
INTRODUCTION
In 1988, the phosphatidylinositol kinase that copurified with tyrosine kinase receptors was found to phosphorylate phosphatidylinositol lipids at the 3’ hydroxyl position [1, 2]. This enzyme, class I phosphatidylinositol 3’ kinase (PI3K), was subsequently found to be responsible for converting phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol 3,4,5-trisphosphate (PIP3), and has been implicated in biological processes from insulin-mediated regulation of glucose uptake and metabolism to transformation and even metastatic dissemination of tumor cells [1]. Class IA PI3Ks are composed of one regulatory and one catalytic subunit. The most frequently expressed and commonly studied regulatory subunit is p85α. This binds to the p110α catalytic subunit to control its stability and activity [3]. PI3K signaling is stimulated in response to activation of many growth factor receptors, most potently by the insulin receptor tyrosine kinase (InsR) or related insulin-like growth factor 1 receptor (IGF-1R)(Figure 1). In either case, receptor activation leads to tyrosine phosphorylation of a large adaptor protein from the insulin receptor substrate family (IRS1, 2, 3 or 4) [4]. The IRS proteins contain a number of YxxM motifs that, when phosphorylated on tyrosine (Y), form high affinity binding sites for certain SH2 domains such as those found within the p85 regulatory subunit [5]. Recruitment of the p85;p110 PI3K complex to tyrosine-phosphorylated IRS overrides the inhibitor influence of p85 on its catalytic partner [3, 5, 6]. Binding of GTP-loaded Ras to p110 also increases kinase activity [7]. These effects synergize, and the resulting activated PI3K converts PIP2 into PIP3 [7, 8]. PIP3 subsequently recruits, and in some cases activates, a series of signaling proteins, most of which contain PIP3-binding pleckstrin homology (PH) domains. Best studied among these PIP3 targets are the Akt (1, 2 3)/PKB (α, β or γ) AGC-family serine/threonine kinases [9-11], as well as PDK1 which phosphorylates Akt at threonine 308 (Akt1), thereby activating it with respect to a number of substrates including PRAS40 and TSC2. Phosphorylation of PRAS40 by Akt induces sequestration of p-PRAS40 by 14-3-3 proteins, which prevents it from inhibiting the mTOR, Raptor, mLST8 and Deptor-containing TORC1 serine/threonine kinase complex [12-14]. Similarly, Akt-mediated phosphorylation of TSC2 leads to suppression of the TSC1/TSC2 Rheb GAP activity with subsequent accumulation of GTP on the Rheb small GTPase [15]. Rheb-GTP activates TORC1. Thus, phosphorylation of PRAS40 and TSC2 lead to activation of TORC1, which blocks autophagy while increasing cap-dependent protein translation, glucose uptake, glycolysis, activation of the pentose phosphate pathway as well as fatty acid and sterol synthesis [16-20]. TORC1 is also regulated by Rag-family GTPases that respond to amino acid levels, AMP kinase that is regulated by the AMP:ATP ratio as a readout of cellular energy levels, as well as by the Rac GTPase that functions downstream of growth factor signaling [21]. Full activation of Akt also requires phosphorylation at serine 473 by TORC2, a TORC1-related complex containing mTOR, Rictor, mLST8, Deptor, mSIN and Proctor [22]. TORC2, or a related Rictor-containing complex, also contains integrin linked kinase (ILK) [23-26]. ILK is a kinase/adaptor protein that binds to β1-integrins as well as to PIP3, and is required for recruitment of caveolae to the plasma membrane [23, 25, 27, 28]. AktpT308/pS473 phosphorylates many signaling proteins, including GSK3 serine/threonine kinases and Hdm2 E3 ubiquitin ligases, inhibiting the former and activating the latter [10, 29]. Akt also phosphorylates FOXO1a/3a transcription factors, which causes FOXO-14-3-3 complex formation and nuclear exclusion, thereby blocking the ability of FOXO proteins to activate a pro-apoptotic transcriptional program [22, 30]. Indeed, Akt regulates survival on the level of transcription, through FOXO, and also through phosphorylation of cytoplasmic proteins including Bad [10].
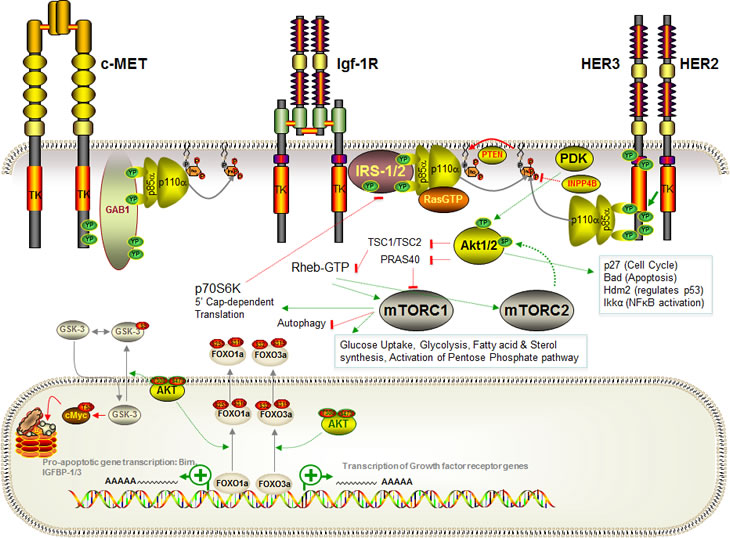
Figure 1: Schematic diagram of the PI3K pathway and its activation in human breast cancer. The pathway can be activated at multiple levels including at the level of InsR/Igf-1R, c-MET or HEH2/HER3 receptors, at the level of adaptor proteins, PI3K regulatory or catalytic subunits, at the level of downstream Akt kinases or through deletion/inactivation of inhibitors of the pathway. Note, for simplicity, alternative receptor-activated signaling pathways as well as the full spectrum of PIP3 targets and Akt and TORC targets are not shown.
Both constitutive and inducible negative regulators act to antagonize the PI3K pathway, maintaining the system in an off state under resting conditions and returning it to this state once stimulation is relaxed (Figure 1). These include ligand-sequestering proteins as well as tyrosine phosphatases and endocytic regulators that function to shut down receptor signaling. In addition, PIP3 levels are held in check by phosphatases that remove the 3’ phosphate, the 4’ phosphate or the 5’ phosphate. The most direct negative regulator of this pathway is PTEN, a phosphatidylinositol 3’ phosphatase and limited specificity protein phosphatase [31-35]. PTEN was discovered as a tumor suppressor that, when deleted, leads to elevated levels of PIP3. Interestingly, PTEN also functions in the nucleus as a cell cycle inhibitor via its ability to positively regulate the anaphase promoting complex (APC-CDH1) [36, 37]. Of note, type II phosphatidylinositol 4’ phosphatase also functions to limit signaling though PI3K pathway targets and is deleted in some tumors (see below) [38]. Other inhibitors of this pathway include PHLDA3, a p53-inducible PH domain protein that sequesters PIP3 and blocks activation of Akt [39], as well as the PH-domain containing PHLPP serine/threonine phosphatases that dephosphorylate Akt at serine 473 [40]. In addition to PRAS40 and TSC2, which limit TORC1 activation as noted above, the Dep and PDZ domain protein, Deptor, is a potent inhibitor of TORC1 and TORC2 [40, 41]. Interestingly, Deptor can enhance the TORC2/TORC1 activity ratio in cells [41]. Finally, several PI3K pathway kinases function as feedback inhibitors to shut down signaling. For example, activated Akt can inhibit FOXO-mediated expression of growth factor receptors including InsR, IGF-1R and HER3 [30, 42]. The TORC1 target, p70S6 Kinase (S6K1) can phosphorylate and target IRS proteins for degradation, and thereby choke off further insulin-mediated activation of PI3K [4, 43].
THE PI3K PATHWAY IS ONCOGENIC
The PI3K pathway has been linked to growth control and transformation in many tissues [19]. Indeed, genes coding for most of the proteins described above are oncogenes or tumor suppressor genes, depending on whether they function to activate signaling through the PI3K pathway or act to inhibit it. The gene coding for p110α, PIK3CA, was identified as a viral oncogene in Avian Sarcoma Virus 16 [44, 45]. This discovery presaged identification of PIK3CA mutations in many human tumors, most prominently in tumors of the breast, colon, endometrium and thyroid gland [11, 45-47]. Mutations in this gene map to two hotspots, one coding for a centrally located helical domain, typically E542K or E545K, and one in the C-terminal kinase domain, most commonly H1047R [46]. Surprisingly, while helical domain and kinase domain mutations both increase the catalytic activity of p110α, they show differing requirements for full activation in vivo. Specifically, helical domain mutants still depend on Ras-GTP for activation, but not on p85. In contrast, full activation of the H1047R kinase domain mutant is Ras-independent, but dependent on p85-phosphotyrosine interaction [48]. Indeed, helical and kinase domain mutations cooperate when present in the same cDNA, generating an allele capable of activating Akt/PKB to a level 1000-fold higher than observed downstream of either single mutant [48]. These mutations are not commonly seen together in the same tumor. However, PIK3CA mutations do occur with mutations that activate tyrosine kinases, activate Ras or inactivate PTEN [49]. Thus, greatly enhanced PI3K signaling may only be achieved through cooperating oncogenic mutations that override negative regulation of this biologically powerful pathway. In this regard, p53 and the PI3K pathway intersect at multiple levels. For example, Akt activates the Hdm2 E3 ligase that targets p53 for destruction [50, 51], and conversely, p53 induces expression of PI3K pathway inhibitors, PTEN and PHLDA3 [39, 52]. These interactions may well explain the coincidence of mutations that affect both pathways in many tumors (see below).
MUTATIONS IN PI3K PATHWAY IN HUMAN BREAST CANCER
With the advent of tumor re-sequencing, commonly mutated oncogenes, tumor suppressor genes and defective signaling pathways involved in many tumor types have been identified. This analysis yielded a somewhat disappointingly complex picture for breast cancer, where a large number of mutations have been identified, each in a small percentage of tumors [53]. There were, however, two genes mutated in a large fraction of breast tumors: TP53 and PIK3CA [53]. For example, mutant alleles of PIK3CA were identified in approximately 30% of breast tumors [46, 54-56]. The specific alleles found include both helical and kinase domain mutants, each occurring with approximately the same frequency. In some cases, PIK3CA gene amplification was also noted [57]. This was more common in tumors with helical domain mutant alleles [58].
The PI3K pathway is also activated in breast cancer through copy number changes and/or mutations or deletions in several other genes [59-63] (Figure 1). For example, the gene coding for HER2/Neu is amplified and frequently associated with high-level expression of HER3, a pseudokinase receptor, HER2-binding partner and substrate with multiple YxxM sites for recruitment of p85 [64]. Some breast carcinomas show gene amplification at the MET/CAVEOLIN gene locus [65-68]. This results in elevated tyrosine kinase signaling from MET through to Gab adaptor proteins [69, 70] and potentially to HER3 [71], both of which bind p85 when phosphorylated [64, 69, 70]. Caveolin proteins enhance InsR and IGF-1R signaling [72-74]. Indeed, the InsR and IGF-1R genes are amplified in some breast tumors [75, 76]. Genes coding for cytoplasmic adaptor proteins like IRS-4 as well as Gab1 and Gab2 are mutated or amplified in a small percentage of breast cancers [53, 77-79]. PIK3R1, the gene coding for p85α, is also mutated in some cases [53, 78]. Downstream of PIK3CA, gene amplifications occur in PDPK1, the gene coding for PDK1 [80]. Also, gain-of-function, activating mutations were found in the PH domain of AKT1 (AKT1E17K) [81, 82].
Mutations that disrupt negative regulators of the PI3K pathway have also been detected in breast cancer. For example, the gene coding for PTPN12/PTP-Pest, a non-receptor tyrosine phosphatase, is commonly disrupted, leading to enhanced tyrosine phosphorylation of multiple growth factor receptors, with resulting downstream PI3K pathway activation [83]. Heterozygous loss-of-function germline mutations in PTEN cause PTEN hamartoma tumor syndromes (PHTS) including Cowden’s syndrome that is associated with a high incidence of breast cancer [84]. Accordingly, approximately 30% of sporadic breast tumors show PTEN inactivation, either through mutation or epigenetic suppression [31, 32, 84-88]. miR-21 has been shown to suppress PTEN gene expression in response to IL6/Stat3 signaling in many breast tumors [89-92]. HER2-mediated activation of the Src tyrosine kinase causes phosphorylation of PTEN and its dissociation from the plasma membrane, thereby enhancing PI3K pathway signaling [93, 94]. Finally, as noted above, loss-of-function mutations in type II phosphatidylinositol 4’ phosphatase (INPP4B) also occur in breast cancer [38, 95]. As more breast cancer genomes are sequenced, it is not unreasonable to expect evidence for each and every PI3K pathway regulatory gene to be implicated in a subset of breast tumors.
THE PI3K PATHWAY AND BREAST CANCER SUBTYPES
The diagnosis of breast cancer describes a collection of diseases. The distinction between hormone receptor positive and negative forms dates back many years, and pathologists have long noted a wide range of histological and clinical features in breast cancer. However, with advances in transcriptional profiling, a relationship between pathological subtype and what is now called molecular subtype has emerged [96]. Equally exciting is the realization that less common mutations in breast cancer as a whole can be quite common when individual breast cancer subtypes are considered [97]. The major molecular subtypes are: luminal A and B, HER2+, basal and claudin-low. Luminal A and B are both estrogen receptor (ERα) positive subtypes, whereas basal and claudin-low are triple negative tumors (ERα-negative, progesterone receptor negative and HER2 negative) [98].
PIK3CA mutations are found in tumors from most subtypes, which explains why this gene scored as one of the two most commonly mutated genes in breast cancer [53-56]. For example, 35% of estrogen receptor (ERα) positive tumors, 23% of HER2/Neu positive tumors and 8% of basal tumors have PIK3CA mutations [99]. PIK3CA mutations either do not occur, or occur at a very low frequency, in claudin-low breast cancer [99]. In addition to the major subtypes, there are a number of rare pathological variants of breast cancer that are not represented in most studies. Indeed, metaplastic breast cancer, a relatively rare form [100], shows the highest frequency of PIK3CA mutations (47%) [99]. Finally, PIK3CA mutations are found in many papillary breast tumors and in androgen receptor positive apocrine breast tumors, as well as in premalignant lesions such as DCIS [101-104]. Remarkably, breast cancers with helical and kinase domain mutant alleles show widely differing prognoses. Helical domain mutations are associated with dramatically reduced overall and disease-free survival, whereas patients with kinase domain mutant breast tumors show enhanced survival as compared to patients with either wildtype or helical mutant PIK3CA [105]. In line with this finding, expression of an E545K helical domain mutant of PIK3CA in MDA-MB-231 cells induced a highly motile and malignant state, in contrast to the effect of expressing an H1047R allele, which caused more limited transformation [106].
With additional transcriptional profiling, the 5 molecular subtypes have been further subdivided on the basis of signaling pathway activation to 17 identifiable groups of tumors [97]. This analysis has shown that PI3K pathway activity is elevated in over half of the luminal subtypes, in 1 of 2 HER2/Neu subtypes and 3 of 3 basal subtypes. Particularly striking is the very high level of PI3K pathway activation observed in luminal B subtype 6, suggesting that mutations in several genes may cooperate to hyperactivate the pathway in these tumors [97].
In contrast to PIK3CA mutations, some PI3K pathway mutations are found in a limited group of breast tumors because they are associated with one or few subtypes. For example, and by definition, high-level expression of HER2/Neu activates PI3K signaling in HER2+ subtype tumors. Loss of PTEN gene function or expression is frequently observed in basal-like breast tumors [107, 108]. In addition, INPP4B is preferentially lost in basal breast cancers [95, 109]. In contrast, activating mutations in Akt1 are most frequently observed in luminal tumors and specifically in papillary tumors [101].
Some forms of breast cancer show very high level PI3K pathway activation [97], and this situation is associated with poor survival [61, 62]. Initially, it was thought that different mutations in the pathway would be mutually exclusive and unnecessary to achieve transformation. However, this idea has proved to be incorrect and cooperation between several oncogenic mutations on the pathway is relatively common, especially in poor prognosis tumors [49]. For example, amplification of HER2 and mutational activation of PIK3CA or PTEN inactivation occur together in many breast tumors [56]. This situation has been associated with resistance to HER2 targeted therapy [93, 94, 110-112]. Also, in MCF10A cells expressing high levels of HER2/Neu, a kinase domain mutant of PIK3CA (H1047R) induced expression of Heregulin, the ligand that activates HER2/HER3 signaling through the PI3K pathway [113]. In contrast, a helical domain mutant (E545K) enhanced transformation without inducing Heregulin expression [113]. Therefore, in HER2 subtype breast cancers with PIK3CAH1047R or other kinase domain mutant alleles, therapy with Herceptin/Trastuzumab together with an antibody that blocks Heregulin could be particularly effective. As noted above, the gene coding for PDK1 is amplified in many breast tumors with PI3K pathway activation, including tumors with HER2 amplification, activating mutations in PIK3CA or PTEN inactivation [80]. PTEN inactivation and PIK3CA mutation occur together in a subset of tumors [1], as do PTEN inactivation and inositol polyphosphate 4-phosphatase II gene deletion [109].
MOUSE MODELS
Mouse models of breast cancer have been refined through use of gene targeting to generate conditional mutants and transgenics that mimic pathological features of specific breast cancer subtypes [114-118]. To model PIK3CA-mutant breast cancer we generated mice with an H1047R mutant Pik3ca cDNA targeted to the ubiquitously expressed ROSA26 locus (R26) [119]. This cDNA was preceded by 5’ loxP-flanked transcriptional stop sequences and, when mated to MMTV-Cre mice, approximately 70% of the resulting female R26-Pik3caH1047R;MMTV-Cre mice developed mammary adenosquamous carcinoma or adenomyoepithelioma starting at about 5 months of age. Control R26-Pik3cawt;MMTV-Cre females were also generated but these animals did not develop mammary tumors. Glandular regions of the H1047R mutant tumors included cells expressing luminal and basal epithelial markers, whereas squamous regions expressed mesenchymal markers such as vimentin, desmin and/or N-cadherin. A subset of glandular cells expressed the estrogen receptor, which matches molecular subtype data in humans, where PIK3CA mutations are commonly found in ERα-positive luminal breast cancers. As expected, tumors from this mouse model showed evidence of PI3K/Akt pathway activation. To test for cooperation between PIK3CA and TP53, the two most common mutations in breast cancer [53], R26-Pik3caH1047R;MMTV-Cre mice were also crossed with p53loxP conditional mutants [120]. The resulting double mutant females showed accelerated tumor onset as well as an altered spectrum of mammary tumors [119]. A second model of PIK3CAH1047R-induced breast cancer has also been reported. In this study, a ROSA-targeted H1047R mutant cDNA was activated by expression of either WAP-Cre or MMTV-Cre. Mammary tumors in this model, which were ERα-positive and contained cells expressing either cytokeratin type as above, also showed evidence of enhanced PI3K/Akt pathway signaling. These mice developed mammary adenosquamous carcinomas and adenomyoepitheliomas, as well as adenocarcinomas with squamous metaplasia, adenocarcinomas and adenocarcinmatosis with invasive periductal cords of neoplastic cells [121]. Thus, in both cases, Pik3caH1047R induced a heterogeneous mixture of ERα-positive mammary tumors, some of which showed metasplastic differentiation. These data fit with the wide spectrum of PIK3CA mutant breast tumors observed in humans. However, these results contrast studies with activated Akt. Several groups have generated transgenic mice expressing mutationally activated Akt1 in the mammary gland. In each case, mutant Akt1 delayed involution but did not induce tumor formation [122, 123]. As with R26-Pik3caH1047R-Cre model mice, but unlike Akt1 transgenics, Pten loss-of-function mutants develop mammary tumors. This was first observed in Pten heterozygous mice that model Cowden’s syndrome [124], but also confirmed in PtenloxP;MMTV-Cre conditional mutants which developed mammary tumors starting at 2 months of age [125]. These tumors ranged from benign fibroadenomas to pleiomorphic adenocarcinomas [125]. Finally, mammary tumors were also induced in transgenic mice overexpressing Igf1R in mammary epithelium [126]. A heterogeneous mixture of tumor types was also observed in this model, with adenosquamous carcinoma and adenomyoepithelioma occurring at a high frequency. In addition, more homogenous HER2/Neu-like tumors were also noted as were metaplastic Wnt-like tumors [126, 127].
Mouse studies have also revealed cooperative interaction between PI3K pathway genes and other genes or pathways implicated in human breast cancer. As noted above, TP53 deletion showed cooperative interactions with Pik3caH1047R [119]. Also, a dominant Akt1 mutant reduced latency of tumor formation in MMTV-Neu mice, while decreasing invasion and metastasis in this model [128]. In contrast, deletion of Pten decreased tumor latency in MMTV-Neu mice but induced development of heterogeneous basal-like mammary tumors with enhanced metastatic dissemination [129]. These results were somewhat surprising and revealed further complexity in PI3K pathway signaling. Indeed, in vitro studies have shown that, like p110α [130], Akt2 activation is associated with enhanced β1-integrin mediated migration [131]. In fact Akt1 and Akt2 appear to play opposite roles in this context, with Akt1 suppressing Akt2-dependent migration [132]. In vivo, Akt1 enhanced growth of HER2/Neu primary tumors, but suppresses their dissemination, whereas Akt2 impairs local growth but stimulated metastasis [133-135]. The mechanism by which Akt isoforms regulate migration and metastasis in opposite directions may involve differential regulation of Pak kinase by Akt1 and Akt2 and/or distinct subcellular localization [134, 136, 137]. Also, Akt1 signaling maintains high expression of miR-200-family microRNAs that suppress epithelial mesenchymal transition and Akt2 upregulates miR-21, which inhibits PTEN expression as noted above [138, 139]. On the other hand, differential activation of Akt1 and 2 may be achieved through PHLPP serine/threonine phosphatases. PHLPP1 dephosphorylates and thereby inactivates Akt2, whereby PHLPP2 targets Akt1 for dephosphorylation [140].
USING THE MOUSE TO GUIDE EFFECTIVE THERAPY
These data highlight a series of questions that can be resolved through the study of mouse models of PI3K-pathway activated breast cancer. Furthermore, the answers to these questions can help guide development of effective therapy. First of all, breast cancer patients with helical and kinase domain mutants show dramatically different survival [105]. Why is this? Perhaps helical mutants activate pro-migratory Akt2 dependent signaling and metastasis, whereas kinase domain mutants activate Akt1? If so, how does this operate at the molecular level? Alternatively, helical or kinase domain mutants may activate a different set of PIP3 targets such as ILK or SGK3, an estrogen-regulated AGC family kinase that is required for survival of the ERα-positive breast cancer cell line MCF7 [141, 142]. These questions can be readily addressed with mouse models. Firstly, a mouse model of Pik3caE545K-induced breast cancer would have to be generated, analyzed and compared to an isogenic model of Pik3caH1047R-induced disease. As helical domain mutants are more frequent associated with infiltrating lobular carcinoma and patients with these mutants show relatively poor survival [105], it would be interesting to determine whether tumors in a mouse model of Pik3caE545K-induced breast cancer are lobular and metastatic, in contrast to the essentially non-metastatic tumors typically observed in Pik3caH1047R-model mice [119].
Helical and kinase domain mutants are dependent on distinct signaling inputs for full PI3K pathway activation [48]. Consequently, a different network of oncogenic mutations may cooperate with each. Perhaps, mutations that activate Ras signaling will be selected for in tumors with helical domain mutants of PIK3CA, whereas tyrosine kinase receptor genes will be activated or amplified in tumors with kinase domain mutants. By using retroviral- or transposon-based insertional mutagenesis screens in Pik3ca(E545K vs H1047R)-model mice, or deep sequencing of Pik3ca(E545K vs H1047R)-model tumors, the cooperative network associated with each Pik3ca mutant can be defined and new combination therapies developed for patients with analogous PIK3CA mutant breast tumors. To test whether helical and kinase mutant alleles activate different PIP3 targets, such as Akt1, Akt2, ILK, SGK3 or guanine nucleotide exchange factors for Rho family proteins including Rac, signaling experiments can be performed on mouse tumors induced by each mutant. These experiments are particularly important as activated Akt1 does not induce tumors in mice but overexpression of ILK does [143], and ILK has been shown to play a critical role in MMTV-Neu mouse mammary tumors [144]. Thus, with the genomic tools available today, sophisticated mouse models can be used to define how PI3K pathway genes cooperate with other mutations to control tumor dissemination.
Another unresolved question is the relationship between specific PI3K pathway mutations and breast tumor subtype. As noted above, PIK3CA mutations occur in many breast cancer subtypes but are rarely, if ever, seen in claudin-low tumors [99]. In contrast, PTEN mutations tend to cluster in triple negative tumors, a designation including basal-like and claudin-low breast cancers [107, 108, 145]. Potentially, this could be due to a specific biological response to each mutation type in the same cell-of-origin, and/or to distinct sensitivity of mammary stem cells or luminal progenitors to transformation by PIK3CA gain-of-function mutation versus PTEN deletion [146]. With respect to different biological response, activation of the PI3K signaling pathway at the level of HER2/Neu or IGF1 will result in activation of PI3K signaling together with activation of other signaling pathways stimulated by these receptors. This would not occur when the pathway is activated through PIK3CA mutation or PTEN deletion. Similarly, stimulation of the pathway by PIK3CA mutation will necessarily have different consequences than stimulation through PTEN inactivation, since PTEN protein also functions to inhibit the Src tyrosine kinase [35, 147, 148] and activate the APC-CDH1 complex [36, 37]. Once again, this question can be addressed using existing mouse models of breast cancer. Recently described Pik3caH1047R breast cancer models are Cre-dependent [119, 121]. Therefore, by using multiple mammary specific Cre driver lines (eg. Wap-Cre versus MMTV-Cre [121] or K14-Cre) it will be possible to compare tumors that arise through expression of a Pik3ca mutant or deletion of Pten within the same mammary stem or progenitor cell.
Mouse models of breast cancer have been used to define signaling proteins and pathways that are required to initiate tumor formation, to sustain tumors and/or to promote metastasis. Indeed, as discussed above, this approach has shown that Akt1 and Akt2 perform very different roles with respect to growth and dissemination of HER2/Neu subtype tumors [134]. This approach can also be used to define the role or function of Akt1 and 2 downstream of mutant Pik3ca or Pten deletion in mice. The specific PI3K pathway involved in transformation may be different in tumors with amplified HER2/Neu, amplified InsR/Igf-1R, with mutant PIK3CA, with PTEN inactivation or with Akt1E17K. With the exception of activated Akt1, there are now mouse models for each of these, and with loxP/Cre-mediated deletion or even gene knockdown, the role of other components on the PI3K pathway can be determined. For example, the p110β subunit of PI3K may play an important role in PTEN inactivated tumors [149-152].
New pathways that activate PI3K signaling have been discovered. For example, the non-canonical IκB Kinase, IKKϵ, can phosphorylate Akt in a PI3K-dependent, but mTOR-independent, manner [153]. IKKϵ is overexpressed in most breast cancers and in 30% of cases this is associated with amplification of sequences on the long arm of human chromosome 1, including the IKKϵ gene, IKBKE [154]. As RTK/PIK3CA/AKT1/PTEN and IKK/NFκB are the two most frequently mutated pathways in breast cancer, any crosstalk between them may represent a critical therapeutic target [53]. Once again, a mouse model would help to probe this issue. Would a mouse model of Ikbke-induced breast cancer show cooperation with mouse models of activated Pik3ca? Indeed, IKBKE is amplified and overexpressed together with PIK3CA mutation in some human breast tumors suggesting that these mutations can cooperate (e.g. MCF7 cells show IKBKE amplification/overexpression and PIK3CAE545K mutation [141, 154]). By generating a mouse model of IKBKE overexpressing and PIK3CAE545K mutant breast cancer, it would be possible to define which PI3K pathway components are involved in growth and invasion in this context, and whether the specific oncogenic pathway is different from tumors with Pik3caE545K and other cooperating mutations.
Finally, with the development of new mouse models to mimic specific forms of human breast cancer, it should be possible to perform high-throughput screens for chemicals or shRNAs that target mouse mammary tumor initiating cells while sparing normal mammary stem cells as well as other normal cells and tissues throughout the body. Thus, models that mimic each breast cancer subtype, with specific molecular lesions or combinations of lesions, can be developed and used to identify targets for combination therapy that will justify clinical trials on patients with analogous breast tumors. As activation of the mTOR pathway occurs in most breast tumors, it may also be possible to exploit this feature [155]. For example, in a mouse model of basal breast cancer with activated Ras, Igf1r signaling is required for survival [156]. Also, while direct mutational activation of the PI3K pathway in many breast tumors will preclude therapy based solely on caloric restriction [157], a number of reports have identified metabolic sensitivities associated with activation of specific oncogenic signaling pathways. For example, melanomas with activation of the Ras/Mapk pathway undergo apoptosis in response to leucine depletion. This effect is related to maintenance of activated TORC1 at lysosomes where it blocks autophagy, even in the absence of leucine [158]. As most breast tumors have sustained mutations that affect the very pathway used to sense nutrient availability, it is plausible that mouse models could prove useful in designing and testing nutrition based therapies.
ACKNOWLEDGEMENTS
The authors would like to thank Drs. Jim Woodgett and Greg Hannigan for valuable comments on the manuscript. We also gratefully acknowledge support from the Canadian Breast Cancer Foundation – Ontario Division and the Komen Foundation for the Cure.
REFERENCES
1. Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008; 27:5497-5510.
2. Whitman M, Downes CP, Keeler M, Keller T, Cantley L. Type I phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol-3-phosphate. Nature. 1988; 332:644-646.
3. Yu J, Zhang Y, McIlroy J, Rordorf-Nikolic T, Orr GA, Backer JM. Regulation of the p85/p110 phosphatidylinositol 3’-kinase: stabilization and inhibition of the p110alpha catalytic subunit by the p85 regulatory subunit. Mol Cell Biol. 1998; 18:1379-1387.
4. Sun XJ, Liu F. Phosphorylation of IRS proteins Yin-Yang regulation of insulin signaling. Vitam Horm. 2009; 80:351-387.
5. Yenush L, White MF. The IRS-signalling system during insulin and cytokine action. Bioessays. 1997; 19:491-500.
6. Ueki K, Fruman DA, Brachmann SM, Tseng YH, Cantley LC, Kahn CR. Molecular balance between the regulatory and catalytic subunits of phosphoinositide 3-kinase regulates cell signaling and survival. Mol Cell Biol. 2002; 22:965-977.
7. Rodriguez-Viciana P, Warne PH, Khwaja A, Marte BM, Pappin D, Das P, Waterfield MD, Ridley A, Downward J. Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell. 1997; 89:457-467.
8. Gupta S, Ramjaun AR, Haiko P, Wang Y, Warne PH, Nicke B, Nye E, Stamp G, Alitalo K, Downward J. Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell. 2007; 129:957-968.
9. Woodgett JR. Recent advances in the protein kinase B signaling pathway. Curr Opin Cell Biol. 2005; 17:150-157.
10. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007; 129:1261-1274.
11. Franke TF. PI3K/Akt: getting it right matters. Oncogene. 2008; 27:6473-6488.
12. Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009; 122:3589-3594.
13. Laplante M, Sabatini DM. An emerging role of mTOR in lipid biosynthesis. Curr Biol. 2009; 19:R1046-1052.
14. Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, Carr SA, Sabatini DM. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007; 25:903-915.
15. Huang J, Manning BD. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J. 2008; 412:179-190.
16. Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, Vander Heiden MG, MacKeigan JP, Finan PM, Clish CB, Murphy LO, Manning BD. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010; 39:171-183.
17. Yecies JL, Manning BD. Transcriptional Control of Cellular Metabolism by mTOR Signaling. Cancer Res. 2011; 71:2815-2820.
18. Yecies JL, Manning BD. mTOR links oncogenic signaling to tumor cell metabolism. J Mol Med. 2011; 89:221-228.
19. Efeyan A, Sabatini DM. mTOR and cancer: many loops in one pathway. Curr Opin Cell Biol. 2010; 22:169-176.
20. Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011; 12:21-35.
21. Saci A, Cantley LC, Carpenter CL. Rac1 Regulates the Activity of mTORC1 and mTORC2 and Controls Cellular Size. Mol Cell. 2011; 42:50-61.
22. Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ, Sabatini DM. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell. 2006; 11:859-871.
23. McDonald PC, Oloumi A, Mills J, Dobreva I, Maidan M, Gray V, Wederell ED, Bally MB, Foster LJ, Dedhar S. Rictor and integrin-linked kinase interact and regulate Akt phosphorylation and cancer cell survival. Cancer Res. 2008; 68:1618-1624.
24. Hannigan GE, McDonald PC, Walsh MP, Dedhar S. Integrin-linked kinase: Not so ‘pseudo’ after all. Oncogene. 2011.
25. Maydan M, McDonald PC, Sanghera J, Yan J, Rallis C, Pinchin S, Hannigan GE, Foster LJ, Ish-Horowicz D, Walsh MP, Dedhar S. Integrin-linked kinase is a functional Mn2+-dependent protein kinase that regulates glycogen synthase kinase-3beta (GSK-3beta) phosphorylation. PLoS One. 2010; 5:e12356.
26. Delcommenne M, Tan C, Gray V, Rue L, Woodgett J, Dedhar S. Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc Natl Acad Sci U S A. 1998; 95:11211-11216.
27. Wickstrom SA, Lange A, Hess MW, Polleux J, Spatz JP, Kruger M, Pfaller K, Lambacher A, Bloch W, Mann M, Huber LA, Fassler R. Integrin-linked kinase controls microtubule dynamics required for plasma membrane targeting of caveolae. Dev Cell. 2010; 19:574-588.
28. Hannigan G, Troussard AA, Dedhar S. Integrin-linked kinase: a cancer therapeutic target unique among its ILK. Nat Rev Cancer. 2005; 5:51-63.
29. Voskas D, Ling LS, Woodgett JR. Does GSK-3 provide a shortcut for PI3K activation of Wnt signalling? F1000 Biol Rep. 2010; 2:82.
30. Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, Majumder PK, Baselga J, Rosen N. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011; 19:58-71.
31. Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997; 275:1943-1947.
32. Rhei E, Kang L, Bogomolniy F, Federici MG, Borgen PI, Boyd J. Mutation analysis of the putative tumor suppressor gene PTEN/MMAC1 in primary breast carcinomas. Cancer Res. 1997; 57:3657-3659.
33. Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, Mak TW. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998; 95:29-39.
34. Myers MP, Pass I, Batty IH, Van der Kaay J, Stolarov JP, Hemmings BA, Wigler MH, Downes CP, Tonks NK. The lipid phosphatase activity of PTEN is critical for its tumor supressor function. Proc Natl Acad Sci U S A. 1998; 95:13513-13518.
35. Dey N, Crosswell HE, De P, Parsons R, Peng Q, Su JD, Durden DL. The protein phosphatase activity of PTEN regulates SRC family kinases and controls glioma migration. Cancer Res. 2008; 68:1862-1871.
36. Gao D, Inuzuka H, Tseng A, Chin RY, Toker A, Wei W. Phosphorylation by Akt1 promotes cytoplasmic localization of Skp2 and impairs APCCdh1-mediated Skp2 destruction. Nat Cell Biol. 2009; 11:397-408.
37. Song MS, Carracedo A, Salmena L, Song SJ, Egia A, Malumbres M, Pandolfi PP. Nuclear PTEN regulates the APC-CDH1 tumor-suppressive complex in a phosphatase-independent manner. Cell. 2011; 144:187-199.
38. Gewinner C, Wang ZC, Richardson A, Teruya-Feldstein J, Etemadmoghadam D, Bowtell D, Barretina J, Lin WM, Rameh L, Salmena L, Pandolfi PP, Cantley LC. Evidence that inositol polyphosphate 4-phosphatase type II is a tumor suppressor that inhibits PI3K signaling. Cancer Cell. 2009; 16:115-125.
39. Kawase T, Ohki R, Shibata T, Tsutsumi S, Kamimura N, Inazawa J, Ohta T, Ichikawa H, Aburatani H, Tashiro F, Taya Y. PH domain-only protein PHLDA3 is a p53-regulated repressor of Akt. Cell. 2009; 136:535-550.
40. Gao T, Furnari F, Newton AC. PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell. 2005; 18:13-24.
41. Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, Gray NS, Sabatini DM. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009; 137:873-886.
42. Chakrabarty A, Sanchez V, Kuba MG, Rinehart C, Arteaga CL. Breast Cancer Special Feature: Feedback upregulation of HER3 (ErbB3) expression and activity attenuates antitumor effect of PI3K inhibitors. Proc Natl Acad Sci U S A. 2011.
43. Zick Y. Ser/Thr phosphorylation of IRS proteins: a molecular basis for insulin resistance. Sci STKE. 2005; 2005:pe4.
44. Vogt PK, Bader AG, Kang S. Phosphoinositide 3-kinase: from viral oncoprotein to drug target. Virology. 2006; 344:131-138.
45. Zhao L, Vogt PK. Class I PI3K in oncogenic cellular transformation. Oncogene. 2008; 27:5486-5496.
46. Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JK, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004; 304:554.
47. Isakoff SJ, Engelman JA, Irie HY, Luo J, Brachmann SM, Pearline RV, Cantley LC, Brugge JS. Breast cancer-associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer Res. 2005; 65:10992-11000.
48. Zhao L, Vogt PK. Helical domain and kinase domain mutations in p110alpha of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc Natl Acad Sci U S A. 2008; 105:2652-2657.
49. Oda K, Okada J, Timmerman L, Rodriguez-Viciana P, Stokoe D, Shoji K, Taketani Y, Kuramoto H, Knight ZA, Shokat KM, McCormick F. PIK3CA cooperates with other phosphatidylinositol 3’-kinase pathway mutations to effect oncogenic transformation. Cancer Res. 2008; 68:8127-8136.
50. Mayo LD, Donner DB. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci U S A. 2001; 98:11598-11603.
51. Zhou BP, Liao Y, Xia W, Zou Y, Spohn B, Hung MC. HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat Cell Biol. 2001; 3:973-982.
52. Stambolic V, MacPherson D, Sas D, Lin Y, Snow B, Jang Y, Benchimol S, Mak TW. Regulation of PTEN transcription by p53. Mol Cell. 2001; 8:317-325.
53. Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, Silliman N, Szabo S, Dezso Z, Ustyanksky V, Nikolskaya T, Nikolsky Y et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007; 318:1108-1113.
54. Bachman KE, Argani P, Samuels Y, Silliman N, Ptak J, Szabo S, Konishi H, Karakas B, Blair BG, Lin C, Peters BA, Velculescu VE, Park BH. The PIK3CA gene is mutated with high frequency in human breast cancers. Cancer Biol Ther. 2004; 3:772-775.
55. Campbell IG, Russell SE, Choong DY, Montgomery KG, Ciavarella ML, Hooi CS, Cristiano BE, Pearson RB, Phillips WA. Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res. 2004; 64:7678-7681.
56. Saal LH, Holm K, Maurer M, Memeo L, Su T, Wang X, Yu JS, Malmstrom PO, Mansukhani M, Enoksson J, Hibshoosh H, Borg A, Parsons R. PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer Res. 2005; 65:2554-2559.
57. Wu G, Xing M, Mambo E, Huang X, Liu J, Guo Z, Chatterjee A, Goldenberg D, Gollin SM, Sukumar S, Trink B, Sidransky D. Somatic mutation and gain of copy number of PIK3CA in human breast cancer. Breast Cancer Res. 2005; 7:R609-616.
58. Kadota M, Sato M, Duncan B, Ooshima A, Yang HH, Diaz-Meyer N, Gere S, Kageyama S, Fukuoka J, Nagata T, Tsukada K, Dunn BK, Wakefield LM, Lee MP. Identification of novel gene amplifications in breast cancer and coexistence of gene amplification with an activating mutation of PIK3CA. Cancer Res. 2009; 69:7357-7365.
59. Dillon RL, White DE, Muller WJ. The phosphatidyl inositol 3-kinase signaling network: implications for human breast cancer. Oncogene. 2007; 26:1338-1345.
60. Leary RJ, Lin JC, Cummins J, Boca S, Wood LD, Parsons DW, Jones S, Sjoblom T, Park BH, Parsons R, Willis J, Dawson D, Willson JK, Nikolskaya T, Nikolsky Y, Kopelovich L et al. Integrated analysis of homozygous deletions, focal amplifications, and sequence alterations in breast and colorectal cancers. Proc Natl Acad Sci U S A. 2008; 105:16224-16229.
61. Lopez-Knowles E, O’Toole SA, McNeil CM, Millar EK, Qiu MR, Crea P, Daly RJ, Musgrove EA, Sutherland RL. PI3K pathway activation in breast cancer is associated with the basal-like phenotype and cancer-specific mortality. Int J Cancer. 2010; 126:1121-1131.
62. Capodanno A, Camerini A, Orlandini C, Baldini E, Resta ML, Bevilacqua G, Collecchi P. Dysregulated PI3K/Akt/PTEN pathway is a marker of a short disease-free survival in node-negative breast carcinoma. Hum Pathol. 2009; 40:1408-1417.
63. Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, Neve RM, Kuo WL, Davies M, Carey M, Hu Z, Guan Y, Sahin A, Symmans WF, Pusztai L, Nolden LK, Horlings H, Berns K, Hung MC et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008; 68:6084-6091.
64. Baselga J, Swain SM. Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nat Rev Cancer. 2009; 9:463-475.
65. Elsheikh SE, Green AR, Rakha EA, Samaka RM, Ammar AA, Powe D, Reis-Filho JS, Ellis IO. Caveolin 1 and Caveolin 2 are associated with breast cancer basal-like and triple-negative immunophenotype. Br J Cancer. 2008; 99:327-334.
66. Gastaldi S, Comoglio PM, Trusolino L. The Met oncogene and basal-like breast cancer: another culprit to watch out for? Breast Cancer Res. 2010; 12:208.
67. Ponzo MG, Park M. The met receptor tyrosine kinase and basal breast cancer. Cell Cycle. 2010; 9.
68. Savage K, Lambros MB, Robertson D, Jones RL, Jones C, Mackay A, James M, Hornick JL, Pereira EM, Milanezi F, Fletcher CD, Schmitt FC, Ashworth A, Reis-Filho JS. Caveolin 1 is overexpressed and amplified in a subset of basal-like and metaplastic breast carcinomas: a morphologic, ultrastructural, immunohistochemical, and in situ hybridization analysis. Clin Cancer Res. 2007; 13:90-101.
69. Wohrle FU, Daly RJ, Brummer T. Function, regulation and pathological roles of the Gab/DOS docking proteins. Cell Commun Signal. 2009; 7:22.
70. Gu H, Neel BG. The “Gab” in signal transduction. Trends Cell Biol. 2003; 13:122-130.
71. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, Lee C et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007; 316:1039-1043.
72. Lu X, Kambe F, Cao X, Yamauchi M, Seo H. Insulin-like growth factor-I activation of Akt survival cascade in neuronal cells requires the presence of its cognate receptor in caveolae. Exp Cell Res. 2008; 314:342-351.
73. Ravid D, Maor S, Werner H, Liscovitch M. Caveolin-1 inhibits cell detachment-induced p53 activation and anoikis by upregulation of insulin-like growth factor-I receptors and signaling. Oncogene. 2005; 24:1338-1347.
74. Salani B, Briatore L, Garibaldi S, Cordera R, Maggi D. Caveolin-1 down-regulation inhibits insulin-like growth factor-I receptor signal transduction in H9C2 rat cardiomyoblasts. Endocrinology. 2008; 149:461-465.
75. Almeida A, Muleris M, Dutrillaux B, Malfoy B. The insulin-like growth factor I receptor gene is the target for the 15q26 amplicon in breast cancer. Genes Chromosomes Cancer. 1994; 11:63-65.
76. Shah SP, Morin RD, Khattra J, Prentice L, Pugh T, Burleigh A, Delaney A, Gelmon K, Guliany R, Senz J, Steidl C, Holt RA, Jones S, Sun M, Leung G, Moore R et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature. 2009; 461:809-813.
77. Chan BT, Lee AV. Insulin receptor substrates (IRSs) and breast tumorigenesis. J Mammary Gland Biol Neoplasia. 2008; 13:415-422.
78. Jaiswal BS, Janakiraman V, Kljavin NM, Chaudhuri S, Stern HM, Wang W, Kan Z, Dbouk HA, Peters BA, Waring P, Dela Vega T, Kenski DM, Bowman KK, Lorenzo M, Li H, Wu J et al. Somatic mutations in p85alpha promote tumorigenesis through class IA PI3K activation. Cancer Cell. 2009; 16:463-474.
79. Bentires-Alj M, Gil SG, Chan R, Wang ZC, Wang Y, Imanaka N, Harris LN, Richardson A, Neel BG, Gu H. A role for the scaffolding adapter GAB2 in breast cancer. Nat Med. 2006; 12:114-121.
80. Maurer M, Su T, Saal LH, Koujak S, Hopkins BD, Barkley CR, Wu J, Nandula S, Dutta B, Xie Y, Chin YR, Kim DI, Ferris JS, Gruvberger-Saal SK, Laakso M, Wang X et al. 3-Phosphoinositide-dependent kinase 1 potentiates upstream lesions on the phosphatidylinositol 3-kinase pathway in breast carcinoma. Cancer Res. 2009; 69:6299-6306.
81. Carpten JD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, Hostetter G, Boguslawski S, Moses TY, Savage S, Uhlik M, Lin A, Du J, Qian YW, Zeckner DJ, Tucker-Kellogg G et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007; 448:439-444.
82. Brugge J, Hung MC, Mills GB. A new mutational AKTivation in the PI3K pathway. Cancer Cell. 2007; 12:104-107.
83. Sun T, Aceto N, Meerbrey KL, Kessler JD, Zhou C, Migliaccio I, Nguyen DX, Pavlova NN, Botero M, Huang J, Bernardi RJ, Schmitt E, Hu G, Li MZ, Dephoure N, Gygi SP et al. Activation of Multiple Proto-oncogenic Tyrosine Kinases in Breast Cancer via Loss of the PTPN12 Phosphatase. Cell. 2011; 144:703-718.
84. Hollander MC, Blumenthal GM, Dennis PA. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat Rev Cancer. 2011; 11:289-301.
85. Garcia JM, Silva JM, Dominguez G, Gonzalez R, Navarro A, Carretero L, Provencio M, Espana P, Bonilla F. Allelic loss of the PTEN region (10q23) in breast carcinomas of poor pathophenotype. Breast Cancer Res Treat. 1999; 57:237-243.
86. Garcia JM, Silva J, Pena C, Garcia V, Rodriguez R, Cruz MA, Cantos B, Provencio M, Espana P, Bonilla F. Promoter methylation of the PTEN gene is a common molecular change in breast cancer. Genes Chromosomes Cancer. 2004; 41:117-124.
87. Depowski PL, Rosenthal SI, Ross JS. Loss of expression of the PTEN gene protein product is associated with poor outcome in breast cancer. Mod Pathol. 2001; 14:672-676.
88. Perren A, Weng LP, Boag AH, Ziebold U, Thakore K, Dahia PL, Komminoth P, Lees JA, Mulligan LM, Mutter GL, Eng C. Immunohistochemical evidence of loss of PTEN expression in primary ductal adenocarcinomas of the breast. Am J Pathol. 1999; 155:1253-1260.
89. Sansone P, Storci G, Tavolari S, Guarnieri T, Giovannini C, Taffurelli M, Ceccarelli C, Santini D, Paterini P, Marcu KB, Chieco P, Bonafe M. IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J Clin Invest. 2007; 117:3988-4002.
90. Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell. 2009; 139:693-706.
91. Iliopoulos D, Jaeger SA, Hirsch HA, Bulyk ML, Struhl K. STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol Cell. 2010; 39:493-506.
92. Hartman ZC, Yang XY, Glass O, Lei G, Osada T, Dave SS, Morse MA, Clay TM, Lyerly HK. HER2 overexpression elicits a pro-inflammatory IL-6 autocrine signaling loop that is critical for tumorigenesis. Cancer Res. 2011.
93. Lu Y, Yu Q, Liu JH, Zhang J, Wang H, Koul D, McMurray JS, Fang X, Yung WK, Siminovitch KA, Mills GB. Src family protein-tyrosine kinases alter the function of PTEN to regulate phosphatidylinositol 3-kinase/AKT cascades. J Biol Chem. 2003; 278:40057-40066.
94. Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, Klos KS, Li P, Monia BP, Nguyen NT, Hortobagyi GN, Hung MC, Yu D. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004; 6:117-127.
95. Agoulnik IU, Hodgson MC, Bowden WA, Ittmann MM. INPP4B: the New Kid on the PI3K Block. Oncotarget. 2011; 2:321-328.
96. Perou CM, Parker JS, Prat A, Ellis MJ, Bernard PS. Clinical implementation of the intrinsic subtypes of breast cancer. Lancet Oncol. 2010; 11:718-719; author reply 720-711.
97. Gatza ML, Lucas JE, Barry WT, Kim JW, Wang Q, Crawford MD, Datto MB, Kelley M, Mathey-Prevot B, Potti A, Nevins JR. A pathway-based classification of human breast cancer. Proc Natl Acad Sci U S A. 2010; 107:6994-6999.
98. Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, He X, Perou CM. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010; 12:R68.
99. Hennessy BT, Gonzalez-Angulo AM, Stemke-Hale K, Gilcrease MZ, Krishnamurthy S, Lee JS, Fridlyand J, Sahin A, Agarwal R, Joy C, Liu W, Stivers D, Baggerly K, Carey M, Lluch A, Monteagudo C et al. Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res. 2009; 69:4116-4124.
100. Weigelt B, Kreike B, Reis-Filho JS. Metaplastic breast carcinomas are basal-like breast cancers: a genomic profiling analysis. Breast Cancer Res Treat. 2009; 117:273-280.
101. Troxell ML, Levine J, Beadling C, Warrick A, Dunlap J, Presnell A, Patterson J, Shukla A, Olson NR, Heinrich MC, Corless CL. High prevalence of PIK3CA/AKT pathway mutations in papillary neoplasms of the breast. Mod Pathol. 2010; 23:27-37.
102. Li H, Zhu R, Wang L, Zhu T, Li Q, Chen Q, Wang H, Zhu H. PIK3CA mutations mostly begin to develop in ductal carcinoma of the breast. Exp Mol Pathol. 2010; 88:150-155.
103. Dunlap J, Le C, Shukla A, Patterson J, Presnell A, Heinrich MC, Corless CL, Troxell ML. Phosphatidylinositol-3-kinase and AKT1 mutations occur early in breast carcinoma. Breast Cancer Res Treat. 2010; 120:409-418.
104. Lehmann BD, J.A. B, Chen X, Sanders ME, Bapsi Chakravarthy A, Shyr Y, Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. Journal of Clinical Investigation. 2011.
105. Barbareschi M, Buttitta F, Felicioni L, Cotrupi S, Barassi F, Del Grammastro M, Ferro A, Dalla Palma P, Galligioni E, Marchetti A. Different prognostic roles of mutations in the helical and kinase domains of the PIK3CA gene in breast carcinomas. Clin Cancer Res. 2007; 13:6064-6069.
106. Pang H, Flinn R, Patsialou A, Wyckoff J, Roussos ET, Wu H, Pozzuto M, Goswami S, Condeelis JS, Bresnick AR, Segall JE, Backer JM. Differential enhancement of breast cancer cell motility and metastasis by helical and kinase domain mutations of class IA phosphoinositide 3-kinase. Cancer Res. 2009; 69:8868-8876.
107. Marty B, Maire V, Gravier E, Rigaill G, Vincent-Salomon A, Kappler M, Lebigot I, Djelti F, Tourdes A, Gestraud P, Hupe P, Barillot E, Cruzalegui F, Tucker GC, Stern MH, Thiery JP et al. Frequent PTEN genomic alterations and activated phosphatidylinositol 3-kinase pathway in basal-like breast cancer cells. Breast Cancer Res. 2008; 10:R101.
108. Saal LH, Gruvberger-Saal SK, Persson C, Lovgren K, Jumppanen M, Staaf J, Jonsson G, Pires MM, Maurer M, Holm K, Koujak S, Subramaniyam S, Vallon-Christersson J, Olsson H, Su T, Memeo L et al. Recurrent gross mutations of the PTEN tumor suppressor gene in breast cancers with deficient DSB repair. Nat Genet. 2008; 40:102-107.
109. Fedele CG, Ooms LM, Ho M, Vieusseux J, O’Toole SA, Millar EK, Lopez-Knowles E, Sriratana A, Gurung R, Baglietto L, Giles GG, Bailey CG, Rasko JE, Shields BJ, Price JT, Majerus PW et al. Inositol polyphosphate 4-phosphatase II regulates PI3K/Akt signaling and is lost in human basal-like breast cancers. Proc Natl Acad Sci U S A. 2010; 107:22231-22236.
110. Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, Linn SC, Gonzalez-Angulo AM, Stemke-Hale K, Hauptmann M, Beijersbergen RL, Mills GB, van de Vijver MJ, Bernards R. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007; 12:395-402.
111. Gong C, Yao Y, Wang Y, Liu B, Wu W, Chen J, Su F, Yao H, Song E. Upregulation of MIR-21 mediates resistance to trastuzumab therapy for breast cancer. J Biol Chem. 2011.
112. O’Brien NA, Browne BC, Chow L, Wang Y, Ginther C, Arboleda J, Duffy MJ, Crown J, O’Donovan N, Slamon DJ. Activated phosphoinositide 3-kinase/AKT signaling confers resistance to trastuzumab but not lapatinib. Mol Cancer Ther. 2010; 9:1489-1502.
113. Chakrabarty A, Rexer BN, Wang SE, Cook RS, Engelman JA, Arteaga CL. H1047R phosphatidylinositol 3-kinase mutant enhances HER2-mediated transformation by heregulin production and activation of HER3. Oncogene. 2010; 29:5193-5203.
114. Herschkowitz JI, Simin K, Weigman VJ, Mikaelian I, Usary J, Hu Z, Rasmussen KE, Jones LP, Assefnia S, Chandrasekharan S, Backlund MG, Yin Y, Khramtsov AI, Bastein R, Quackenbush J, Glazer RI et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007; 8:R76.
115. Jiang Z, Deng T, Jones R, Li H, Herschkowitz JI, Liu JC, Weigman VJ, Tsao MS, Lane TF, Perou CM, Zacksenhaus E. Rb deletion in mouse mammary progenitors induces luminal-B or basal-like/EMT tumor subtypes depending on p53 status. J Clin Invest. 2010; 120:3296-3309.
116. Cardiff RD. The pathology of EMT in mouse mammary tumorigenesis. J Mammary Gland Biol Neoplasia. 2010; 15:225-233.
117. Cardiff RD. Mouse models of human breast cancer. Comp Med. 2003; 53:250-253.
118. Ursini-Siegel J, Schade B, Cardiff RD, Muller WJ. Insights from transgenic mouse models of ERBB2-induced breast cancer. Nat Rev Cancer. 2007; 7:389-397.
119. Adams JR, Xu K, Liu JC, Agamez NM, Loch AJ, Wong RG, Wang W, Wright KL, Lane TF, Zacksenhaus E, Egan SE. Cooperation between Pik3ca and p53 Mutations in Mouse Mammary Tumor Formation. Cancer Res. 2011; 71:2706-2717.
120. Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet. 2001; 29:418-425.
121. Meyer DS, Brinkhaus H, Muller U, Muller M, Cardiff RD, Bentires-Alj M. Luminal Expression of PIK3CA Mutant H1047R in the Mammary Gland Induces Heterogeneous Tumors. Cancer Res. 2011.
122. Schwertfeger KL, Richert MM, Anderson SM. Mammary gland involution is delayed by activated Akt in transgenic mice. Mol Endocrinol. 2001; 15:867-881.
123. Hutchinson J, Jin J, Cardiff RD, Woodgett JR, Muller WJ. Activation of Akt (protein kinase B) in mammary epithelium provides a critical cell survival signal required for tumor progression. Mol Cell Biol. 2001; 21:2203-2212.
124. Stambolic V, Tsao MS, Macpherson D, Suzuki A, Chapman WB, Mak TW. High incidence of breast and endometrial neoplasia resembling human Cowden syndrome in pten+/- mice. Cancer Res. 2000; 60:3605-3611.
125. Li G, Robinson GW, Lesche R, Martinez-Diaz H, Jiang Z, Rozengurt N, Wagner KU, Wu DC, Lane TF, Liu X, Hennighausen L, Wu H. Conditional loss of PTEN leads to precocious development and neoplasia in the mammary gland. Development. 2002; 129:4159-4170.
126. Jones RA, Campbell CI, Gunther EJ, Chodosh LA, Petrik JJ, Khokha R, Moorehead RA. Transgenic overexpression of IGF-IR disrupts mammary ductal morphogenesis and induces tumor formation. Oncogene. 2007; 26:1636-1644.
127. Jones RA, Petrik JJ, Moorehead RA. Preneoplastic changes persist after IGF-IR downregulation and tumor regression. Oncogene. 2010; 29:4779-4786.
128. Hutchinson JN, Jin J, Cardiff RD, Woodgett JR, Muller WJ. Activation of Akt-1 (PKB-alpha) can accelerate ErbB-2-mediated mammary tumorigenesis but suppresses tumor invasion. Cancer Res. 2004; 64:3171-3178.
129. Schade B, Rao T, Dourdin N, Lesurf R, Hallett M, Cardiff RD, Muller WJ. PTEN deficiency in a luminal ErbB-2 mouse model results in dramatic acceleration of mammary tumorigenesis and metastasis. J Biol Chem. 2009; 284:19018-19026.
130. Zeller KS, Idevall-Hagren O, Stefansson A, Velling T, Jackson SP, Downward J, Tengholm A, Johansson S. PI3-kinase p110alpha mediates beta1 integrin-induced Akt activation and membrane protrusion during cell attachment and initial spreading. Cell Signal. 2010; 22:1838-1848.
131. Arboleda MJ, Lyons JF, Kabbinavar FF, Bray MR, Snow BE, Ayala R, Danino M, Karlan BY, Slamon DJ. Overexpression of AKT2/protein kinase Bbeta leads to up-regulation of beta1 integrins, increased invasion, and metastasis of human breast and ovarian cancer cells. Cancer Res. 2003; 63:196-206.
132. Irie HY, Pearline RV, Grueneberg D, Hsia M, Ravichandran P, Kothari N, Natesan S, Brugge JS. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition. J Cell Biol. 2005; 171:1023-1034.
133. Maroulakou IG, Oemler W, Naber SP, Tsichlis PN. Akt1 ablation inhibits, whereas Akt2 ablation accelerates, the development of mammary adenocarcinomas in mouse mammary tumor virus (MMTV)-ErbB2/neu and MMTV-polyoma middle T transgenic mice. Cancer Res. 2007; 67:167-177.
134. Dillon RL, Muller WJ. Distinct biological roles for the akt family in mammary tumor progression. Cancer Res. 2010; 70:4260-4264.
135. Dillon RL, Marcotte R, Hennessy BT, Woodgett JR, Mills GB, Muller WJ. Akt1 and akt2 play distinct roles in the initiation and metastatic phases of mammary tumor progression. Cancer Res. 2009; 69:5057-5064.
136. Zhou GL, Tucker DF, Bae SS, Bhatheja K, Birnbaum MJ, Field J. Opposing roles for Akt1 and Akt2 in Rac/Pak signaling and cell migration. J Biol Chem. 2006; 281:36443-36453.
137. Gonzalez E, McGraw TE. The Akt kinases: isoform specificity in metabolism and cancer. Cell Cycle. 2009; 8:2502-2508.
138. Iliopoulos D, Polytarchou C, Hatziapostolou M, Kottakis F, Maroulakou IG, Struhl K, Tsichlis PN. MicroRNAs differentially regulated by Akt isoforms control EMT and stem cell renewal in cancer cells. Sci Signal. 2009; 2:ra62.
139. Polytarchou C, Iliopoulos D, Hatziapostolou M, Kottakis F, Maroulakou I, Struhl K, Tsichlis PN. Akt2 regulates all Akt isoforms and promotes resistance to hypoxia through induction of miR-21 upon oxygen deprivation. Cancer Res. 2011.
140. Brognard J, Sierecki E, Gao T, Newton AC. PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol Cell. 2007; 25:917-931.
141. Vasudevan KM, Barbie DA, Davies MA, Rabinovsky R, McNear CJ, Kim JJ, Hennessy BT, Tseng H, Pochanard P, Kim SY, Dunn IF, Schinzel AC, Sandy P, Hoersch S, Sheng Q, Gupta PB et al. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell. 2009; 16:21-32.
142. Wang Y, Zhou D, Phung S, Masri S, Smith D, Chen S. SGK3 is an estrogen-inducible kinase promoting estrogen-mediated survival of breast cancer cells. Mol Endocrinol. 2011; 25:72-82.
143. White DE, Cardiff RD, Dedhar S, Muller WJ. Mammary epithelial-specific expression of the integrin-linked kinase (ILK) results in the induction of mammary gland hyperplasias and tumors in transgenic mice. Oncogene. 2001; 20:7064-7072.
144. Pontier SM, Huck L, White DE, Rayment J, Sanguin-Gendreau V, Hennessy B, Zuo D, St-Arnaud R, Mills GB, Dedhar S, Marshall CJ, Muller WJ. Integrin-linked kinase has a critical role in ErbB2 mammary tumor progression: implications for human breast cancer. Oncogene. 2010; 29:3374-3385.
145. Saal LH, Johansson P, Holm K, Gruvberger-Saal SK, She QB, Maurer M, Koujak S, Ferrando AA, Malmstrom P, Memeo L, Isola J, Bendahl PO, Rosen N, Hibshoosh H, Ringner M, Borg A et al. Poor prognosis in carcinoma is associated with a gene expression signature of aberrant PTEN tumor suppressor pathway activity. Proc Natl Acad Sci U S A. 2007; 104:7564-7569.
146. Visvader JE. Cells of origin in cancer. Nature. 2011; 469:314-322.
147. Poon JS, Eves R, Mak AS. Both lipid- and protein-phosphatase activities of PTEN contribute to the p53-PTEN anti-invasion pathway. Cell Cycle. 2010; 9:4450-4454.
148. Zhang S, Huang WC, Li P, Guo H, Poh SB, Brady SW, Xiong Y, Tseng LM, Li SH, Ding Z, Sahin AA, Esteva FJ, Hortobagyi GN, Yu D. Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat Med. 2011; 17:461-469.
149. Marques M, Kumar A, Poveda AM, Zuluaga S, Hernandez C, Jackson S, Pasero P, Carrera AC. Specific function of phosphoinositide 3-kinase beta in the control of DNA replication. Proc Natl Acad Sci U S A. 2009; 106:7525-7530.
150. Jiang X, Chen S, Asara JM, Balk SP. Phosphoinositide 3-kinase pathway activation in phosphate and tensin homolog (PTEN)-deficient prostate cancer cells is independent of receptor tyrosine kinases and mediated by the p110beta and p110delta catalytic subunits. J Biol Chem. 2010; 285:14980-14989.
151. Carvalho S, Milanezi F, Costa JL, Amendoeira I, Schmitt F. PIKing the right isoform: the emergent role of the p110beta subunit in breast cancer. Virchows Arch. 2010; 456:235-243.
152. Dbouk HA, Backer JM. A beta version of life: p110beta takes center stage. Oncotarget. 2010; 1:729-733.
153. Xie X, Zhang D, Zhao B, Lu MK, You M, Condorelli G, Wang CY, Guan KL. I{kappa}B kinase {varepsilon} and TANK-binding kinase 1 activate AKT by direct phosphorylation. Proc Natl Acad Sci U S A. 2011; 108:6474-6479.
154. Boehm JS, Zhao JJ, Yao J, Kim SY, Firestein R, Dunn IF, Sjostrom SK, Garraway LA, Weremowicz S, Richardson AL, Greulich H, Stewart CJ, Mulvey LA, Shen RR, Ambrogio L, Hirozane-Kishikawa T et al. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell. 2007; 129:1065-1079.
155. Markman B, Dienstmann R, Tabernero J. Targeting the PI3K/Akt/mTOR pathway--beyond rapalogs. Oncotarget. 2010; 1:530-543.
156. Klinakis A, Szabolcs M, Chen G, Xuan S, Hibshoosh H, Efstratiadis A. Igf1r as a therapeutic target in a mouse model of basal-like breast cancer. Proc Natl Acad Sci U S A. 2009; 106:2359-2364.
157. Kalaany NY, Sabatini DM. Tumours with PI3K activation are resistant to dietary restriction. Nature. 2009; 458:725-731.
158. Sheen JH, Zoncu R, Kim D, Sabatini DM. Defective Regulation of Autophagy upon Leucine Deprivation Reveals a Targetable Liability of Human Melanoma Cells In Vitro and In Vivo. Cancer Cell. 2011; 19:613-628.