPK pathways that is essential for appropriate response to proliferative and survival stimuli [68–71]. In turn, the double mutant tumors exhibited decreased response to BRAF inhibitors and more prompt resumption of tumor growth after cessation of therapy compared to single mutant mice. Moreover, the double mutants were noted on histologic examination to have increased size of anaplastic foci compared to untreated mice, suggesting paradoxical ERK signaling via the Pik3ca mutation may also drive the development of more aggressive histology. Paradoxical PI3K-dependent ERK activation was also noted in similar experiments using human ATC cell lines [24].
Further supporting the functional relevance of ERK signaling to anaplastic transformation, Ingenuity Pathway Analyses (Figure 2) of the PTC and ATC samples showed enrichment in the PI3K-AKT and mTOR pathways, as well as in an overall measure of thyroid-specific ERK signaling described by Agrawal et al. [9]. Downstream targets such as eIF4E binding protein and p70 ribosomal protein S6 kinase were also enriched. Taken together, these findings support a growing body of literature on the role of compensatory pathways such as PI3K-AKT in bypassing BRAF inhibition, emphasizing the limitations of monotherapy and need for further investigations on multi-modality treatments. These findings suggest that in some cases, selective pressure induced by BRAF inhibition could promote outgrowth of, for example, PIK3CA-mutated subclones, and ultimate anaplastic transformation, although it is impossible to say for sure that this would not have happened in the absence of therapeutic BRAF inhibition.
Other than PIK3CA, additional mutations associated with BRAF inhibitor resistance and/or anaplastic transformation have been described in the literature [10, 72–78]. These were also observed to be enriched in thyroid carcinomas that dedifferentiated after treatment with vemurafenib or dabrafenib (Figure 3). Compared to tumors that did not dedifferentiate (N = 9), these previously described mutations were found in greater frequency in patients who experienced tumor dedifferentiation (N = 7) after treatment (33% vs. 86%). Mutations observed in this cohort include one involved in the PI3K/AKT/mTOR pathway (MTOR) as well as additional mutations affecting the MAPK/ERK pathway (MET amplifications, NF2, NRAS, and RASA1). Two alternative mechanisms identified in this cohort include the SWI/SNF chromatin remodeling complex (ARID2 and PBRM1) and the JAK/STAT pathway (JAK1). The SWI/SNF chromatin remodeling complex has been described to bypass BRAF inhibition by promoting stem-cell-like properties and reestablishing regular thyroid iodine metabolism [10, 75], while JAK1 mutations mediate resistance to BRAF inhibitors through reduced RN125 expression leading to overexpression of receptor tyrosine kinases such as EGFR [78]. Findings from this expanded cohort highlight the varied number of mechanisms used by tumors to bypass BRAF blockade, again suggesting the need for alternative strategies to address compensatory mechanisms.
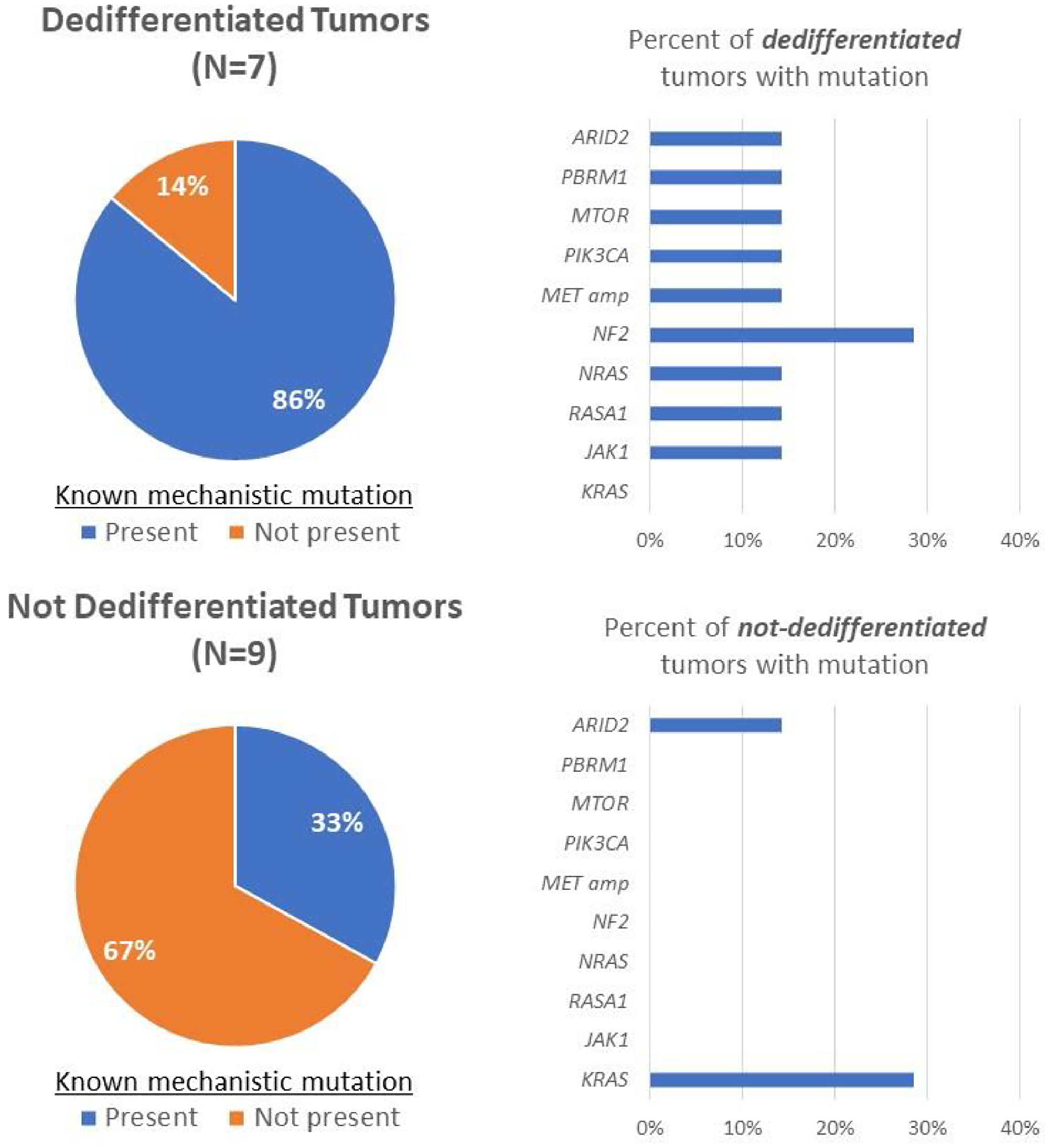
Figure 3: Rates of mutations with previously described mechanistic roles in BRAF inhibitor resistance and/or anaplastic transformation were compared between patients who dedifferentiated after BRAF inhibitor targeted therapy and those who did not using the MSK-IMPACT targeted next-generation sequencing platform.
Genetic alterations in 834 thyroid carcinomas along the spectrum of de-differentiation
To assess the role of these mutations in thyroid dedifferentiation more broadly, we examined genetic data from a cohort of 639 advanced, recurrent, and/or metastatic thyroid tumors spanning the spectrum of dedifferentiation that had undergone targeted next-generation sequencing. Here, we provide updated data from an expanded cohort of 834 tumors along the spectrum of thyroid cancer differentiation profiled with targeted next-generation sequencing on the MSK-IMPACT platform (469 PTC, 225 PDTC, and 140 ATC) and compared these to 496 primary PTCs from The Cancer Genome Atlas [9]. The full methodology for MSK-IMPACT was described in a prior article [79]. Briefly, informed consent was collected for analysis of tumor and paired-normal blood samples. DNA was extracted from 834 formalin-fixed, paraffin-embedded thyroid tumors treated at MSKCC between April 2015 and September 2023. Slides were prepared by board-certified subspecialty pathologists with hematoxylin and eosin stains per standard protocol. Histologic type of thyroid cancer was determined according to the World Health Organization’s classification of endocrine tumors [80]. Average target coverage for the PTC, PDTC, and ATC groups were and 530×, 553×, and 646×, respectively. Mutational data for these cohorts are provided in the Supplementary Table 1.
Consistent with prior analyses, the previously identified mutations with described roles in BRAF inhibitor resistance were markedly more prevalent in de-differentiated thyroid carcinoma histologies (Table 1). Mutations in ARID2, JAK1, MTOR, NF2, NRAS, PBRM1, PIK3CA, and TERT promotor increased to a statistically significant degree in PDTCs and ATCs relative to the TCGA primary PTCs. While not statistically significant, MET amplifications are an overall rare event and were only seen in PDTCs and ATCs and not in either PTC cohort.
Table 1: Frequency of mutations with described roles in BRAF inhibitor resistance and/or anaplastic evolution across the spectrum of thyroid tumor dedifferentiation
| Primary PTCs† | R/M PTCs‡ | R/M PDTCs‡ | ATCs‡ | |
|---|---|---|---|---|
| ARID2 mutation | 1/496 (0.2%) | 7/469 (1.5%)* | 8/225 (3.6%)*** | 9/140 (6.4%)**** |
| JAK1 mutation | 1/496 (0.2%) | 0/469 (0.0%) | 6/225 (2.7%)** | 2/140 (1.4%) |
| MET amplification | 0/496 (0.0%) | 0/469 (0.0%) | 1/225 (0.4%) | 1/140 (0.7%) |
| MTOR mutation | 0/496 (0.0%) | 4/469 (0.9%) | 2/225 (0.9%) | 6/140 (4.3%)**** |
| NF2 mutation | 1/496 (0.2%) | 0/469 (0.0%) | 3/225 (1.3%) | 19/140 (13.6%)**** |
| NRAS mutation | 34/496 (6.9%) | 36/469 (7.7%) | 70/225 (31.1%)**** | 23/140 (16.4%)** |
| PBRM1 mutation | 0/496 (0.0%) | 1/469 (0.2%) | 1/225 (0.4%) | 8/140 (5.7%)**** |
| PIK3CA mutation | 2/496 (0.4%) | 18/469 (3.8%)**** | 10/225 (4.4%)*** | 20/140 (14.3%)**** |
| RASA1 mutation | 1/496 (0.2%) | 2/469 (0.4%) | 2/225 (0.9%) | 1/140 (0.7%) |
| TERT promotor | 0/496 (0.0%) | 262/469 (55.9%)**** | 126/225 (56.0%)**** | 113/140 (80.7%)**** |
In conclusion, a number of compensatory mechanisms are used by tumors to bypass BRAF blockade. Mutations involving these pathways are noted in higher frequency in tumors that dedifferentiate after BRAF inhibitor treatment. The functional relevance of these pathways is supported by increased expression in alternative pathways such as PI3K-AKT and mTOR after tumor dedifferentiation and BRAF inhibitor treatment.
The immune microenvironment in anaplastic evolution and BRAF inhibitor resistance
Another area of active investigation is the tumor immune microenvironment because of its known role in thyroid cancer pathogenesis and drug resistance [81–83]. While generally thought to increase anti-tumor immunity [84], BRAF inhibitors may also exert competing effects such as driving tumor infiltration by macrophages, which can mediate resistance to BRAF inhibition through TNFα and VEGF [85]. The development of ATC has also been associated with changes to the immune milieu, including increased infiltration by macrophages [86, 87] and fibroblasts [88, 89], a finding reflected in our prior immune deconvolution analyses comparing ATC samples transformed from PTC after treatment with vemurafenib (Figure 2). In ATC, macrophages form dense nests with elongated cytoplasmic extensions surrounding the undifferentiated tumor cells [87]. The full significance of this interaction is still to be further elucidated. Fibroblasts are enriched in PDTC and are thought to alter the extracellular matrix including enrichment of stromal-derived fibrillar collagen which in turn facilitates mobility of tumor cells [88]. The ATC samples in our study also exhibited increased CD8 T-cell exclusion and dysfunction compared to pre-treatment PTC. Similar changes were observed and further extrapolated on in a recent study by Lu et al. [90] that assessed ATC with single-cell sequencing. The authors described a progression from stress-responsive differentiated tumor cells to a diploid stage with anaplastic cells exhibiting an inflammatory phenotype and lastly to an aneuploid stage with tumor cells gaining mesenchymal properties. This progression parallels a shift from M1 macrophages to M2 and T cells from cytotoxic to an exhausted state. Overall, these changes represent an immune-suppressed microenvironment seen in both BRAF inhibitor resistance and anaplastic evolution, and may represent a potential target for future multi-modality therapy.
Current practice and future directions in targeted therapy for thyroid cancer
Locally advanced, recurrent, metastatic and/or dedifferentiated tumors occur in a subset of thyroid cancers. The standard therapeutic approach remains surgical resection when feasible, sometimes followed by adjuvant radioactive iodine therapy. There is a limited role for cytotoxic chemotherapy given low response rates, short duration of tumor control, and toxic effects [91]. However, chemotherapy with surgery and radiation can provide marginal survival benefit in stage IVA and IVB ATC [92], and select cases of ATC may be amenable to neoadjuvant use of chemotherapy followed by surgical excision if able [93]. For metastatic disease harboring the BRAF V600E mutation, BRAF inhibitors are an available treatment; however, their utility as single-modality therapy is limited by near inevitable disease progression. Recent investigations on BRAF inhibitors have shifted to concurrent blockade of ancillary signaling pathways or promoting anti-tumor immune activity.
Targeted agents have been discovered for several of the resistance mechanisms described above. For patients with ATCs harboring the BRAF V600E mutation that are not initially surgically resectable but may be borderline resectable, neoadjuvant combination kinase inhibition with dabrafenib plus trametinib is recommended [94, 95]. This regimen can also be considered for systemic therapy of metastatic ATC based on data from the phase II ROAR trial [96]. The drug everolimus inhibits MTOR and in a limited phase I trial of everolimus with vemurafenib in 20 patients, including 4 with thyroid cancer, it was shown to be well tolerated with partial response and stable disease rates of 22% and 50%, respectively [97]. Crizotinib targets MET and a phase I study including an arm for vemurafenib with crizotinib (N = 11) showed a partial response rate of 27% and stable disease of 9%, although none of the groups with clinical benefit included patients with thyroid cancer [98]. PI3K can be targeted with alpelisib or buparlisib, and while no results are available to date for combination use with BRAF inhibitors, they have been tested with the MEK inhibitors trametinib [99] and binimetinib [100] and demonstrated antitumor activity in cancer types such as ovarian cancer. The agent momelotinib (JAK1/2 inhibitor) also has yet to be trialed with BRAF inhibitors, but a study using it in combination with trametinib demonstrated no benefit in KRAS-mutated non-small cell lung cancer [101]. Despite the mechanistic rational for multi-target therapies, long-term disease control with these agents remain elusive as demonstrated by pooled analyses with larger samples. For example, the VEM-PLUS study showed no meaningful benefit of vemurafenib with RAF or mTOR-targeted agents compared to vemurafenib alone [102]. Another study using data from 5 phase I and II trials showed no benefit with vemurafenib combined with crizotinib, sorafenib or everolimus [103]. Moreover, these studies found a high rate of adverse effects with combination therapy, requiring dose adjustments that further limit anti-tumor activity. Findings from these recent studies highlight the complex, multifactorial nature of these pathways and the limits of precision oncology platforms.
Based on observations of the immunosuppressive microenvironment of advanced thyroid tumors and prior studies of the possible immune-potentiating effects of BRAF inhibitors, several studies have evaluated the efficacy of combined BRAF blockade with immunotherapy. Indeed, pre-clinical studies of this combination demonstrated possible complimentary activities. A study by Zhi and colleagues [104] using mouse models found that treatment with the BRAF inhibitor PLX4032 in combination with anti-PD-1 antibody reversed TGF-β1/SMAD3 facilitated repression of tumor-specific major histocompatibility complex class II, increased CD4 T-cell infiltration, and suppressed tumor growth. Other novel immunotherapy strategies have also been evaluated for complimentary effects to BRAF inhibitors, including oncolytic HSV [105] and CAR T-cell therapy [106], both of were shown to enhance tumor control in pre-clinical models. However, the pre-clinical literature includes conflicting results, such as a study by Gunda et al. [107] that showed short (2–3 week) tumor control and re-establishment of an immune suppressive environment in ATC mice treated with combination BRAF and immune checkpoint blockade. Investigations into the real-world efficacy of these combinations are ongoing, however early results show promising anti-tumor effects. For example, a phase II clinical trial (ATLEP) on lenvatinib with pembrolizumab including 27 and 8 patients with ATC and PDTC respectively yielded 51.9% partial response and 44.4% stable disease in ATC and 75% partial response and 25% stable disease in PDTC [108]. While promising, additional clinical studies with long-term follow-up are needed to evaluate the real-world efficacy of combined BRAF blockade with these novel immune-modulating therapies.
In conclusion, mutations in thyroid cancer associated with BRAF inhibitor resistance may also be associated with thyroid tumor dedifferentiation. This may be relevant to rare cases of tumors observed to dedifferentiate after BRAF blockade. While early studies of BRAF inhibitors demonstrated anti-tumor activity in advanced thyroid cancers harboring the BRAF V600E mutation, subsequent experience and research showed limited long-term disease control due to abundant compensatory mechanisms. One mechanism of resistance is the transition to an alternative cell state driven by selective pressures from BRAF blockade, which on rare occasion can result in tumor dedifferentiation. These mechanisms include mutations affecting the PI3K/AKT (PIK3CA, MTOR), MAPK/ERK pathway (MET amplifications, NF2, NRAS, RASA1), SWI/SNF chromatin remodeling complex (ARID2, PBMR1) and JAK/STAT pathways (JAK1). Dual-target therapies have been trialed but with continued limitations to long-term disease control. Thyroid tumor dedifferentiation and BRAF inhibitor resistance are also found to be associated with a transition to an immunosuppressed state. Early studies on combined targeted and immune-modulated therapy have demonstrated promising outcomes. Further clinical studies are needed to test real-world effectiveness of these novel immunotherapies with targeted therapy.
CONFLICTS OF INTEREST
Authors have no conflicts of interest to declare.
ETHICAL STATEMENT AND CONSENT
This research was approved by the Institutional Review Board of Memorial Sloan Kettering Cancer Center (IRB protocol #11-195). Informed consent was collected for analysis of tumor and paired-normal blood samples.
FUNDING
This study was supported in part by NIH R01 DE027738, US Department of Defense CA210784, The Geoffrey Beene Cancer Research Center, The MSK Population Science Research Program, the Jayme and Peter Flowers Fund, and the Sebastian Nativo Fund (to LGTM), and the NIH/NCI Cancer Center Support Grant P30 CA008748 (to MSKCC).
References
1. Fagin JA, Wells SA Jr. Biologic and Clinical Perspectives on Thyroid Cancer. N Engl J Med. 2016; 375:1054–67. https://doi.org/10.1056/NEJMra1501993. [PubMed].
2. Fan D, Ma J, Bell AC, Groen AH, Olsen KS, Lok BH, Leeman JE, Anderson E, Riaz N, McBride S, Ganly I, Shaha AR, Sherman EJ, et al. Outcomes of multimodal therapy in a large series of patients with anaplastic thyroid cancer. Cancer. 2020; 126:444–52. https://doi.org/10.1002/cncr.32548. [PubMed].
3. Maniakas A, Dadu R, Busaidy NL, Wang JR, Ferrarotto R, Lu C, Williams MD, Gunn GB, Hofmann MC, Cote G, Sperling J, Gross ND, Sturgis EM, et al. Evaluation of Overall Survival in Patients With Anaplastic Thyroid Carcinoma, 2000-2019. JAMA Oncol. 2020; 6:1397–404. https://doi.org/10.1001/jamaoncol.2020.3362. [PubMed].
4. Raman M, Chen W, Cobb MH. Differential regulation and properties of MAPKs. Oncogene. 2007; 26:3100–12. https://doi.org/10.1038/sj.onc.1210392. [PubMed].
5. Qi M, Elion EA. MAP kinase pathways. J Cell Sci. 2005; 118:3569–72. https://doi.org/10.1242/jcs.02470. [PubMed].
6. Keshet Y, Seger R. The MAP kinase signaling cascades: a system of hundreds of components regulates a diverse array of physiological functions. Methods Mol Biol. 2010; 661:3–38. https://doi.org/10.1007/978-1-60761-795-2_1. [PubMed].
7. Plotnikov A, Zehorai E, Procaccia S, Seger R. The MAPK cascades: signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim Biophys Acta. 2011; 1813:1619–33. https://doi.org/10.1016/j.bbamcr.2010.12.012. [PubMed].
8. Kimura ET, Nikiforova MN, Zhu Z, Knauf JA, Nikiforov YE, Fagin JA. High prevalence of BRAF mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res. 2003; 63:1454–57. [PubMed].
9. Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell. 2014; 159:676–90. https://doi.org/10.1016/j.cell.2014.09.050. [PubMed].
10. Landa I, Ibrahimpasic T, Boucai L, Sinha R, Knauf JA, Shah RH, Dogan S, Ricarte-Filho JC, Krishnamoorthy GP, Xu B, Schultz N, Berger MF, Sander C, et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J Clin Invest. 2016; 126:1052–66. https://doi.org/10.1172/JCI85271. [PubMed].
11. Hu S, Liu D, Tufano RP, Carson KA, Rosenbaum E, Cohen Y, Holt EH, Kiseljak-Vassiliades K, Rhoden KJ, Tolaney S, Condouris S, Tallini G, Westra WH, et al. Association of aberrant methylation of tumor suppressor genes with tumor aggressiveness and BRAF mutation in papillary thyroid cancer. Int J Cancer. 2006; 119:2322–29. https://doi.org/10.1002/ijc.22110. [PubMed].
12. Brose MS, Troxel AB, Redlinger M, Harlacker K, Redlinger C, Chalian KT, Flaherty AA, Loevner LA, Mandel SJ, O’Dwyer PJ. Effect of BRAFV600E on response to sorafenib in advanced thyroid cancer patients. J Clin Oncol. 2009 (15_suppl); 27:6002. https://doi.org/10.1200/jco.2009.27.15_suppl.6002.
13. Schlumberger M, Tahara M, Wirth LJ, Robinson B, Brose MS, Elisei R, Habra MA, Newbold K, Shah MH, Hoff AO, Gianoukakis AG, Kiyota N, Taylor MH, et al. Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N Engl J Med. 2015; 372:621–30. https://doi.org/10.1056/NEJMoa1406470. [PubMed].
14. Ahmed M, Barbachano Y, Riddell A, Hickey J, Newbold KL, Viros A, Harrington KJ, Marais R, Nutting CM. Analysis of the efficacy and toxicity of sorafenib in thyroid cancer: a phase II study in a UK based population. Eur J Endocrinol. 2011; 165:315–22. https://doi.org/10.1530/EJE-11-0129. [PubMed].
15. Cabanillas ME, Takahashi S. Managing the adverse events associated with lenvatinib therapy in radioiodine-refractory differentiated thyroid cancer. Semin Oncol. 2019; 46:57–64. https://doi.org/10.1053/j.seminoncol.2018.11.004. [PubMed].
16. Brose MS, Nutting CM, Jarzab B, Elisei R, Siena S, Bastholt L, de la Fouchardiere C, Pacini F, Paschke R, Shong YK, Sherman SI, Smit JW, Chung J, et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: a randomised, double-blind, phase 3 trial. Lancet. 2014; 384:319–28. https://doi.org/10.1016/S0140-6736(14)60421-9. [PubMed].
17. Falchook GS, Long GV, Kurzrock R, Kim KB, Arkenau TH, Brown MP, Hamid O, Infante JR, Millward M, Pavlick AC, O’Day SJ, Blackman SC, Curtis CM, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet. 2012; 379:1893–901. https://doi.org/10.1016/S0140-6736(12)60398-5. [PubMed].
18. Dunn LA, Sherman EJ, Baxi SS, Tchekmedyian V, Grewal RK, Larson SM, Pentlow KS, Haque S, Tuttle RM, Sabra MM, Fish S, Boucai L, Walters J, et al. Vemurafenib Redifferentiation of BRAF Mutant, RAI-Refractory Thyroid Cancers. J Clin Endocrinol Metab. 2019; 104:1417–28. https://doi.org/10.1210/jc.2018-01478. [PubMed].
19. Brose MS, Cabanillas ME, Cohen EE, Wirth LJ, Riehl T, Yue H, Sherman SI, Sherman EJ. Vemurafenib in patients with BRAF(V600E)-positive metastatic or unresectable papillary thyroid cancer refractory to radioactive iodine: a non-randomised, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016; 17:1272–82. https://doi.org/10.1016/S1470-2045(16)30166-8. [PubMed].
20. Falchook GS, Millward M, Hong D, Naing A, Piha-Paul S, Waguespack SG, Cabanillas ME, Sherman SI, Ma B, Curtis M, Goodman V, Kurzrock R. BRAF inhibitor dabrafenib in patients with metastatic BRAF-mutant thyroid cancer. Thyroid. 2015; 25:71–77. https://doi.org/10.1089/thy.2014.0123. [PubMed].
21. Rothenberg SM, McFadden DG, Palmer EL, Daniels GH, Wirth LJ. Redifferentiation of iodine-refractory BRAF V600E-mutant metastatic papillary thyroid cancer with dabrafenib. Clin Cancer Res. 2015; 21:1028–35. https://doi.org/10.1158/1078-0432.CCR-14-2915. [PubMed].
22. Scheffel RS, Dora JM, Maia AL. BRAF mutations in thyroid cancer. Curr Opin Oncol. 2022; 34:9–18. https://doi.org/10.1097/CCO.0000000000000797. [PubMed].
23. Crispo F, Notarangelo T, Pietrafesa M, Lettini G, Storto G, Sgambato A, Maddalena F, Landriscina M. BRAF Inhibitors in Thyroid Cancer: Clinical Impact, Mechanisms of Resistance and Future Perspectives. Cancers (Basel). 2019; 11:1388. https://doi.org/10.3390/cancers11091388. [PubMed].
24. Roelli MA, Ruffieux-Daidié D, Stooss A, ElMokh O, Phillips WA, Dettmer MS, Charles RP. PIK3CA(H1047R)-induced paradoxical ERK activation results in resistance to BRAF(V600E) specific inhibitors in BRAF(V600E) PIK3CA(H1047R) double mutant thyroid tumors. Oncotarget. 2017; 8:103207–22. https://doi.org/10.18632/oncotarget.21732. [PubMed].
25. Danysh BP, Rieger EY, Sinha DK, Evers CV, Cote GJ, Cabanillas ME, Hofmann MC. Long-term vemurafenib treatment drives inhibitor resistance through a spontaneous KRAS G12D mutation in a BRAF V600E papillary thyroid carcinoma model. Oncotarget. 2016; 7:30907–23. https://doi.org/10.18632/oncotarget.9023. [PubMed].
26. Montero-Conde C, Ruiz-Llorente S, Dominguez JM, Knauf JA, Viale A, Sherman EJ, Ryder M, Ghossein RA, Rosen N, Fagin JA. Relief of feedback inhibition of HER3 transcription by RAF and MEK inhibitors attenuates their antitumor effects in BRAF-mutant thyroid carcinomas. Cancer Discov. 2013; 3:520–33. https://doi.org/10.1158/2159-8290.CD-12-0531. [PubMed].
27. Byeon HK, Na HJ, Yang YJ, Kwon HJ, Chang JW, Ban MJ, Kim WS, Shin DY, Lee EJ, Koh YW, Yoon JH, Choi EC. c-Met-mediated reactivation of PI3K/AKT signaling contributes to insensitivity of BRAF(V600E) mutant thyroid cancer to BRAF inhibition. Mol Carcinog. 2016; 55:1678–87. https://doi.org/10.1002/mc.22418. [PubMed].
28. Sos ML, Levin RS, Gordan JD, Oses-Prieto JA, Webber JT, Salt M, Hann B, Burlingame AL, McCormick F, Bandyopadhyay S, Shokat KM. Oncogene mimicry as a mechanism of primary resistance to BRAF inhibitors. Cell Rep. 2014; 8:1037–48. https://doi.org/10.1016/j.celrep.2014.07.010. [PubMed].
29. Landriscina M, Pannone G, Piscazzi A, Toti P, Fabiano A, Tortorella S, Occhini R, Ambrosi A, Bufo P, Cignarelli M. Epidermal growth factor receptor 1 expression is upregulated in undifferentiated thyroid carcinomas in humans. Thyroid. 2011; 21:1227–34. https://doi.org/10.1089/thy.2011.0172. [PubMed].
30. Konieczkowski DJ, Johannessen CM, Garraway LA. A Convergence-Based Framework for Cancer Drug Resistance. Cancer Cell. 2018; 33:801–15. https://doi.org/10.1016/j.ccell.2018.03.025. [PubMed].
31. Lee M, Untch BR, Xu B, Ghossein R, Han C, Kuo F, Valero C, Nadeem Z, Patel N, Makarov V, Dogan S, Wong RJ, Sherman EJ, et al. Genomic and Transcriptomic Correlates of Thyroid Carcinoma Evolution after BRAF Inhibitor Therapy. Mol Cancer Res. 2022; 20:45–55. https://doi.org/10.1158/1541-7786.MCR-21-0442. [PubMed].
32. Venkatesh YS, Ordonez NG, Schultz PN, Hickey RC, Goepfert H, Samaan NA. Anaplastic carcinoma of the thyroid. A clinicopathologic study of 121 cases. Cancer. 1990; 66:321–30. https://doi.org/10.1002/1097-0142(19900715)66:2%3c321::aid-cncr2820660221%3e3.0.co;2-a. [PubMed].
33. Demeter JG, De Jong SA, Lawrence AM, Paloyan E. Anaplastic thyroid carcinoma: risk factors and outcome. Surgery. 1991; 110:956. [PubMed].
34. Carcangiu ML, Steeper T, Zampi G, Rosai J. Anaplastic thyroid carcinoma. A study of 70 cases. Am J Clin Pathol. 1985; 83:135–58. https://doi.org/10.1093/ajcp/83.2.135. [PubMed].
35. Passler C, Scheuba C, Prager G, Kaserer K, Flores JA, Vierhapper H, Niederle B. Anaplastic (undifferentiated) thyroid carcinoma (ATC). A retrospective analysis. Langenbecks Arch Surg. 1999; 384:284–93. https://doi.org/10.1007/s004230050205. [PubMed].
36. Junor EJ, Paul J, Reed NS. Anaplastic thyroid carcinoma: 91 patients treated by surgery and radiotherapy. Eur J Surg Oncol. 1992; 18:83–88. [PubMed].
37. Ain KB. Anaplastic thyroid carcinoma: a therapeutic challenge. Semin Surg Oncol. 1999; 16:64–9. https://doi.org/10.1002/(sici)1098-2388(199901/02)16:1<64::aid-ssu10>3.0.co;2-u. [PubMed].
38. Ibanez ML, Russell WO, Albores-Saavedra J, Lampertico P, White EC, Clark RL. Thyroid carcinoma--biologic behavior and mortality. Postmortem findings in 42 cases, including 27 in which the disease was fatal. Cancer. 1966; 19:1039–52. https://doi.org/10.1002/1097-0142(196608)19:8%3c1039::aid-cncr2820190802%3e3.0.co;2-1. [PubMed].
39. Wiseman SM, Loree TR, Hicks WL Jr, Rigual NR, Winston JS, Tan D, Anderson GR, Stoler DL. Anaplastic thyroid cancer evolved from papillary carcinoma: demonstration of anaplastic transformation by means of the inter-simple sequence repeat polymerase chain reaction. Arch Otolaryngol Head Neck Surg. 2003; 129:96–100. https://doi.org/10.1001/archotol.129.1.96. [PubMed].
40. Hunt JL, Tometsko M, LiVolsi VA, Swalsky P, Finkelstein SD, Barnes EL. Molecular evidence of anaplastic transformation in coexisting well-differentiated and anaplastic carcinomas of the thyroid. Am J Surg Pathol. 2003; 27:1559–64. https://doi.org/10.1097/00000478-200312000-00009. [PubMed].
41. Dumitriu L, Stefăneanu L, Taşcă C. The anaplastic transformation of differentiated thyroid carcinoma. An ultrastructural study. Endocrinologie. 1984; 22:91–96. [PubMed].
42. Galera-Davidson H, Bibbo M, Dytch HE, González-Cámpora R, Fernández A, Wied GL. Nuclear DNA in anaplastic thyroid carcinoma with a differentiated component. Histopathology. 1987; 11:715–22. https://doi.org/10.1111/j.1365-2559.1987.tb02685.x. [PubMed].
43. Wallin G, Bäckdahl M, Tallroth-Ekman E, Lundell G, Auer G, Löwhagen T. Co-existent anaplastic and well differentiated thyroid carcinomas: a nuclear DNA study. Eur J Surg Oncol. 1989; 15:43–48. [PubMed].
44. Hemmer S, Wasenius VM, Knuutila S, Franssila K, Joensuu H. DNA copy number changes in thyroid carcinoma. Am J Pathol. 1999; 154:1539–47. https://doi.org/10.1016/S0002-9440(10)65407-7. [PubMed].
45. Moretti F, Farsetti A, Soddu S, Misiti S, Crescenzi M, Filetti S, Andreoli M, Sacchi A, Pontecorvi A. p53 re-expression inhibits proliferation and restores differentiation of human thyroid anaplastic carcinoma cells. Oncogene. 1997; 14:729–40. https://doi.org/10.1038/sj.onc.1200887. [PubMed].
46. Zou M, Shi Y, Farid NR. p53 mutations in all stages of thyroid carcinomas. J Clin Endocrinol Metab. 1993; 77:1054–58. https://doi.org/10.1210/jcem.77.4.8408453. [PubMed].
47. Nakamura T, Yana I, Kobayashi T, Shin E, Karakawa K, Fujita S, Miya A, Mori T, Nishisho I, Takai S. p53 gene mutations associated with anaplastic transformation of human thyroid carcinomas. Jpn J Cancer Res. 1992; 83:1293–8. https://doi.org/10.1111/j.1349-7006.1992.tb02761.x. [PubMed].
48. Fagin JA, Matsuo K, Karmakar A, Chen DL, Tang SH, Koeffler HP. High prevalence of mutations of the p53 gene in poorly differentiated human thyroid carcinomas. J Clin Invest. 1993; 91:179–84. https://doi.org/10.1172/JCI116168. [PubMed].
49. Donghi R, Longoni A, Pilotti S, Michieli P, Della Porta G, Pierotti MA. Gene p53 mutations are restricted to poorly differentiated and undifferentiated carcinomas of the thyroid gland. J Clin Invest. 1993; 91:1753–60. https://doi.org/10.1172/JCI116385. [PubMed].
50. Fagin JA. Tumor suppressor genes in human thyroid neoplasms: p53 mutations are associated undifferentiated thyroid cancers. J Endocrinol Invest. 1995; 18:140–42. https://doi.org/10.1007/BF03349723. [PubMed].
51. Battista S, Martelli ML, Fedele M, Chiappetta G, Trapasso F, De Vita G, Battaglia C, Santoro M, Viglietto G, Fagin JA. A mutated p53 gene alters thyroid cell differentiation. Oncogene. 1995; 11:2029–37. [PubMed].
52. Matias-Guiu X, Villanueva A, Cuatrecasas M, Capella G, De Leiva A, Prat J. p53 in a thyroid follicular carcinoma with foci of poorly differentiated and anaplastic carcinoma. Pathol Res Pract. 1996; 192:1242. https://doi.org/10.1016/S0344-0338(96)80159-2. [PubMed].
53. Blagosklonny MV, Giannakakou P, Wojtowicz M, Romanova LY, Ain KB, Bates SE, Fojo T. Effects of p53-expressing adenovirus on the chemosensitivity and differentiation of anaplastic thyroid cancer cells. J Clin Endocrinol Metab. 1998; 83:2516–22. https://doi.org/10.1210/jcem.83.7.4984. [PubMed].
54. Dobashi Y, Sugimura H, Sakamoto A, Mernyei M, Mori M, Oyama T, Machinami R. Stepwise participation of p53 gene mutation during dedifferentiation of human thyroid carcinomas. Diagn Mol Pathol. 1994; 3:9–14. https://doi.org/10.1097/00019606-199403010-00003. [PubMed].
55. Ito T, Seyama T, Mizuno T, Tsuyama N, Hayashi T, Hayashi Y, Dohi K, Nakamura N, Akiyama M. Unique association of p53 mutations with undifferentiated but not with differentiated carcinomas of the thyroid gland. Cancer Res. 1992; 52:1369–71. [PubMed].
56. Fagin JA, Tang SH, Zeki K, Di Lauro R, Fusco A, Gonsky R. Reexpression of thyroid peroxidase in a derivative of an undifferentiated thyroid carcinoma cell line by introduction of wild-type p53. Cancer Res. 1996; 56:765–71. [PubMed].
57. Pilotti S, Collini P, Rilke F, Cattoretti G, Del Bo R, Pierotti MA. Bcl-2 protein expression in carcinomas originating from the follicular epithelium of the thyroid gland. J Pathol. 1994; 172:337–42. https://doi.org/10.1002/path.1711720408. [PubMed].
58. Wang S, Lloyd RV, Hutzler MJ, Safran MS, Patwardhan NA, Khan A. The role of cell cycle regulatory protein, cyclin D1, in the progression of thyroid cancer. Mod Pathol. 2000; 13:882–87. https://doi.org/10.1038/modpathol.3880157. [PubMed].
59. Garcia-Rostan G, Tallini G, Herrero A, D’Aquila TG, Carcangiu ML, Rimm DL. Frequent mutation and nuclear localization of beta-catenin in anaplastic thyroid carcinoma. Cancer Res. 1999; 59:1811–15. [PubMed].
60. Bergström JD, Hermansson A, Diaz de Ståhl T, Heldin NE. Non-autocrine, constitutive activation of Met in human anaplastic thyroid carcinoma cells in culture. Br J Cancer. 1999; 80:650–56. https://doi.org/10.1038/sj.bjc.6690406. [PubMed].
61. Hoang-Vu C, Dralle H, Scheumann G, Maenhaut C, Horn R, von zur Mühlen A, Brabant G. Gene expression of differentiation- and dedifferentiation markers in normal and malignant human thyroid tissues. Exp Clin Endocrinol. 1992; 100:51–56. https://doi.org/10.1055/s-0029-1211176. [PubMed].
62. Zou M, Shi Y, al-Sedairy S, Farid NR. High levels of Nm23 gene expression in advanced stage of thyroid carcinomas. Br J Cancer. 1993; 68:385–88. https://doi.org/10.1038/bjc.1993.345. [PubMed].
63. Stringer BM, Rowson JM, Parkar MH, Seid JM, Hearn PR, Wynford-Thomas D, Ingemansson S, Woodhouse N, Goyns MH. Detection of the H-RAS oncogene in human thyroid anaplastic carcinomas. Experientia. 1989; 45:372–76. https://doi.org/10.1007/BF01957483. [PubMed].
64. Suárez HG, Du Villard JA, Caillou B, Schlumberger M, Tubiana M, Parmentier C, Monier R. Detection of activated ras oncogenes in human thyroid carcinomas. Oncogene. 1988; 2:403–6. [PubMed].
65. Tsoi J, Robert L, Paraiso K, Galvan C, Sheu KM, Lay J, Wong DJL, Atefi M, Shirazi R, Wang X, Braas D, Grasso CS, Palaskas N, et al. Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell. 2018; 33:890–904.e5. https://doi.org/10.1016/j.ccell.2018.03.017. [PubMed].
66. Konieczkowski DJ, Johannessen CM, Abudayyeh O, Kim JW, Cooper ZA, Piris A, Frederick DT, Barzily-Rokni M, Straussman R, Haq R, Fisher DE, Mesirov JP, Hahn WC, et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov. 2014; 4:816–27. https://doi.org/10.1158/2159-8290.CD-13-0424. [PubMed].
67. Patham B, Bible K, Waguespack S, Cabanillas M. Anaplastic transformation (ATC-t) of papillary thyroid cancer (PTC) after treatment with BRAF inhibitors (BRAFi). Endocr Rev. 2015; 36:SAT-011.
68. Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci. 2011; 36:320–28. https://doi.org/10.1016/j.tibs.2011.03.006. [PubMed].
69. Guan KL, Figueroa C, Brtva TR, Zhu T, Taylor J, Barber TD, Vojtek AB. Negative regulation of the serine/threonine kinase B-Raf by Akt. J Biol Chem. 2000; 275:27354–59. https://doi.org/10.1074/jbc.M004371200. [PubMed].
70. Candido S, Salemi R, Piccinin S, Falzone L, Libra M. The PIK3CA H1047R Mutation Confers Resistance to BRAF and MEK Inhibitors in A375 Melanoma Cells through the Cross-Activation of MAPK and PI3K-Akt Pathways. Pharmaceutics. 2022; 14:590. https://doi.org/10.3390/pharmaceutics14030590. [PubMed].
71. Guri Y, Hall MN. mTOR Signaling Confers Resistance to Targeted Cancer Drugs. Trends Cancer. 2016; 2:688–97. https://doi.org/10.1016/j.trecan.2016.10.006. [PubMed].
72. Garcia-Rendueles ME, Ricarte-Filho JC, Untch BR, Landa I, Knauf JA, Voza F, Smith VE, Ganly I, Taylor BS, Persaud Y, Oler G, Fang Y, Jhanwar SC, et al. NF2 Loss Promotes Oncogenic RAS-Induced Thyroid Cancers via YAP-Dependent Transactivation of RAS Proteins and Sensitizes Them to MEK Inhibition. Cancer Discov. 2015; 5:1178–93. https://doi.org/10.1158/2159-8290.CD-15-0330. [PubMed].
73. Knauf JA, Luckett KA, Chen KY, Voza F, Socci ND, Ghossein R, Fagin JA. Hgf/Met activation mediates resistance to BRAF inhibition in murine anaplastic thyroid cancers. J Clin Invest. 2018; 128:4086–97. https://doi.org/10.1172/JCI120966. [PubMed].
74. Pozdeyev N, Gay LM, Sokol ES, Hartmaier R, Deaver KE, Davis S, French JD, Borre PV, LaBarbera DV, Tan AC, Schweppe RE, Fishbein L, Ross JS, et al. Genetic Analysis of 779 Advanced Differentiated and Anaplastic Thyroid Cancers. Clin Cancer Res. 2018; 24:3059–68. https://doi.org/10.1158/1078-0432.CCR-18-0373. [PubMed].
75. Saqcena M, Leandro-Garcia LJ, Maag JLV, Tchekmedyian V, Krishnamoorthy GP, Tamarapu PP, Tiedje V, Reuter V, Knauf JA, de Stanchina E, Xu B, Liao XH, Refetoff S, et al. SWI/SNF Complex Mutations Promote Thyroid Tumor Progression and Insensitivity to Redifferentiation Therapies. Cancer Discov. 2021; 11:1158–75. https://doi.org/10.1158/2159-8290.CD-20-0735. [PubMed].
76. Jouenne F, Reger de Moura C, Lorillon G, Meignin V, Dumaz N, Lebbe C, Mourah S, Tazi A. RASA1 loss in a BRAF-mutated Langerhans cell sarcoma: a mechanism of resistance to BRAF inhibitor. Ann Oncol. 2019; 30:1170–72. https://doi.org/10.1093/annonc/mdz125. [PubMed].
77. Kaplan FM, Kugel CH 3rd, Dadpey N, Shao Y, Abel EV, Aplin AE. SHOC2 and CRAF mediate ERK1/2 reactivation in mutant NRAS-mediated resistance to RAF inhibitor. J Biol Chem. 2012; 287:41797–807. https://doi.org/10.1074/jbc.M112.390906. [PubMed].
78. Kim H, Frederick DT, Levesque MP, Cooper ZA, Feng Y, Krepler C, Brill L, Samuels Y, Hayward NK, Perlina A, Piris A, Zhang T, Halaban R, et al. Downregulation of the Ubiquitin Ligase RNF125 Underlies Resistance of Melanoma Cells to BRAF Inhibitors via JAK1 Deregulation. Cell Rep. 2015; 11:1458–73. https://doi.org/10.1016/j.celrep.2015.04.049. [PubMed].
79. Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, Chandramohan R, Liu ZY, Won HH, Scott SN, Brannon AR, O’Reilly C, Sadowska J, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn. 2015; 17:251–64. https://doi.org/10.1016/j.jmoldx.2014.12.006. [PubMed].
80. Lloyd R, Osamura R, Klöppel G, Rosai J. WHO Classification of tumours of endocrine organs, ed 4. Geneva. World Health Organization. 2017.
81. Bergdorf K, Ferguson DC, Mehrad M, Ely K, Stricker T, Weiss VL. Papillary thyroid carcinoma behavior: clues in the tumor microenvironment. Endocr Relat Cancer. 2019; 26:601–14. https://doi.org/10.1530/ERC-19-0074. [PubMed].
82. Bastman JJ, Serracino HS, Zhu Y, Koenig MR, Mateescu V, Sams SB, Davies KD, Raeburn CD, McIntyre RC Jr, Haugen BR, French JD. Tumor-Infiltrating T Cells and the PD-1 Checkpoint Pathway in Advanced Differentiated and Anaplastic Thyroid Cancer. J Clin Endocrinol Metab. 2016; 101:2863–73. https://doi.org/10.1210/jc.2015-4227. [PubMed].
83. Prete A, Lo AS, Sadow PM, Bhasin SS, Antonello ZA, Vodopivec DM, Ullas S, Sims JN, Clohessy J, Dvorak AM, Sciuto T, Bhasin M, Murphy-Ullrich JE, et al. Pericytes Elicit Resistance to Vemurafenib and Sorafenib Therapy in Thyroid Carcinoma via the TSP-1/TGFβ1 Axis. Clin Cancer Res. 2018; 24:6078–97. https://doi.org/10.1158/1078-0432.CCR-18-0693. [PubMed].
84. Callahan MK, Masters G, Pratilas CA, Ariyan C, Katz J, Kitano S, Russell V, Gordon RA, Vyas S, Yuan J, Gupta A, Wigginton JM, Rosen N, et al. Paradoxical activation of T cells via augmented ERK signaling mediated by a RAF inhibitor. Cancer Immunol Res. 2014; 2:70–79. https://doi.org/10.1158/2326-6066.CIR-13-0160. [PubMed].
85. Smith MP, Sanchez-Laorden B, O’Brien K, Brunton H, Ferguson J, Young H, Dhomen N, Flaherty KT, Frederick DT, Cooper ZA, Wargo JA, Marais R, Wellbrock C. The immune microenvironment confers resistance to MAPK pathway inhibitors through macrophage-derived TNFα. Cancer Discov. 2014; 4:1214–29. https://doi.org/10.1158/2159-8290.CD-13-1007. [PubMed].
86. Varricchi G, Loffredo S, Marone G, Modestino L, Fallahi P, Ferrari SM, de Paulis A, Antonelli A, Galdiero MR. The Immune Landscape of Thyroid Cancer in the Context of Immune Checkpoint Inhibition. Int J Mol Sci. 2019; 20:3934. https://doi.org/10.3390/ijms20163934. [PubMed].
87. Caillou B, Talbot M, Weyemi U, Pioche-Durieu C, Al Ghuzlan A, Bidart JM, Chouaib S, Schlumberger M, Dupuy C. Tumor-associated macrophages (TAMs) form an interconnected cellular supportive network in anaplastic thyroid carcinoma. PLoS One. 2011; 6:e22567. https://doi.org/10.1371/journal.pone.0022567. [PubMed].
88. Jolly LA, Novitskiy S, Owens P, Massoll N, Cheng N, Fang W, Moses HL, Franco AT. Fibroblast-Mediated Collagen Remodeling Within the Tumor Microenvironment Facilitates Progression of Thyroid Cancers Driven by BrafV600E and Pten Loss. Cancer Res. 2016; 76:1804–13. https://doi.org/10.1158/0008-5472.CAN-15-2351. [PubMed].
89. Fozzatti L, Alamino VA, Park S, Giusiano L, Volpini X, Zhao L, Stempin CC, Donadio AC, Cheng SY, Pellizas CG. Interplay of fibroblasts with anaplastic tumor cells promotes follicular thyroid cancer progression. Sci Rep. 2019; 9:8028. https://doi.org/10.1038/s41598-019-44361-6. [PubMed].
90. Lu L, Wang JR, Henderson YC, Bai S, Yang J, Hu M, Shiau CK, Pan T, Yan Y, Tran TM, Li J, Kieser R, Zhao X, et al. Anaplastic transformation in thyroid cancer revealed by single-cell transcriptomics. J Clin Invest. 2023; 133:e169653. https://doi.org/10.1172/JCI169653. [PubMed].
91. Sherman SI. Cytotoxic chemotherapy for differentiated thyroid carcinoma. Clin Oncol (R Coll Radiol). 2010; 22:464–68. https://doi.org/10.1016/j.clon.2010.03.014. [PubMed].
92. Haymart MR, Banerjee M, Yin H, Worden F, Griggs JJ. Marginal treatment benefit in anaplastic thyroid cancer. Cancer. 2013; 119:3133–39. https://doi.org/10.1002/cncr.28187. [PubMed].
93. Higashiyama T, Ito Y, Hirokawa M, Fukushima M, Uruno T, Miya A, Matsuzuka F, Miyauchi A. Induction chemotherapy with weekly paclitaxel administration for anaplastic thyroid carcinoma. Thyroid. 2010; 20:7–14. https://doi.org/10.1089/thy.2009.0115. [PubMed].
94. National Comprehensive Clinical Network. Thyroid Carcinoma (Version 4.2023). https://www.nccn.org/professionals/physician_gls/pdf/thyroid.pdf. Accessed Oct 30, 2023, 2023.
95. Wang JR, Zafereo ME, Dadu R, Ferrarotto R, Busaidy NL, Lu C, Ahmed S, Gule-Monroe MK, Williams MD, Sturgis EM, Goepfert RP, Gross ND, Lai SY, et al. Complete Surgical Resection Following Neoadjuvant Dabrafenib Plus Trametinib in BRAF(V600E)-Mutated Anaplastic Thyroid Carcinoma. Thyroid. 2019; 29:1036–43. https://doi.org/10.1089/thy.2019.0133. [PubMed].
96. Subbiah V, Kreitman RJ, Wainberg ZA, Cho JY, Schellens JHM, Soria JC, Wen PY, Zielinski CC, Cabanillas ME, Boran A, Ilankumaran P, Burgess P, Romero Salas T, Keam B. Dabrafenib plus trametinib in patients with BRAF V600E-mutant anaplastic thyroid cancer: updated analysis from the phase II ROAR basket study. Ann Oncol. 2022; 33:406–15. https://doi.org/10.1016/j.annonc.2021.12.014. [PubMed].
97. Subbiah V, Sen S, Hess KR, Janku F, Hong DS, Khatua S, Karp DD, Munoz J, Falchook GS, Groisberg R, Tsimberidou AM, Sherman SI, Hwu P, Meric-Bernstam F. Phase I Study of the BRAF Inhibitor Vemurafenib in Combination With the Mammalian Target of Rapamycin Inhibitor Everolimus in Patients With BRAF-Mutated Malignancies. JCO Precis Oncol. 2018; 2:PO.18.00189. https://doi.org/10.1200/PO.18.00189. [PubMed].
98. Kato S, Naing A, Falchook G, Holley VR, Velez-Bravo VM, Patel S, Zinner RG, Piha-Paul SA, Apostolia M. Tsimberidou AM, Hong DS, Kurzrock R, Meric-Bernstam F, Janku F. Overcoming BRAF/MEK resistance using vemurafenib with crizotinib or sorafenib in patients with BRAF-mutant advanced cancers: Phase I study. Cancer Res. 2015; 75:2689. https://doi.org/10.1158/1538-7445.AM2015-2689.
99. Bedard PL, Tabernero J, Janku F, Wainberg ZA, Paz-Ares L, Vansteenkiste J, Van Cutsem E, Pérez-García J, Stathis A, Britten CD, Le N, Carter K, Demanse D, et al. A phase Ib dose-escalation study of the oral pan-PI3K inhibitor buparlisib (BKM120) in combination with the oral MEK1/2 inhibitor trametinib (GSK1120212) in patients with selected advanced solid tumors. Clin Cancer Res. 2015; 21:730–38. https://doi.org/10.1158/1078-0432.CCR-14-1814. [PubMed].
100. Bardia A, Gounder M, Rodon J, Janku F, Lolkema MP, Stephenson JJ, Bedard PL, Schuler M, Sessa C, LoRusso P, Thomas M, Maacke H, Evans H, et al. Phase Ib Study of Combination Therapy with MEK Inhibitor Binimetinib and Phosphatidylinositol 3-Kinase Inhibitor Buparlisib in Patients with Advanced Solid Tumors with RAS/RAF Alterations. Oncologist. 2020; 25:e160–69. https://doi.org/10.1634/theoncologist.2019-0297. [PubMed].
101. Barbie DA, Spira A, Kelly K, Humeniuk R, Kawashima J, Kong S, Koczywas M. Phase 1B Study of Momelotinib Combined With Trametinib in Metastatic, Kirsten Rat Sarcoma Viral Oncogene Homolog-Mutated Non-Small-Cell Lung Cancer After Platinum-Based Chemotherapy Treatment Failure. Clin Lung Cancer. 2018; 19:e853–59. https://doi.org/10.1016/j.cllc.2018.07.004. [PubMed].
102. Nelson BE, Roszik J, Janku F, Hong DS, Kato S, Naing A, Piha-Paul S, Fu S, Tsimberidou A, Cabanillas M, Busaidy NL, Javle M, Byers LA, et al. BRAF v600E-mutant cancers treated with vemurafenib alone or in combination with everolimus, sorafenib, or crizotinib or with paclitaxel and carboplatin (VEM-PLUS) study. NPJ Precis Oncol. 2023; 7:19. https://doi.org/10.1038/s41698-022-00341-0. [PubMed].
103. Nelson BE, Roszik J, Janku F, Hong D, Kato S, Naing A, Piha-Paul S, Fu S, Tsimberidou A, Cabanillas M, Busaidy N, Javle M, Byers L. B-Raf V600E harboring non-melanoma cancers treated with Vemurafenib monotherapy and in combination with Everolimus/Sorafenib/Crizotinib/Paclitaxel+ Carboplatin: a pooled analysis of five phase 1/2 studies. Cancer Res. 2022; 82:5237. https://doi.org/10.1158/1538-7445.AM2022-5237.
104. Zhi J, Zhang P, Zhang W, Ruan X, Tian M, Guo S, Zhang W, Zheng X, Zhao L, Gao M. Inhibition of BRAF Sensitizes Thyroid Carcinoma to Immunotherapy by Enhancing tsMHCII-mediated Immune Recognition. J Clin Endocrinol Metab. 2021; 106:91–107. https://doi.org/10.1210/clinem/dgaa656. [PubMed].
105. Crespo-Rodriguez E, Bergerhoff K, Bozhanova G, Foo S, Patin EC, Whittock H, Buus R, Haider S, Muirhead G, Thway K, Newbold K, Coffin RS, Vile RG, et al. Combining BRAF inhibition with oncolytic herpes simplex virus enhances the immune-mediated antitumor therapy of BRAF-mutant thyroid cancer. J Immunother Cancer. 2020; 8:e000698. https://doi.org/10.1136/jitc-2020-000698. [PubMed].
106. Copland J, Schick K, Gleba J, Huynh T, Miller J, Miller E, Demirer AA, Tapper E, Sakemura R, Siegler E, Cox M, Stewart C, Can I. 324 Sensitizing poorly differentiated thyroid cancers to TSHR-CART cell therapy with MEK inhibitors. J Immunother Cancer; 2022; 10. https://doi.org/10.1136/jitc-2022-SITC2022.0324.
107. Gunda V, Gigliotti B, Ndishabandi D, Ashry T, McCarthy M, Zhou Z, Amin S, Freeman GJ, Alessandrini A, Parangi S. Combinations of BRAF inhibitor and anti-PD-1/PD-L1 antibody improve survival and tumour immunity in an immunocompetent model of orthotopic murine anaplastic thyroid cancer. Br J Cancer. 2018; 119:1223–32. https://doi.org/10.1038/s41416-018-0296-2. [PubMed].
108. Dierks C, Ruf J, Seufert J, Kreissl M, Klein C, Spitzweg C, Kroiss M, Thomusch O, Lorenz K, Zielke A, Miething C. 1646MO Phase II ATLEP trial: final results for lenvatinib/pembrolizumab in metastasized anaplastic and poorly differentiated thyroid carcinoma. Ann Oncol. 2022; 33:S1295. https://doi.org/10.1016/j.annonc.2022.07.1726.