
INTRODUCTION
The etiology of oncogenic mutations in colorectal cancer (CRC) is only explainable in cancers with microsatellite instability (MSI) [1]. MSI is originated by defects in the DNA mismatch repair (MMR) system and is the hallmark of the Hereditary Non-Polyposis Colon Cancer (HNPCC) syndrome [2, 3]. Germ line mutations in MMR genes, combined with other somatic alterations in the remaining allele impair MMR and, as a result, hundreds of thousands of spontaneous DNA replication errors accumulate in the genome in the course of multiple consecutive cell replications. MSI is also manifested in approximately 10–15% and 15–20% of non-hereditary CRC and endometrial cancer (EC) [1–4], predominantly as a result of epigenetic silencing of MLH1 linked to promoter hypermethylation [5, 6].
MSI tumors display a mutator phenotype that raises the tumor cell mutation rate two to three orders of magnitude over that of normal cells [2, 7]. Paradoxically, despite their mutator phenotype, the frequency of mutations in the prototypical cancer genes for CRC, KRAS and TP53, is lower in MSI tumors than in tumors without MSI [1]. This can be explained as the MSI mutator phenotype leads to biallelic mutations in other oncogenic target genes such as TGFRBII and BAX [8, 9].
Activation of ras oncogenes is implicated in human carcinogenesis [10], mainly by promoting cellular proliferation and inhibiting apoptosis [11]. KRAS mutations occur frequently in tumors of the pancreas and the lung, and colorectal adenomas and carcinomas [12, 13]. Point mutations of KRAS also occur in 10% to 40% of EC [14].
The tumor suppressor gene TP53 is a checkpoint regulator that has been named the “guardian of the genome” [15]. The frequency of TP53 somatic point mutations in CRC is estimated to be above 50%. The majority (approximately 80%) are G > A missense transition mutations at CpG dinucleotides that mainly occur in five hotspot codons (175, 245, 248, 273, and 282) [16].
O6-methylguanine-DNA methyltransferase (MGMT) is an ubiquitous DNA repair enzyme that removes mutagenic and cytotoxic adducts from the O6-guanine in DNA, the preferred point of attack by many carcinogens and alkylating chemotherapeutic agents [17]. Alkylating agents are also provided by N-nitrosation of amines derived from protein catabolism that occurs primarily in the acid environment of the stomach [18]. Thus, lack of MGMT function has a mutagenic effect that leads to G to A transition mutations. The human MGMT gene has a normally unmethylated promoter CpG island that, when hypermethylated, correlates with transcriptional silencing [19, 20].
A causal relationship between MGMT silencing and somatic mutations in KRAS and TP53 in CRC was initially proposed based on the association between MGMT hypermethylation and G > A transitions in KRAS and TP53 [21, 22]. This view remains widely accepted [23–25], despite some reports with contradictory results [26–29].
SNS can be transition or transversion mutations. Transitions change one purine for another purine (G→A or A→G), or pyrimidine for another pyrimidine (C→T or T→C). G→A transition is the same as C→T transition (G→A in one strand and C→T in the other). For simplification we refer it as G > A transition. Most SNS at codons 12 (GGT) and 13 (GGC) of KRAS, activate its oncogenic activity. The most frequent KRAS mutations in CRC are G > A transitions at the second G of these triplets, leading to a glycine→aspartic acid substitution: GGT→GAT or GGC→GAC. In TP53, SNS are spread over the gene coding region. Of the G > A transitions, those occurring in CpG dinucleotides CG → TG (GC →AC in the reverse strand) were the most abundant in TP53 (81%).
In this study, we analyzed the association between KRAS and TP53 missense mutations with MGMT methylation in CRC with and without MSI. The relationship between these genetic and epigenetic somatic alterations was also expanded by exome sequencing and by in silico analysis of the publicly available data from The Cancer Genome Atlas (TCGA) consortium, (http://cancergenome.nih.gov/). As MSI CRC is very different from CRC without MSI (MSS) in genotype and phenotype, the inclusion of MSI in CRC analysis is a confounding factor. We then focused on the analysis of MSS cancers for the studies on methylation and mutation.
We also analyzed the methylation of ADAMTS14, a member of the ADAM/ADAMTS gene family that plays a role in cell migration and invasion, recently found altered genetically and epigenetically in carcinogenesis in general and CRC in particular [30–33]. We explored the relationship between ADAMTS14 and MGMT methylation in normal and tumor tissues of CRC patients and correlated these findings with tumor genotype and phenotype, including clinico-histological parameters.
RESULTS
MGMT methylation, MSI, KRAS and TP53 mutations in CRC
In the complete series of 735 CRC 89 (12.1%) were classified as MSI, and 432 were analyzed for single base substitution (SNS) mutations in KRAS and 392 for SNS mutations in TP53. A subset of 261 cases was also analyzed for MGMT methylation. The associations between MSI, KRAS and TP53 mutations with CRC genotype (oncogenic mutations) and phenotype (clinico-pathological features) are summarized in supporting Figures S1–S3. MGMT methylation associated with African Americans ( p = 0.015) and there was a trend for older patients (p < 0.1) in CRCs without MSI (Figure 1). No association was found between methylation and location, stage and grade in tumors without (Figure 1) or with (Figure S4) MSI. All subsequent results describe tumors without MSI unless otherwise specified.
MGMT methylation and type of KRAS and TP53 mutations
In CRC, MGMT methylation associated positively with KRAS mutations (28% vs. 52% p = 0.0006) and negatively with TP53 mutations (47% vs 27%, p = 0.002) (Figure 2 top). MGMT methylation had no significant association with G > A transitions in KRAS (48% vs. 54%, p = 0.66) and TP53 (18% vs. 22% p = 1.0) (Figure 2a). There was no association between MGMT methylation and TP53 G > A mutations in CpG sites (Figure 2a).
Multivariate logistic regression analysis including all the genotypic and phenotypic characteristics of CRCs (Figures 1 and 2a), confirmed the statistically significant association of MGMT methylation with African Americans (OR = 3.11, p = 0.013) and KRAS mutations (OR = 2.62, p = 0.013), and the negative association with TP53 mutations (OR = 0.41, p = 0.022) (Figure S5).
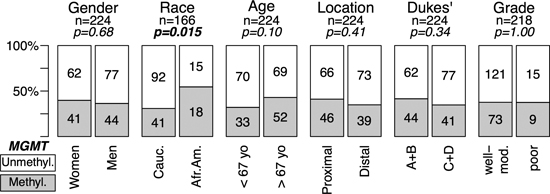
Figure 1: MGMT methylation and genotype/phenotype of CRC without MSI. Data for MSI positive tumors are in Supplementary Figure S4. MGMT-hypermethylated tumors, grey; tumors without hypermethylation, white. Gender, woman vs. men; Race, Caucasian (Cauc.) vs. African American (Afr.Am.); Age, below median (<67 years) vs. above median (>67 years); Location, Proximal (proxim.) includes cecum, ascending and transversal colon vs. Distal, descending, sigmoid and rectum; Invasiveness, Dukes’ A+B vs. Dukes’ C+D; Differentiation, moderate to well differentiated (Mod-Well) vs. poorly differentiated (Poor). MGMT methylation was assessed by MSP (see Materials and Methods). p-values were calculated by univariate Fisher’s tests. In bold, statistically significant values.
To further assess our findings, we analyzed the matching normal and tumor samples from 36 CRCs, using Illumina HM450K arrays. Samples were classified into MGMT-methylated or demethylated based on the values of three probes located within the MGMT 5’ CpG island and enhancer region that exhibits a bimodal distribution in tumor samples (Materials and Methods, Figures S6 and S7). The concordance of the Illumina HM450K results with the previous MSP-based classification was 93%. We therefore used these three probes to explore the available public data from the Cancer Genome Atlas (TCGA) consortium on CRC (COAD plus READ databases, Figure 2b) [34]. There was no correlation between MGMT methylation and KRAS SNS in codons 12 and 13 (p = 0.60), and non-silent TP53 SNS were fewer in tumors with MGMT methylation ( p = 9 × 10−4). There was no significant difference in the relative frequency of G > A transitions in tumors with and without MGMT methylation in either gene.
MGMT methylation and exome mutation spectrum
To study the relationship between MGMT hypermethylation and the overall incidence of somatic G > A transitions, we performed exome sequencing in 18 CRCs and their matched normal tissues, 9 of them with MGMT hypermethylation. To reduce confounding factors, we selected only proximal colon cancers without MSI from Caucasian patients.
The exome sequencing data was used to estimate the mutation spectrum in cancers with and without MGMT methylation. Figure 3a shows that there was no difference in the frequency of G > A transitions regardless of MGMT methylation status. Similar result was derived in our in silico analysis of the TCGA consortium data. Only a weak positive association between MGMT methylation and G > A transitions in non-CpG sites was detected for both the entire series of cancers including the rectum (p = 0.07) and proximal cancers ( p = 0.035) (Figure 3b and 3c). Multivariate regression analysis also revealed that MGMT methylation had no detectable effect in the frequency of SNS, including G > A transitions, in CRC without MSI (Figure S9). Instead, patient age showed a significant association with mutation for all SNS (p = 0.009) or G > A transitions ( p = 1.2 × 10−4), especially those taking place in CpG sites (p = 1.0 × 10−5), but not for non-G > A SNS ( p = 0.665) or G > A transitions in non CpG sites (p = 0.131) (Figure S9).
MGMT methylation and mRNA expression
MGMT methylation assessed by Illumina microarrays in the TCGA CRC samples showed a clear bimodal distribution (Figures 4c and S6). We classified samples into MGMT methylated or MGMT unmethylated by setting the ß-value threshold of 0.2, at the valley of the bimodal distribution of methylation. Notably, the second peak of this distribution was centered at a ß-value 0.5, suggestive of monoallelic methylation (Figure 4c and S6). Similar results were derived from our sample collection (Figure S7). In contrast, the methylation distribution of the MLH1 gene, which is known to undergo transcriptional silencing by promoter methylation in CRC with MSI, showed a methylation peak above ß-value of 0.8, indicating biallelic methylation or monoallelic methylation accompanied by loss of the unmethylated allele (Figure 4d). MGMT expression of the TCGA CRC samples (as assessed by Agilent expression microarrays), also exhibited a bimodal distribution, with 81.3% of the samples with an expression level above 0.36 (relative to the average expression of MGMT in 22 normal colon samples, Figure 4a), used as cut-off for MGMT-silencing because it coincided with the valley separating the two expression peaks.
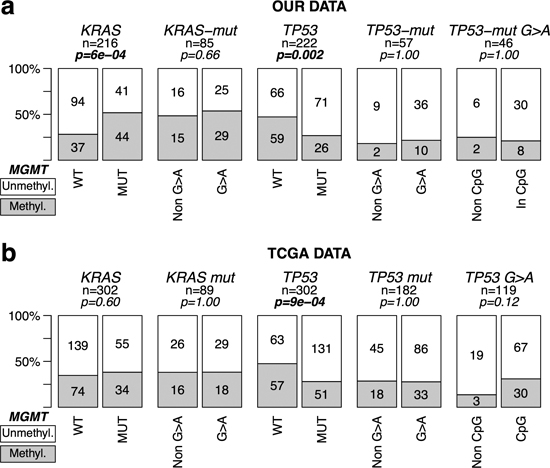
Figure 2: MGMT methylation and mutations in KRAS & TP53 in CRC without MSI. Symbols and codes as in Figure 1. WT; wild type; MUT, mutant. In the upper row, data from our tumor collection. Mutations refer to SNS at codons 12 and 13 of KRAS and in exons 4–9 of TP53. In the lower row, TCGA COAD + READ datasets, excluding tumors classified as hypermutated (MMR defective, MSI). Only non-silent SNS mutations occurring in the whole coding sequence of TP53, and in codons 12 and 13 of KRAS were considered for the analysis.
MGMT methylation associated with lower expression (r = −0.67, p < 2.2 × 10−16, Figure 4b). However, in the majority of MGMT methylated cases mRNA levels were roughly equal to 50% of the levels in the negative cases, again suggesting that epigenetic silencing affected only one allele (Figure 4b). A minority of MGMT methylated cases exhibited lower expression, despite their methylation being also around ß = 0.5 (Figure 4b), suggesting that other mechanism, in addition to promoter methylation, was required for full MGMT transcriptional silencing in these tumors.
MGMT and ADAMTS14 methylation in normal mucosa of CRC
MGMT has also been found methylated in normal colon mucosa, having been associated with an epigenetic field defect for cancerization in CRC [24, 35, 36]. Albeit reduced compared with the tumors, there was a significant level of methylation in normal mucosa from 7% (11/157) of CRC. In a parallel study we also identified significant methylation of ADAMTS14 in normal colon mucosa in 14.6% (23/157) of CRC patients (Figure 5a and 5b). ADAMTS14 methylation in normal mucosa was significantly associated with old age and was more frequent among African-Americans (Figure 5c). These associations remained statistically significant in multivariate logistic regression analysis (Figure 5d), with race being the most significant.
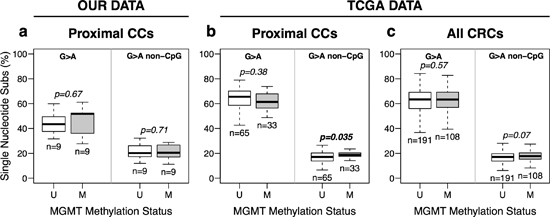
Figure 3: MGMT methylation and somatic G > A transition mutations in CRC without MSI. (a) Data from exome sequencing of eighteen pairs of matching Normal-Tumor samples, 9 with (M) and without (U) MGMT hypermethylation, from our tumor collection. (b and c) data from proximal (b) and all (c) CRCs cancers from the TCGA, excluding hypermutated (MSI) tumors. Box plots represent the median percent of G > A transition mutations vs. all other single base substitutions. p-values were calculated by Student’s t-test.
There was a significant clustering of methylation of MGMT and ADAMTS14 in normal tissue (Figure 6a). We then reclassified the 144 CRCs without MSI for which complete methylation information for the two genes was available into three groups: i) no methylation in either gene, ii) methylation in one or both genes in tumor, but not in normal tissue, and iii) methylation in normal tissue in at least one of these two genes. Methylation in normal tissue occurred preferentially in elder CRC patients (p = 0.001) and African Americans ( p = 1 × 10−5) with CRC (Figure 6b).
DISCUSSION
Accumulation of mutations in KRAS and TP53 cancer genes is critical for CRC pathogenesis. MGMT hypermethylation was reported to associate with KRAS and TP53 G > A transitions, the most frequent single base substitutions in both genes [21, 22]. Thus, critical oncogenic mutations for CRC could be explained by the previous MGMT epigenetic silencing. This report shows the absence of association between MGMT methylation and G > A transition mutations in KRAS and TP53 genes in CRC without MSI. Because this conclusion was validated by the massive TCGA consortium data, the results do not appear to be due to tumor sample variation.
Next, we performed exome sequencing to determine the somatic mutational spectra of 18 CRC with and without MGMT methylation. Again, no association was found between methylation and the proportion of G > A somatic transitions. Identical conclusion was reached by in silico analysis of the TCGA consortium: there was no association between MGMT methylation and G > A transition mutations in KRAS and TP53 or in the whole exome of 386 CRC without MSI. Therefore, epigenetic silencing of MGMT cannot explain the origin of the majority of oncogenic mutations in KRAS or TP53, in CRC without MSI. In support of this conclusion are our unpublished results with endometrial cancer (EC). Although KRAS mutations were identified in 34.1% MSI and 9.2% non-MSI EC (p < 0.0002), none of the 204 EC samples exhibited MGMT methylation (data not shown).
While in MSI CRC missense mutations in KRAS and TP53 (and in any gene) can be accounted for by the strong mutator phenotype displayed by these tumors, in CRC without MSI the origin of these oncogenic mutations remains unclear [14]. While the spontaneous mutation rate may account for mutations in several cancer genes in stem cells [37, 38], it is very unlikely that the same gene accumulates two mutations. Thus, TP53 somatic biallelic mutations in cancer cannot be explained by the spontaneous mutation rate of normal cells [39]. This also applies to all tumor suppressors and mutators as the MMR genes themselves. Ras, however, are oncogenes and in principle a mono-allelic mutation may be sufficient for their activation. Nevertheless, the common occurrence in CRC is that several copies of the mutated allele coexist in tumors with the wild type allele, indicating that the gene acts in a dominant, but dose dependent manner [40–42].
As biallelic missense mutations are virtually absent in CRC without MSI, a plausible hypothesis is that the SNS common for both KRAS and TP53 occur spontaneously (or induced by uncharacterized mutagens) and that subsequent mutations in the other allele typically involve another type of alteration, the occurrence of which is enhanced over the spontaneous rate. Deletions and amplifications of chromosomal segments are the most common, although not unique, mutational events that affect the gene dosage of KRAS and TP53 in CRC. It is uncertain whether these alterations are the consequence of a defect involved in the active generation of chromosomal instability, or just the result of a stepwise augmentation of the probability of incurring into a mitotic error during clonal selection. In the case of KRAS, this may be simply due to the increased mitotic activity presumably caused by the mono-allelic mutation. Whether some TP53 monoallelic missense mutations may also work in a haplo-insufficiency manner increasing the chances of subsequent loss of heterozygosity (LOH) events is also a possibility, especially since the heterogeneity of these mutations [16]. Alternatively, the second event may be influenced by other independent events already present in the tumor cell at that stage of tumor progression.
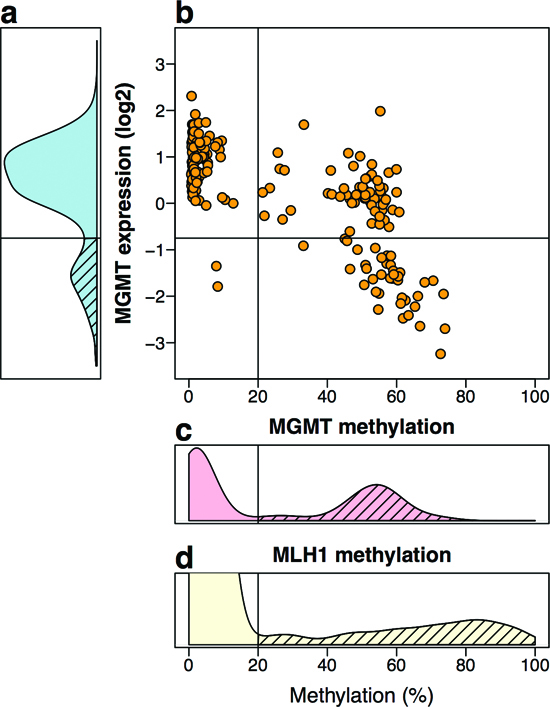
Figure 4: MGMT methylation and expression in silico TCGA data of 222 CRC. (a) Distribution of MGMT expression levels by Agilent two-color expression microarrays, and normalized relative to the average expression level of 22 normal samples. Yellow, orange and red lines indicate the mean expression values of the groups defined in b. (b) Dot-plot of methylation levels vs. expression levels. By cutting at a relative expression of 0.36, and methylation level of 20% (ß-value = 0.2), three groups of tumors are defined: 54% of tumors with no MGMT methylation and average expression level similar to that of the normal tissues (yellow circles), 27.5% of tumors with methylation and a moderate reduction in expression (orange squares), 18.5% of tumors with reduction in expression, most but not all with methylation (red diamonds). (c) Distribution of the average methylation of probes cg12434587, cg12981137 and cg02941816, located in the promoter region of MGMT. In the normal tissues available at the TCGA, these probes never showed methylation above 20%. Of the 222 cases analyzed, 45.05% exhibited an average methylation above 20% (dashed area) with a peak centered around 50%. Association of MGMT methylation with lower expression was very significant (r = −0.67, 95%CI = [−0.73,−0.59], p < 2.2 × 10−16, by Pearson’s product-moment correlation test). (d) Methylation status of hMLH1 promoter (probe cg13846866) is shown for comparison from a comprehensive larger dataset of CRC from the TCGA. About 12% of the tumor samples (62/514) exhibited methylation above 20% (dashed area), with a peak centered around 80%.
The weak association of G > A mutations occurring in non-CpG sites in the cancer cell exome (Figures 3 and S9) shows that MGMT deficiency may indeed contribute to the accumulation of some of these mutations. However, the difference in mutation frequency is very small, implying that many of the mutations have been already generated prior to the epigenetic silencing of the MGMT gene. In fact, the principal SNS in CRC are G > A transitions in CpG sites that accumulate in an age dependent manner (Figure S9), which is consistent with the hypothesis that most SNS take place throughout the entire life of the patient and prior to tumorigenesis, mainly by spontaneous deamination of methylated cytosines [43, 44]. Thus, mutations occurring after MGMT silencing may be a minority compared with the previous mutations accumulated during the life of the tumor precursor cells. The vast majority of these “passenger” mutations do not contribute to tumorigenesis, but they are exposed by the clonal expansion of the carrying precursor cells.
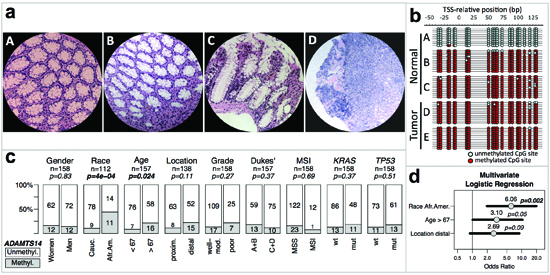
Figure 5: ADAMTS14 methylation and genotype/phenotype of CRC without MSI. (a) Micrographs at 200X of hematoxylin-eosin stained representative sections of normal colonic tissue from a patient without (A) and a patient with (B and C) ADAMTS14 hypermethylation, and tumor tissue from the latter patient (D) (b) Bisulfite sequencing profiles of the ADAMTS14 5’ region from –50 to 130 bp, relative to the transcriptional start site (TSS) from DNA isolated from normal and tumor microdissected areas equivalent to the areas in micrographs A–D, and another tumor area from the same patient (E) Red and blue circles represent methylated and unmethylated CpG sites, respectively. (c) Associations of ADAMTS14 hypermethylation in normal mucosa of CRC patients with their phenotype/genotype. Symbols and codes as in Figure 1. (d) Forest plot of multivariate logistic regression analysis of the association of ADAMTS14 hypermethylation with race, age and location. The horizontal bars represent the 95% confidence intervals with the centered circles representing the estimated odds ratio, with values indicated above. p-values of the multivariate analysis are also indicated for every parameter included in the model.
Further analysis of exome deep sequencing data may shed light on this issue. Non-clonal mutations (after transformation) would be distinguishable from clonal mutations by the frequency recorded in their sequence reads. The hypothesis can be thus tested by a comparative analysis in tumors with and without MGMT methylation of the high and low relative frequency of sequencing reads of G > A vs. non-G > A SNS, and between G > A in CpG vs. non-CpG sites. For instance, in tumors with MGMT methylation, the frequency of non-clonal G > A transitions (with low relative number of mutant vs. wild type sequencing reads) in non CpG sequences, would be higher than that from tumors without methylation.
The lack of significant correlation between methylation and mutation also suggests that there might be no significant contribution by alkylating agents in the large intestine to generate these mutations. However, even if the mutator role of MGMT hypermethylation appears modest, it may have a relevant prognostic value. Recent clinical studies showed that methylation of MGMT is a useful predictor of the responsiveness of tumors to alkylating agents in gliomas [45, 46], and is associated with good survival in patients treated with multidrug regimens [47]. It also predicts the response to the alkylating drug dacarbazine in metastatic CRC [48, 46]. In this context, the data on MGMT methylation and expression reveals a complex pattern of regulation and argues against a complete silencing in a significant proportion of methylated cases. This may have potential impact on current treatment regimens of CRC. We found that methylation of MGMT seems to be predominantly monoallelic in CRCs without MSI, with just a 50% reduction in transcriptional levels in the majority of the cases. It would be interesting to evaluate in future studies the response to these alkylating drugs in patients stratified in three groups (Figure 4), distinguishing between no silencing and mono-allelic or biallelic silencing.
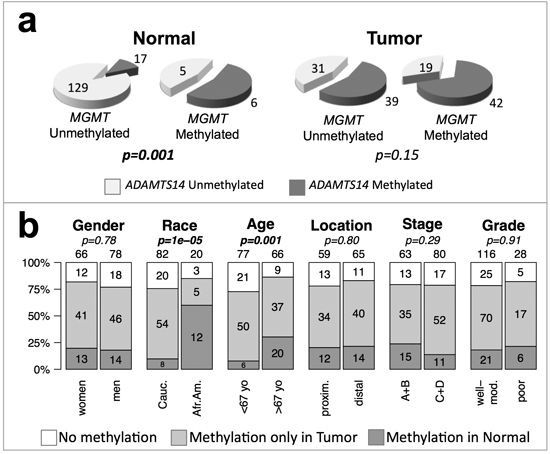
Figure 6: ADAMST14 and MGMT methylation and genotype/phenotype of CRC without MSI. (a) Clustering of MGMT and ADAMTS14 hypermethylation in the normal mucosa (left) and tumor tissue (right) of CRC patients. p-values were calculated by Fisher’s exact test. (b) Patients were divided into three groups according to the methylation status of ADAMTS14 and MGMT: without methylation in either gene (white), with methylation in one or both genes in the tumor sample but not in the non-tumoral mucosa (light grey), or with methylation of at least one of these genes in the non-tumoral mucosa (dark grey). Symbols and codes as in Figure 1. p-values were calculated by χ2 test.
In contrast with the clear asymmetries observed for MSI, KRAS and TP53 mutations, the only significant association found for MGMT methylation was with African-American CRC patients that tended to be older (Figure 1 and Figures S1–S3). In an independent study, we also observed a significant association of hypermethylation of ADAMTS14 in the normal colon mucosa mainly in elder African-Americans with CRC (Figure 5). The tendency of ADAMTS14 hypermethylation in normal mucosa to occur in the distal colon distinguishes this phenomenon from the generalized hypermethylation that is more prominent in tumors of the proximal colon [49–51]. Clustering of methylation of MGMT and ADAMTS14 (Figure 6a) in normal mucosa supports the concept of a field for cancerization in some individuals at high risk for CRC that involves the aberrant methylation of several loci, including, but not restricted to MGMT and ADAMTS14 [24, 36]. We do not suggest, however, that these genes play an active role in this field defect. Probably they are just markers of a more generalized epigenetic dysfunction that underlies an apparent high risk for CRC, preferentially among elderly African-Americans (Figure 6b).
We cannot distinguish at this point between ethnic or environmental causes for this association, and this issue remains the subject for further studies. We also do not know whether this phenomenon is restricted to those individuals already with CRC or if the alteration occurs, as we suspect, in pre-symptomatic individuals. The geography of this field defect also needs to be surveyed to discriminate between systemic or focalized defect. This in turn may have practical consequences for diagnosis of the predictive lesions. Although their detection may be a challenging task, epigenetic alterations can be detected in liquid biopsies. In conclusion, our findings provide mechanistic clues for the ethnic disparities known to affect colon cancer [52], and a rationale for novel CRC predictive tests.
PATIENTS AND METHODS
Study participants and samples
Unselected primary CRC and corresponding non-tumoral tissues were obtained through the Cooperative Human Tissue Network (CHTN) from 751 patients who underwent curative surgery in various hospitals of Philadelphia, Tennessee, Ohio and Alabama between 1985 and 2004. Only patients with adenocarcinomas were included in the study. Clinical information included age at diagnosis, gender, race, tumor location, and surgical stage; and pathology data included grade of differentiation. Tumor staging was based on Duke’s classification. Sanford-Burnham Institutional Review Board approval was obtained for this work. We also retrospectively studied 204 consecutive patients with EC (FIGO stage I-III), diagnosed and treated at the Department of Obstetrics and Gynecology of the Gran Canaria’s Hospital Universitario Materno Infantil (Canary Islands, Spain) between 1990 and 1999. The ethics committees from Gran Canaria’s Hospital Universitario Materno Infantil approved the research protocol, which was in compliance with national legislation and performed according to the ethical guidelines of the Declaration of Helsinki [53]. After surgical resection, samples were rapidly frozen in liquid nitrogen and stored at −80ºC until further use. DNA was isolated by standard procedures involving mechanical disruption of tissue, SDS lysis and proteinase K digestion, phenol-chloroform extraction, and ethanol precipitation [54].
MSI analysis, KRAS and TP53 mutation detection
MSI was determined in 751 CRC as previously described. [55] Codons 12/13 of the KRAS oncogene in 432 CRC, and exons 4 to 8 of the TP53 suppressor gene in 392 CRC were analyzed by single stranded conformation polymorphism (SSCP) and DNA sequencing. PCR primers and conditions for the amplification and analysis of these genes are detailed in supporting materials SM1.
Methylation analysis of MGMT and ADAMTS14
The methylation status of MGMT was determined in 273 CRC samples by methylation-specific single PCR (MSP) [56] of a specific sequence region where methylation invariably correlates with lack of MGMT expression [19, 20]. PCR primers and conditions are described in supporting materials SM2. The promoter region of ADAMTS14 was analyzed by bisulfite sequencing and combined bisulfite and restriction analysis (COBRA) in 158 CRC patients (supporting materials SM3) [57, 58].
Exome sequencing
We sequenced 36 exomes corresponding to tumor tissue and corresponding normal mucosa from 18 patients with proximal colon cancer (9 with and 9 without MGMT methylation), using Illumina TruSeq Enrichment protocol and HiScan-SQ platform. Somatic mutations were determined by comparing the tumor and normal exome from the same individual. Detailed information about the library preparation, sequencing protocol and bioinformatic tools is provided in supporting information SM4. After exome sequencing, over 100 Single nucleotide variants (SNVs) and somatic mutations were manually selected for validation by PCR and sequencing (supporting materials SM5).
In silico analysis of TCGA data
We combined the data from the COAD (colon adenocarcinoma) and READ (rectum adenocarcinoma) databases from the TCGA (https://tcga-data.nci.nih.gov/) [34]. In total, we obtained clinical information from 592 patients, methylation data from 583 primary CRCs, and exome mutational data from 386 CRCs (supporting dataset S1). MGMT methylation status was determined by Illumina HM27/HM450 probes cg12434587, cg12981137 and cg02941816, that interrogate three CpG sites located –239 bp, 128 bp and 248 bp from the MGMT transcriptional start site, respectively, within regions that associate with transcription of MGMT. [20] A detailed explanation of the TCGA datasets we employed in this work and the classification criteria for MSI and MGMT status are provided in supporting materials SM6.
Statistical analysis
Statistical analyses were performed using R environment [59]. Associations in categorical data were analyzed by two-sided Fisher’s exact tests (for 2 × 2 contingency tables) or Chi-squared test with Yates’ correction (for larger contingency tables). Multivariate analyses were performed by logistic regression, with stepwise reduction of non-significant factors and interactions. Forest plots represent the odds ratios (OR) and the 95% confidence intervals for each parameter included in the model. In box plots, the boxes represent the interquantile range (IQR) with a horizontal bar indicating the median. Dispersion bars indicate the highest and lowest datum within 1.5 × IQR of the lower quartile and 1.5 × IQR of the highest quartile. Statistical significance threshold was set at p ≤ 0.05.
ACKNOWLEDGEMENTS
This work was supported by National Institutes of Health Grant R37CA63585, and by Spanish Ministry of Health Grants FIS PI09/2444 and FIS PI12/00511 (to MP) and National Institutes of Health Grants RO1CA83017 and RO1CA157328 (to AYS).
Authors’ contributions
Study concept and design: SA, YD, KY, CB, JCDC, AVC, AYS, MP
KRAS, TP53 and MGMT analyses: YD, KY, TD, RSM, CB, JCDC, MP
ADAMTS14 analysis: SA, SH, AM, MP
Statistical analyses: SA, MP
Exome sequencing and data analysis: SA, AVC, AYS, MP
TCGA data analysis: SA, MP
Manuscript preparation: SA, YD, KY, RSM, CB, JDC, AYS, MP
Obtained funding: JCDC, AYS, MP
Study supervision: MP
REFERENCES
1. Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature. 1993; 363:558–561.
2. Boland CR. Roles of the DNA mismatch repair genes in colorectal tumorigenesis. Int J Cancer. 1996; 69:47–49.
3. Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996; 87:159–170.
4. Black D, Soslow RA, Levine DA, Tornos C, Chen SC, Hummer AJ, Bogomolniy F, Olvera N, Barakat RR, Boyd J. Clinicopathologic significance of defective DNA mismatch repair in endometrial carcinoma. J Clin Oncol. 2006; 24:1745–1753.
5. Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, Jessup JM, Kolodner R. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997; 57:808–811.
6. Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002; 3:415–428.
7. Perucho M. Cancer of the microsatellite mutator phenotype. Biological chemistry. 1996; 377:675–684.
8. Markowitz S, Wang J, Myeroff L, Parsons R, Sun L, Lutterbaugh J, Fan RS, Zborowska E, Kinzler KW, Vogelstein B, et al. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science. 1995; 268:1336–1338.
9. Rampino N, Yamamoto H, Ionov Y, Li Y, Sawai H, Reed JC, Perucho M. Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science. 1997; 275:967–969.
10. Barbacid M. ras oncogenes: their role in neoplasia. Eur J Clin Invest. 1990; 20:225–235.
11. Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003; 3:459–465.
12. Bos JL, Fearon ER, Hamilton SR, Verlaan-de Vries M, van Boom JH, van der Eb AJ, Vogelstein B. Prevalence of ras gene mutations in human colorectal cancers. Nature. 1987; 327:293–297.
13. Forrester K, Almoguera C, Han K, Grizzle WE, Perucho M. Detection of high incidence of K-ras oncogenes during human colon tumorigenesis. Nature. 1987; 327:298–303.
14. Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012; 72:2457–2467.
15. Lane DP. Cancer. p53, guardian of the genome. Nature. 1992; 358:15–16.
16. Russo A, Bazan V, Iacopetta B, Kerr D, Soussi T, Gebbia N. The TP53 colorectal cancer international collaborative study on the prognostic and predictive significance of p53 mutation: influence of tumor site, type of mutation, and adjuvant treatment. J Clin Oncol. 2005; 23:7518–7528.
17. Gerson SL. MGMT: its role in cancer aetiology and cancer therapeutics. Nat Rev Cancer. 2004; 4:296–307.
18. Shuker DE, Margison GP. Nitrosated glycine derivatives as a potential source of O6-methylguanine in DNA. Cancer Res. 1997; 57:366–369.
19. Esteller M, Hamilton SR, Burger PC, Baylin SB, Herman JG. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res. 1999; 59:793–797.
20. Nakagawachi T, Soejima H, Urano T, Zhao W, Higashimoto K, Satoh Y, Matsukura S, Kudo S, Kitajima Y, Harada H, Furukawa K, Matsuzaki H, et al. Silencing effect of CpG island hypermethylation and histone modifications on O6-methylguanine-DNA methyltransferase (MGMT) gene expression in human cancer. Oncogene. 2003; 22:8835–8844.
21. Esteller M, Toyota M, Sanchez-Cespedes M, Capella G, Peinado MA, Watkins DN, Issa JP, Sidransky D, Baylin SB, Herman JG. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is associated with G to A mutations in K-ras in colorectal tumorigenesis. Cancer Res. 2000; 60:2368–2371.
22. Esteller M, Risques RA, Toyota M, Capella G, Moreno V, Peinado MA, Baylin SB, Herman JG. Promoter hypermethylation of the DNA repair gene O(6)-methylguanine-DNA methyltransferase is associated with the presence of G:C to A:T transition mutations in p53 in human colorectal tumorigenesis. Cancer Res. 2001; 61:4689–4692.
23. Qi J, Zhu YQ, Huang MF, Yang D. Hypermethylation of CpG island in O6-methylguanine-DNA methyltransferase gene was associated with K-ras G to A mutation in colorectal tumor. World J Gastroenterol. 2005; 11:2022–2025.
24. Shen L, Kondo Y, Rosner GL, Xiao L, Hernandez NS, Vilaythong J, Houlihan PS, Krouse RS, Prasad AR, Einspahr JG, Buckmeier J, Alberts DS, et al. MGMT promoter methylation and field defect in sporadic colorectal cancer. J Natl Cancer Inst. 2005; 97:1330–1338.
25. Shen H, Laird PW. Interplay between the cancer genome and epigenome. Cell. 2013; 153:38–55.
26. Lind GE, Thorstensen L, Lovig T, Meling GI, Hamelin R, Rognum TO, Esteller M, Lothe RA. A CpG island hypermethylation profile of primary colorectal carcinomas and colon cancer cell lines. Mol Cancer. 2004; 3:28.
27. Halford S, Rowan A, Sawyer E, Talbot I, Tomlinson I. O(6)-methylguanine methyltransferase in colorectal cancers: detection of mutations, loss of expression, and weak association with G:C>A:T transitions. Gut. 2005; 54:797–802.
28. Suehiro Y, Wong CW, Chirieac LR, Kondo Y, Shen L, Webb CR, Chan YW, Chan AS, Chan TL, Wu TT, Rashid A, Hamanaka Y, et al. Epigenetic-genetic interactions in the APC/WNT, RAS/RAF, and P53 pathways in colorectal carcinoma. Clin Cancer Res. 2008; 14:2560–2569.
29. Minoo P. Toward a Molecular Classification of Colorectal Cancer: The Role of MGMT. Frontiers in oncology. 2013; 3:266.
30. Cal S, Obaya AJ, Llamazares M, Garabaya C, Quesada V, Lopez-Otin C. Cloning, expression analysis, and structural characterization of seven novel human ADAMTSs, a family of metalloproteinases with disintegrin and thrombospondin-1 domains. Gene. 2002; 283:49–62.
31. Wagstaff L, Kelwick R, Decock J, Edwards DR. The roles of ADAMTS metalloproteinases in tumorigenesis and metastasis. Front Biosci (Landmark Ed). 2011; 16:1861–1872.
32. Przemyslaw L, Boguslaw HA, Elzbieta S, Malgorzata SM. ADAM, and ADAMTS family proteins and their role in the colorectal cancer etiopathogenesis. BMB Rep. 2013; 46:139–150.
33. Fontanil T, Rua S, Llamazares M, Moncada-Pazos A, Quiros PM, Garcia-Suarez O, Vega JA, Sasaki T, Mohamedi Y, Esteban MM, Obaya AJ, Cal S. Interaction between the ADAMTS-12 metalloprotease and fibulin-2 induces tumor-suppressive effects in breast cancer cells. Oncotarget. 2014; 5:1253–1264.
34. The Cancer Genome Atlas Research NetworkComprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487:330–337.
35. Paun BC, Kukuruga D, Jin Z, Mori Y, Cheng Y, Duncan M, Stass SA, Montgomery E, Hutcheon D, Meltzer SJ. Relation between normal rectal methylation, smoking status, and the presence or absence of colorectal adenomas. Cancer. 2010; 116:4495–4501.
36. Svrcek M, Buhard O, Colas C, Coulet F, Dumont S, Massaoudi I, Lamri A, Hamelin R, Cosnes J, Oliveira C, Seruca R, Gaub MP, et al. Methylation tolerance due to an O6-methylguanine DNA methyltransferase (MGMT) field defect in the colonic mucosa: an initiating step in the development of mismatch repair-deficient colorectal cancers. Gut. 2010; 59:1516–1526.
37. Jackson AL, Loeb LA. The mutation rate and cancer. Genetics. 1998; 148:1483–1490.
38. Loeb LA, Bielas JH, Beckman RA. Cancers exhibit a mutator phenotype: clinical implications. Cancer Res. 2008; 68:3551–3557;discussion 3557.
39. Loeb LA. Human cancers express mutator phenotypes: origin, consequences and targeting. Nat Rev Cancer. 2011; 11:450–457.
40. Winter E, Yamamoto F, Almoguera C, Perucho M. A method to detect and characterize point mutations in transcribed genes: amplification and overexpression of the mutant c-Ki-ras allele in human tumor cells. Proc Natl Acad Sci U S A. 1985; 82:7575–7579.
41. Winter E, Perucho M. Oncogene amplification during tumorigenesis of established rat fibroblasts reversibly transformed by activated human ras oncogenes. Mol Cell Biol. 1986; 6:2562–2570.
42. Finney RE, Bishop JM. Predisposition to neoplastic transformation caused by gene replacement of H-ras1. Science. 1993; 260:1524–1527.
43. Rideout WM 3rd, Coetzee GA, Olumi AF, Jones PA. 5-Methylcytosine as an endogenous mutagen in the human LDL receptor and p53 genes. Science. 1990; 249:1288–1290.
44. Shen JC, Rideout WM 3rd, Jones PA. The rate of hydrolytic deamination of 5-methylcytosine in double-stranded DNA. Nucleic Acids Res. 1994; 22:972–976.
45. Kast RE, Boockvar JA, Bruning A, Cappello F, Chang WW, Cvek B, Dou QP, Duenas-Gonzalez A, Efferth T, Focosi D, Ghaffari SH, Karpel-Massler G, et al. A conceptually new treatment approach for relapsed glioblastoma: coordinated undermining of survival paths with nine repurposed drugs (CUSP9) by the International Initiative for Accelerated Improvement of Glioblastoma Care. Oncotarget. 2013; 4:502–530.
46. Perry C, Agarwal D, Abdel-Fatah TM, Lourdusamy A, Grundy R, Auer DT, Walker D, Lakhani R, Scott IS, Chan S, Ball G, Madhusudan S. Dissecting DNA repair in adult high grade gliomas for patient stratification in the post-genomic era. Oncotarget. 2014; 5:5764–5781.
47. Miyamoto K, Ushijima T. Diagnostic and therapeutic applications of epigenetics. Jpn J Clin Oncol. 2005; 35:293–301.
48. Amatu A, Sartore-Bianchi A, Moutinho C, Belotti A, Bencardino K, Chirico G, Cassingena A, Rusconi F, Esposito A, Nichelatti M, Esteller M, Siena S. Promoter CpG island hypermethylation of the DNA repair enzyme MGMT predicts clinical response to dacarbazine in a phase II study for metastatic colorectal cancer. Clin Cancer Res. 2013; 19:2265–2272.
49. Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A. 1999; 96:8681–8686.
50. Yamashita K, Dai T, Dai Y, Yamamoto F, Perucho M. Genetics supersedes epigenetics in colon cancer phenotype. Cancer Cell. 2003; 4:121–131.
51. Suzuki K, Suzuki I, Leodolter A, Alonso S, Horiuchi S, Yamashita K, Perucho M. Global DNA demethylation in gastrointestinal cancer is age dependent and precedes genomic damage. Cancer Cell. 2006; 9:199–207.
52. Tammana VS, Laiyemo AO. Colorectal cancer disparities: issues, controversies and solutions. World J Gastroenterol. 2014; 20:869–876.
53. World Medical AssociationWorld Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013; 310:2191–2194.
54. Maniatis T, Fritsch EF, Sambrook J. Molecular cloning: a laboratory manual. Cold Spring Harbor, Laboratory Cold Spring Harbor, N.Y: 1982.
55. Yamamoto H, Perez-Piteira J, Yoshida T, Terada M, Itoh F, Imai K, Perucho M. Gastric cancers of the microsatellite mutator phenotype display characteristic genetic and clinical features. Gastroenterology. 1999; 116:1348–1357.
56. Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific, PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A. 1996; 93:9821–9826.
57. Clark SJ, Harrison J, Paul CL, Frommer M. High sensitivity mapping of methylated cytosines. Nucleic Acids Res. 1994; 22:2990–2997.
58. Xiong Z, Laird PW. COBRA: a sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 1997; 25:2532–2534.
59. R Development Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing Vienna, Austria: 2009.