
INTRODUCTION
Cyclins are key regulatory components of the cyclin/CDK (Cyclin-Dependent Kinase) complex that regulate the cell cycle, thus contributing to tumor progression. While cyclin D interacts with CDK4/6, cyclin E interacts with CDK2 to form complexes that play a central role in the G1/S transition of the cell cycle. This complex formed by cyclins and CDKs act by phosphorylating Rb [1, 2], which releases E2F from the complex, allowing it to activate cell cycle progression, Figure 1. Amplifications of cyclin genes are amongst the most common alterations in cancers, with cyclin D1 (CCND1) amplification rates ranging from 15–40% [1].
Cyclin D1 or CCND1 was initially designated PRAD1 or BCL1. PRAD1 was found to be clonally rearranged on chromosome 11q13 with PTH (parathyroid hormone) in parathyroid adenomas, resulting in overexpression of cyclin D1 [3–5]. Similarly, in mantle cell lymphomas, BCL-1 (B-Cell lymphoma-1) is rearranged and juxtaposes BCL-1 and the immunoglobulin heavy chain IGH (t(11;14)(q13;q32)) resulting in overexpression of the BCL-1 (CCND1) protein [6, 7].
CCND1 amplifications have been described in head and neck carcinomas, non-small-cell lung, endometrial, pancreatic, breast, as well as colorectal cancers [1]. Of interest, in addition to direct CCND1 amplification, and rearrangement as described above, CCND1 can be overexpressed through several other mechanisms. Activation of the RAS-MEK-ERK pathway, along with ERBB2 in hormonal-driven cancers (such as breast) and the Wnt pathway [8–10] have also been associated with higher CCND1 expression, that may occur through increasing its transcription [11, 12]. The PI3K-AKT-mTOR pathway also regulates CCND1 translation mechanisms [13–15] via mTOR and phosphorylation of downstream targets such as 4EBP1 (eukaryotic translation initiation factor 4E ) and S6K (ribosomal protein S6 kinase) [16, 17] (Figure 1).
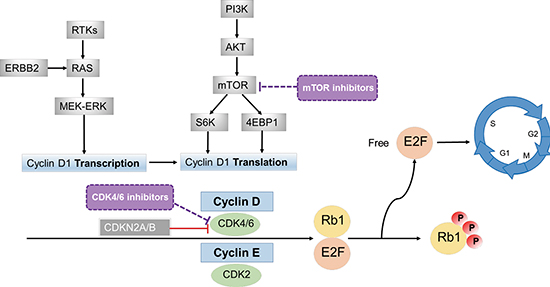
Figure 1: Effects of cyclins on cell cycle. By interacting with CDKs, cyclins form complexes (cyclin D with CDK4/6 and cyclin E with CDK2) that phosphorylate Rb1 (phosphorylated Rb1 is inactive). The Rb protein is a tumor suppressor that plays a pivotal role in the negative control of the cell cycle; Rb1 loss of function is frequently observed in cancer [48]. When Rb1 is phosphorylated, E2F is released and can transcriptionally activate its target genes, enabling the G1/S transition of cell cycle. Cyclin D1 (CCND1) can also be regulated at the transcription level by the RAS-MEK-ERK pathway and at the translation level by mTOR via S6K and 4EBP1 [11, 12, 16, 17]. mTOR inhibitors may attenuate CCND1 action by decreasing translation of CCND1. CDK4/6 inhibitors may attenuate the effects of this pathway as well, especially in the presence of CDK4/6 amplification or CDKN2A/B loss (since CDKN2A/B inhibits CDK4/6 activity). Patients with Rb1 loss or mutations would be expected to be resistant to agents such as mTOR or CDK4/6 inhibitors that act more proximally.
Cyclins have become increasingly relevant in cancer because drugs that can mitigate their effects are now available. Possible approaches to targeting cyclin-dependent cancers include use of CDK4/CDK6 inhibitors, either alone or in combination. Another approach might include use of mTOR inhibitors, since suppressing mTOR would attenuate the translation of CCND1 [18, 19]. Of interest in this regard, the mTOR inhibitor temsirolimus is approved in the European Union for the treatment of mantle cell lymphoma, a disease whose hallmark is CCND1/BCL-1 translocation, resulting in overexpression of CCND1 [20].
Though cyclin gene (CCN) amplifications are frequently observed in different cancers [1], the biological and clinical implications of these alterations have not been comprehensively studied. Hence, we used next generation sequencing in a population comprising 392 patients with cancer, aiming to elucidate the correlation between CCN alterations with clinical characteristics and outcome.
RESULTS
Patient characteristics
The clinical and demographic characteristics of our patient population (N = 392) can be found in Table 1. The median age of patients at diagnosis was 54 years (CI 95%, 53–56). Women comprised 57% (N = 222) of the population. The majority of patients were Caucasian (72%, N = 284). The most represented tumor types were gastro-intestinal (23%, N = 91), breast (21%, N = 81), and brain (14%, N = 56) cancers. The molecular testing was performed mainly on the primary tumor (59% of patients, N = 218).
CCN alterations and associations with clinical features
Overall, altered cyclin genes (CCND1, CCND2, CCND3, and CCNE1) were observed in 50 of the 392 patients tested (13%). There were a total of 53 CCN alterations found in 50 patients (three patients had two alterations), Supplemental Figure 1. All the CCN alterations identified were amplifications; the most frequent were CCND1 amplifications (55%, N = 29/53). In a univariate analysis, CCN alterations were significantly associated with women (17% vs 8%, women:men; P = 0.009). CCN alterations were also associated with breast cancer (N = 22/81 (27%) of breast cancer cases, P < 0.0001). Of note, no CCN alterations was observed in patients with brain cancers (P < 0.001). The biopsy site was more frequently a metastatic site when positive for a CCN amplification (18% vs 10%, metastatic vs. primary; P = 0.029), Table 1.
Table 1: Clinical characteristics of 392 patients with or without CCN amplifications (univariate analysis)
Characteristics |
Total patients, N = 392 |
CCN wild-type, N = 342 |
CCN amplified, N = 50 |
P-value |
Age at diagnosis (years) |
54.3 |
54.4 |
52.5 |
0.860 |
(Median, CI 95%) |
(52.5–56.0) |
(52.6–56.3) |
(48.8–58.1) |
|
Gender |
0.009 |
|||
Women |
222 |
185 (83%) |
37 (17%) |
|
Men |
170 |
157 (92%) |
13 (8%) |
|
Race |
||||
Caucasian |
284 |
250 (88%) |
34 (12%) |
0.498 |
Other |
57 |
47 (82%) |
10 (18%) |
|
Asian |
25 |
22 (88%) |
3 (12%) |
0.281 |
African American |
12 |
12 (100%) |
0 |
|
Unknown |
10 |
8 (80%) |
2 (20%) |
|
Hispanic |
4 |
3 (75%) |
1 (25%) |
|
Type of cancer |
||||
Gastro-intestinal |
91 |
81 (89%) |
10 (11%) |
0.720 |
Breast |
81 |
59 (73%) |
22 (27%) |
< 0.0001 |
Brain |
56 |
56 (100%) |
0 |
0.0004 |
Gynecologic |
33 |
31 (94%) |
2 (6%) |
0.287 |
Head and neck |
30 |
25 (83%) |
5 (17%) |
0.566 |
Liquid |
30 |
28 (93%) |
2 (7%) |
0.402 |
Melanoma |
29 |
27 (93%) |
2 (7%) |
0.560 |
Lung |
26 |
21 (81%) |
5 (19%) |
0.355 |
Othera |
16 |
14 (88%) |
2 (12%) |
1.000 |
Biopsy site used for molecular testingb: |
0.029 |
|||
Primary |
218 |
196 (90%) |
22 (10%) |
|
Metastatic |
149 |
122 (82%) |
27 (18%) |
aEwing sarcoma, carcinoid tumor, sarcomatoid tumor, peripheral nerve sheath tumor, pleiomorphic cell sarcoma (thigh), soft tissue liposarcoma (N = 2), soft tissue rhabdomyosarcoma, pleomorphic liposarcoma, and unknown origin (n = 7).
bFor 25 samples, the origin of the biopsy site used for molecular testing was unavailable.
CCN amplifications and direct co-alterations
In the overall population, the median number of molecular alterations per patient was four (range 0–16), and it was significantly higher in patients harboring CCN alterations (median of eight alterations compared to three in patients without CCN alterations, P < 0.0001). In a univariate analysis, CCN alterations were associated with FGF/FGFR alterations (amplification/mutations identified in FGF3/4/6/10/14/19/23 and FGFR1/2/3/4) (P < 0.0001), ZNF217/ZNF703 (P < 0.0001), AURKA (P = 0.001), ARFRP1(P = 0.003), EMSY (P = 0.007), and MDM2 (P = 0.030) amplifications, as well as amplification/mutations in MET (P = 0.016) or AKT1/2 (P = 0.023), Supplemental Table 1.
Multiple logistic regression analysis of factors associated with CCN amplifications
In a multiple logistic regression model (that included any parameters that were significant (P < 0.05) in univariate analysis), the only variables that remained statistically associated with CCN amplifications were aberrations in FGF/FGFR (P < 0.0001), MET (P = 0.003), and ARFRP1 (P = 0.032). The negative association with brain tumors also remained significant (P = 0.044) Table 2.
CCN extended co-amplification network
We further explored the ramifications of alteration associations, and found, in a multiple logistic regression model, that in addition to its association with CCN amplifications (P < 0.0001), FGF/FGFR associated strongly with RICTOR (P < 0.0001). FGF/FGFR also correlated with ARID1A (P = 0.033), ZNF217/703 (P = 0.019), MYST3 (P = 0.034), and ARID1A (P = 0.033). MET alterations associated with RICTOR (P = 0.011) and NFKBIA (P = 0.032), and ARFRP1 strongly correlated with both ZNF217/703 (P < 0.0001) and AURKA (P = 0.006). Lastly, we investigated the associations with ZNF217/703 amplifications and found strong correlations with AURKA (P < 0.0001), ARFRP1 (P = 0.002), FGF/FGFR (P = 0.004), ERBB2/3/4 (P = 0.044), and MYST3 (P = 0.002), Figure 2A and Supplemental Table 2.
Since the alterations with a very strong association (P < 0.0001) were amplifications, we examined the chromosome location of these genes and found patterns of co-localization in the same chromosome regions. Indeed, the strong association observed between CCN and FGF/FGFR family genes can be partly explained by the co-localization of CCND1 and FGF3/4/19 at the 11q13 locus. Of note, EMSY, that correlated with CCN alterations in the univariate analysis, is also found at the 11q13 locus. Additionally, CCND2 and FGF6/23 are both localized at 12p13. The strong association observed between FGF/FGFR and RICTOR can be explained by the co-localization of FGF10 and RICTOR at the 5p13 genomic location. FGFR1, ZNF703 and MYST3 are localized on 8p11 and ZNF207 was highly co-amplified with ARFRP1 and AURKA at the 20q13 locus, Figure 2B.
Table 2: Multiple logistic regression model for clinical characteristics associated with CCN amplifications
Characteristics |
P-Value |
Odds Ratio |
95% CI |
Gender (women vs. men) |
0.410 |
1.58 |
0.53–4.69 |
Histology |
|||
Breast |
0.913 |
1.06 |
0.36–3.15 |
Braina |
0.044 |
0.26 |
0.05–1.51 |
Site molecular testing (metastatic vs. primary) |
0.248 |
1.75 |
0.68–4.52 |
Genomic alterationsb |
|||
FGF/FGFR |
< 0.0001 |
41.7 |
15.9–111.1 |
MYC |
0.247 |
1.93 |
0.64–5.85 |
ZNF217/703 |
0.475 |
1.70 |
0.40–7.25 |
MDM2 |
0.267 |
2.67 |
0.47–15.15 |
AKT1/2 |
0.271 |
2.55 |
0.48–13.51 |
AURKA |
0.390 |
3.26 |
0.22–47.62 |
ARFRP1 |
0.032 |
18.9 |
1.29–250.0 |
MET |
0.003 |
50.0 |
3.79–689.0 |
EMSY |
0.271 |
6.25 |
0.24–166.7 |
anegative association
bFGF/FGFR comprised amplification/mutations in FGF3/4/6/10/14/19/23 and FGFR1/2/3/4.
MYC, ZNF217/703, MDM2, AURKA, ARFRP1, EMSY alterations were all amplifications. AKT1/2 and MET alterations comprised amplification/mutations.
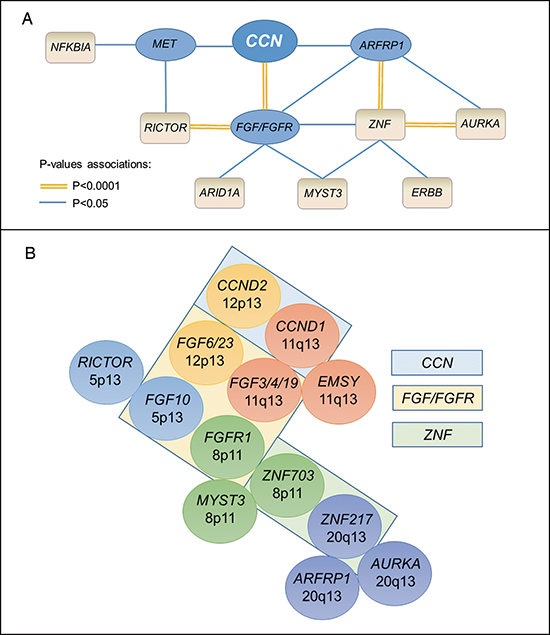
Figure 2: CCN and co-alterations. In panel A, the connectors represent a statistically significant association in a multiple logistic regression model (P < 0.05). The double orange connectors represent a correlation with a P-value < 0.0001; in that case, there was chromosomal co-localization (panel B). Cyclin genes were CCND1, CCND2, CCND3, and CCNE1; FGF/FGFR gene family comprised FGF3/4/6/10/14/19/23 and FGFR1/2/3/4; ERBB comprised ERBB2/3/4; ZNF comprised ZNF217 and ZNF703. In panel B, chromosome co-localizations are represented by the same color. Boxes contain genes that are related family members.
CCN amplifications and clinical outcome
In a univariate analysis, CCN amplifications correlated with the development of metastasis in the liver (P = 0.005), bone (P = 0.036), and brain (P = 0.047), Supplemental Table 3. The only association that remained significant in the multiple logistic regression model was the association with the development of liver metastasis (P = 0.046). Neither the overall survival, nor the time to metastasis/recurrence were statistically different depending on CCN alteration status (HR = 1.17; P = 0.646 and HR = 1.08; P = 0.691, respectively). No differences in median best progression-free survival time (PFS) or PFS analysis by treatment type was observed when patients with, or without, CCN alterations were compared.
Survival analysis in the overall population
In our population of 392 patients, the median follow up time was 37.8 months (95%CI 30.9–44.8 months). In a univariate analysis, the survival was poorer for patients with gastro-intestinal histology (P < 0.0001), men (P = 0.036), or patients harboring a TP53 (P = 0.002), CDK (CDK4/6/2/CDKN2A/2B) (P = 0.005), PTEN (P < 0.0001), or EGFR (P = 0.030) alterations. Of importance, only the gastro-intestinal histology (P < 0.0001), PTEN (P < 0.0001), and CDK alterations (P = 0.041) remained statistically significant independent predictors for poorer survival in a Cox regression model, Table 3. TP53 mutational status was no longer a significant predictor of inferior survival, although a trend persisted with P = 0.087. CCN was not a survival predictor in univariate analysis (P = 0.646).
DISCUSSION
Cyclin amplifications are frequent in cancer. The percentage of cyclin (CCN) alteration varies by tumor type; for instance, in previous studies, CCND1 amplification ranged from 15–40% [1]. Our population had an overall 13% rate of CCN alterations, with most (55%) of them being CCND1 amplifications (Supplemental Figure 1). We found that CCN alterations were associated with breast cancer (27% of cases), which is consistent with the literature [1, 21, 22]. CCND1 overexpression was previously found to be associated with shorter patient survival in many cancers and is often correlated with increased risk of metastasis [23, 24]. In our population, the overall survival was not statistically different in the presence of CCN abnormalities, perhaps because of the small number of death events at the time of analysis (N = 9 deaths amongst 50 patients with cyclin alterations). In a Cox regression model, the variables independently associated with worse survival were gastrointestinal malignancies, consistent with previous publications in advanced cancer [25], and alterations in PTEN as well as CDK family genes (Table 3). Of note, TP53 alterations were no longer associated with a shorter overall survival, perhaps because of its association with gastrointestinal histology (P = 0.024).
Table 3: Overall survival associations in 392 patients with cancer
Univariable |
Multivariable |
|||
Characteristics |
HR (CI 95%) |
P-Value |
HR (CI 95%) |
P-Value |
Gender (men vs. women) |
0.59 (0.36–0.97) |
0.036 |
0.65 (0.38–1.09) |
0.107 |
Histology |
||||
Gastro-intestinal (N = 91) |
0.19 (0.09–0.37) |
< 0.0001 |
0.28 (0.16–0.49) |
< 0.0001 |
Breast (N = 80) |
0.63 (0.37–1.04) |
0.073 |
--- |
--- |
Brain (N = 56) |
1.23 (0.61–2.49) |
0.564 |
--- |
--- |
Gynecologic (N = 33) |
1.47 (0.64–3.38) |
0.359 |
--- |
--- |
Head & Neck (N = 30) |
0.58 (0.25–1.35) |
0.207 |
--- |
--- |
Liquid (N = 29) |
2.37 (0.94–5.93) |
0.065 |
--- |
--- |
Melanoma (N = 29) |
2.23 (0.99–5.02) |
0.053 |
--- |
--- |
Lung (N = 26) |
0.53 (0.17–1.65) |
0.269 |
--- |
--- |
Other (N = 16) |
0.75 (0.16–3.67) |
0.722 |
--- |
--- |
Genetic alteration typea |
||||
TP53 (N = 178) |
0.47 (0.29–0.75) |
0.002 |
0.64 (0.38–1.01) |
0.087 |
CDK (N = 104) |
0.43 (0.24–0.78) |
0.005 |
0.56 (0.32–0.98) |
0.041 |
KRAS (N = 63) |
0.55 (0.27–1.01) |
0.089 |
--- |
--- |
FGF/FGFR (N = 57) |
1.35 (0.74–2.47) |
0.324 |
--- |
--- |
PIK3CA (N = 53) |
1.26 (0.64–2.47) |
0.503 |
--- |
--- |
CCN (N = 50) |
1.17 (0.60–2.26) |
0.645 |
--- |
--- |
MYC (N = 48) |
0.68 (0.33–1.41) |
0.295 |
||
PTEN (N = 42) |
0.09 (0.03–0.23) |
< 0.0001 |
0.19 (0.10–0.36) |
< 0.0001 |
EGFR (N = 31) |
0.29 (0.09–0.89) |
0.030 |
0.95 (0.39–2.29) |
0.900 |
BRCA1/2 (N = 28) |
0.61 (0.24–1.55) |
0.300 |
--- |
--- |
APC (N = 24) |
0.30 (0.09–1.00) |
0.051 |
--- |
--- |
aIncluded alterations with at least 5 events (death) and N = 20 patients with the alteration.
CCN comprised CCND1/2/3 and CCNE1; CDK comprised CDK4/6, CDKN2A/B; FGF/FGFR comprised FGF3/4/6/10/14/19/23 and FGFR1/2/3/4. Only characteristics that were significant (P < 0.05) in the univariable analysis (log-rank test) were included in the multivariable analysis (Cox regression model).
We did not detect any difference in best PFS analysis when patients with, or without CCN amplifications were compared. Types of therapy examined comprised regimens that included gemcitabine, taxanes, platinums, bevacizumab, hormonal treatments, and everolimus (though the number of patients treated with the latter drug was small, thus limiting the power of the analysis) (Supplemental Table 3). Of interest, CCN alterations were more frequent in metastatic sites (p = 0.029; univariate anlaysis), and there was a higher incidence of these alterations in patients who developed liver metastases (multiple logistic regression analysis; p = 0.046) (Tables 1 and Supplemental Table 3). Of interest, some previous studies have also described an association between CCND2 and CCND3 with liver metastasis [26, 27]. Finally, patients harboring CCN amplifications had a median of eight molecular alterations compared to three in patients without CCN amplifications (P < 0.0001).
Of importance, we uncovered an extended network of co-amplifications in our study. Indeed, CCN gene amplifications correlated independently with FGF/FGFR (P < 0.0001), ARFRP1 (membrane associated GTPase) (p = 0.032) and MET alterations (P = 0.003) in a multiple logistic regression analysis. The association between CCN and FGFs is not surprising, considering that CCND1 and FGF3, 4 and 19 co-localize on the same amplicon on chromosome 11q13 [28]. Similarly, CCND2 and FGF6 and 23 co-localize on 12p13. A systematic dissection of the genomic associations at different levels (first and second level of association with CCN amplifications, Figure 2) revealed a more comprehensive network. Indeed, the co-localization involving FGFR1 and ZNF703 has been described [29], consistent with the univariate association between amplifications in CCN (which correlates with FGF/FGFR aberrations) and ZNFs. (ZNF products are zinc finger proteins, which bind DNA and, through this binding, regulate gene transcription). ZNF703, like FGFR1, is localized at the 8p11 locus and there is compelling evidence that ZNF703 is one of the driver genes in this amplified region [30], especially in breast cancers where ZNF703 expression seems to be induced by estrogens [31]. FGFR1 was the first reported oncogene in the 8p11 region, and its amplification and overexpression has been related to poor prognosis and endocrine resistance in breast cancer [32].
Relationships between cyclins and FGF/FGFRs have also been previously reported at the protein level. For instance, a study showed that FGFR4 contributed to the maintenance of CCND1 via the mTOR translation pathway, and several other studies demonstrated cooperation between FGFR and CCND1 [33]. Further, CCND1 can enhance tumor progression, at least in part, by an increased level of transcription of FGFR1 and FGFR2 via E2F [34]. In addition, Kwek et al. [29] suggested that CCND1 induced ZNF703 via the Rb/E2F pathway.
ZNF217 is localized on 20q13 and can be responsible for enhanced AKT phosphorylation [35]. Of interest, ZNF217 was identified as a marker of poor prognosis in breast cancer [36], was shown to drive epithelial-mesenchymal transition and invasion [36], as well as enhance ERBB3 transcription; the latter is required for ERBB2-dependent breast tumor proliferation, since the ERBB2-ERBB3 heterodimer is the functional “oncogenic unit” [37].
While little is known about ADP-ribosylation factor-related protein 1 gene (ARFRP1, anomalies of which correlated with those in CCN in multiple logistic regression analysis) and cancer [38], AURKA, (Aurora A kinase), which co-localizes with ARFRP1 and with ZNF217 on 20q13, appears to be more well characterized and is believed to control the G2/M transition [39]. Anomalies in AURKA and CCN correlated in our study in a univariate analysis. Finally, MET anomalies (which correlated with CCN amplifications; multiple logistic regression analysis) correlated with RICTOR abnormalities (univariate analysis), which in turn co-localized with FGF10 on 5p13. RICTOR is a component of the second mTOR complex, identified as mTORC2. The mTORC2 can phosphorylate protein kinases including AKT and PKCα, turning on a variety of cellular processes such as proliferation and survival [40].
These co-alterations might, to a certain extent, provide a selective advantage to the tumor. One possibility is that the multiple amplifications in this network cooperate synergistically to increase pathway activation and hence the likelihood of proliferation. Another could be that the contribution of each component to functions other than the cell cycle control may offer an additional selective advantage during oncogenesis. In addition, several studies demonstrated that CCND1 amplifications were linked to resistance mechanisms, such as endocrine resistance in breast cancers [41, 42], or BRAF inhibitor resistance in BRAF V600E mutated melanomas [43]. Further, CCND1 has also been implicated in acquired radioresistance [44].
Importantly, cyclins have been correlated with positive estrogen receptor (ER+) in breast cancers [28]. In our population, we observed that 31% (17/53) of breast specimens that were ER+ also harbored a CCN amplification. Due to the availability of anti-estrogen therapies, ER positivity is considered a marker of good prognosis. However, among patients with ER positive tumors, those also harboring CCND1 amplification seem to constitute a subgroup with higher grade and a relatively poor prognosis [45, 46]. Recently, the randomized phase 2 trial PALOMA-1 investigating the CDK4/6 inhibitor palbociclib demonstrated a statistically significant and clinically meaningful improvement in progression-free survival (PFS) for the combination of palbociclib and letrozole compared to letrozole alone (20.2 vs. 10.2 months, P = 0.0004) in women with ER+/HER2- metastatic breast cancer [47].
Our study had several limitations. First, correlations were made via a retrospective review. As such, the timing of response assessment was performed per the discretion of the attending physician and not completely uniform. Second, diverse histologies were included, and in some there were relatively small numbers of patients. On the other hand, these findings could imply that the impact of cyclin amplifications and co-amplification network is important across histologies. Finally, the molecular testing was done on biopsies obtained at different time points during the clinical course (median of nine months after diagnosis, CI 95% 6–14 months).
CCN amplifications appear to have biological implications, including an association with liver metastases, and patterns of molecular correlations, such as those between CCN aberrations and FGF/FGFR amplifications, as well as MET and ARFRP1 alterations. Further, patients harboring CCN amplifications had significantly higher numbers of molecular alterations. The ramifications of a co-amplification network are that these gene products may work together during tumor progression and require tailored combination therapies. As several oncogenic pathways seem to converge on the cyclin-associated pathway and cell cycle progression, the use of drugs to target the cyclin pathway, including CDK4/6 inhibitors, or mTOR inhibitors, have become increasingly relevant, particularly in combinations that can be appropriately customized to attenuate resistance mechanisms.
METHODS
Patients
We retrospectively reviewed the clinicopathology and clinical outcomes of 392 patients with advanced cancer (solid and hematologic tumors were considered) who were seen at the UC San Diego Moores Cancer Center and for whom molecular testing had been performed between October 2012 and April 2014. This study was performed and consents obtained in accordance with UCSD Institutional Review Board guidelines.
Next generation sequencing
Next generation sequencing was performed by Foundation Medicine (FoundationOne™, Cambridge, Massachusetts, http://www.foundationone.com), which is a clinical grade CLIA-approved next-generation sequencing test that sequences the entire coding sequence of 236 cancer-related genes and 47 introns from 19 genes often rearranged in cancer (full list available at http://foundationone.com/genelist1.php). (Nine patients (2.3%) were tested with an earlier version of the panel comprising 182 cancer–related genes). Gene chromosome localization were found using the “genecards” database (http://www.genecards.org/). For the purposes of this analysis, cyclin gene (CCN) status was examined for CCND1, CCND2, CCND3, and CCNE1.
Statistical analysis
Patients’ characteristics were summarized using descriptive statistics. Associations between categorical variables was done using Fisher’s exact tests. Time to metastasis/recurrence was defined as the time interval between diagnosis and first metastasis/recurrence, whichever came first. Overall survival (OS) was defined as the time from diagnosis to death or last follow-up date for patients who were alive. Progression-free survival (PFS) was defined as the time from the beginning of a given therapy to progression or treatment discontinuation for any reason. The data for best progression-free survival was available for 246 patients of 392 (63%). Best PFS was defined as the longest PFS achieved on treatment. Patients who did not progress were censored at the last follow up date. Estimations for OS and PFS were done using a Kaplan-Meier analysis, and were compared among subgroups by the log-rank test. Cox regression model were fit to assess the association between OS and patients’ characteristics and CCN alteration status. Multiple logistic regressions were fit to analyze the association between CCN alterations and other patients’ characteristics. Only variables with P-values less than 0.05 were included in the multiple regression models. All statistical analysis were performed by MS with SPSS version 22.0.
ACKNOWLEDGEMENTS
Funded in part by the Joan and Irwin Jacobs Fund and My Answer To Cancer philanthropic fund.
Dr. Shimabukuro reports personal fees from Genomic Health, outside the submitted work. The other authors have nothing to disclose.
REFERENCES
1. Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer. 2011 Aug; 11:558–72.
2. Caldon CE, Musgrove EA. Distinct and redundant functions of cyclin E1 and cyclin E2 in development and cancer. Cell Div. 2010 Jan 17; 5:2.
3. Arnold A, Kim HG, Gaz RD, Eddy RL, Fukushima Y, Byers MG, Shows TB, Kronenberg HM. Molecular cloning and chromosomal mapping of DNA rearranged with the parathyroid hormone gene in a parathyroid adenoma. J Clin Invest. 1989 Jun; 83:2034–40.
4. Motokura T, Bloom T, Kim HG, Jüppner H, Ruderman JV, Kronenberg HM, Arnold A. A novel cyclin encoded by a bcl1-linked candidate oncogene. Nature. 1991 Apr 11; 350:512–5.
5. Rosenberg CL, Kim HG, Shows TB, Kronenberg HM, Arnold A. Rearrangement and overexpression of D11S287E, a candidate oncogene on chromosome 11q13 in benign parathyroid tumors. Oncogene. 1991 Mar; 6:449–53.
6. Bigoni R, Negrini M, Veronese ML, Cuneo A, Castoldi GL, Croce CM. Characterization of t(11;14) translocation in mantle cell lymphoma by fluorescent in situ hybridization. Oncogene. 1996 Aug 15; 13:797–802.
7. Meeker TC, Sellers W, Harvey R, Withers D, Carey K, Xiao H, Block AM, Dadey B, Han T. Cloning of the t(11;14)(q13;q32) translocation breakpoints from two human leukemia cell lines. Leukemia. 1991 Sep; 5:733–7.
8. Rimerman RA, Gellert-Randleman A, Diehl JA. Wnt1 and MEK1 cooperate to promote cyclin D1 accumulation and cellular transformation. J Biol Chem. 2000 May 12; 275:14736–42.
9. Rhee C-S, Sen M, Lu D, Wu C, Leoni L, Rubin J, Corr M, Carson DA. Wnt and frizzled receptors as potential targets for immunotherapy in head and neck squamous cell carcinomas. Oncogene. 2002 Sep 26; 21:6598–605.
10. Willert J, Epping M, Pollack JR, Brown PO, Nusse R. A transcriptional response to Wnt protein in human embryonic carcinoma cells. BMC Dev Biol. 2002 Jul 2; 2:8.
11. Lee RJ, Albanese C, Fu M, D’Amico M, Lin B, Watanabe G, Haines GK 3rd, Siegel PM, Hung MC, Yarden Y, Horowitz JM, Muller WJ, Pestell RG. Cyclin D1 is required for transformation by activated Neu and is induced through an E2F-dependent signaling pathway. Mol Cell Biol. 2000 Jan; 20:672–83.
12. Desai KV, Xiao N, Wang W, Gangi L, Greene J, Powell JI, Dickson R, Furth P, Hunter K, Kucherlapati R, Simon R, Liu ET, Green JE. Initiating oncogenic event determines gene-expression patterns of human breast cancer models. Proc Natl Acad Sci U S A. 2002 May 14; 99:6967–72.
13. Liang J, Slingerland JM. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle Georget Tex. 2003 Aug; 2:339–45.
14. Averous J, Fonseca BD, Proud CG. Regulation of cyclin D1 expression by mTORC1 signaling requires eukaryotic initiation factor 4E-binding protein 1. Oncogene. 2007 Aug 27; 27:1106–13.
15. Steelman LS, Chappell WH, Abrams SL, Kempf RC, Long J, Laidler P, Mijatovic S, Maksimovic-Ivanic D, Stivala F, Mazzarino MC, Donia M, Fagone P, Malaponte G, Nicoletti F, Libra M, Milella M, Tafuri A, Bonati A, Bäsecke J, Cocco L, Evangelisti C, Martelli AM, Montalto G, Cervello M, McCubrey JA. Roles of the Raf/MEK/ERK and PIK/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging. 2011 Mar; 3:192–222.
16. Inoki K, Ouyang H, Li Y. Signaling by target of rapamycin proteins in cell growth control. Microbiol Mol Biol Rev MMBR. 2005 Mar; 69:79–100.
17. Lang CH, Frost RA. Endotoxin disrupts the leucine-signaling pathway involving phosphorylation of mTOR, 4E-BP1, and S6K1 in skeletal muscle. J Cell Physiol. 2005 Apr; 203:144–55.
18. Vignot S, Faivre S, Aguirre D, Raymond E. mTOR-targeted therapy of cancer with rapamycin derivatives. Ann Oncol. 2005 Apr 1; 16:525–37.
19. Takuwa N, Fukui Y, Takuwa Y. Cyclin D1 Expression Mediated by Phosphatidylinositol 3-Kinase through mTOR-p70S6K-Independent Signaling in Growth Factor-Stimulated NIH 3T3 Fibroblasts. Mol Cell Biol. 1999 Feb; 19:1346–58.
20. Hess G, Herbrecht R, Romaguera J, Verhoef G, Crump M, Gisselbrecht C, Laurell A, Offner F, Strahs A, Berkenblit A, Hanushevsky O, Clancy J, Hewes B, Moore L, Coiffier B. Phase III study to evaluate temsirolimus compared with investigator’s choice therapy for the treatment of relapsed or refractory mantle cell lymphoma. J Clin Oncol Off J Am Soc Clin Oncol. 2009 Aug 10; 27:3822–9.
21. Roy PG, Pratt N, Purdie CA, Baker L, Ashfield A, Quinlan P, Thompson AM. High CCND1 amplification identifies a group of poor prognosis women with estrogen receptor positive breast cancer. Int J Cancer J Int Cancer. 2010 Jul 15; 127:355–60.
22. Arnold A, Papanikolaou A. Cyclin D1 in Breast Cancer Pathogenesis. J Clin Oncol. 2005 Jun 20; 23:4215–24.
23. Thomas GR, Nadiminti H, Regalado J. Molecular predictors of clinical outcome in patients with head and neck squamous cell carcinoma. Int J Exp Pathol. 2005 Dec; 86:347–63.
24. Li Y, Wei J, Xu C, Zhao Z, You T. Prognostic Significance of Cyclin D1 Expression in Colorectal Cancer: A Meta-Analysis of Observational Studies. PLoS ONE. 2014 Apr 11; 9:e94508.
25. Wheler J, Tsimberidou AM, Hong D, Naing A, Falchook G, Piha-Paul S, Fu S, Moulder S, Stephen B, Wen S, Kurzrock R. Survival of 1, 181 patients in a phase I clinic: the MD Anderson Clinical Center for targeted therapy experience. Clin Cancer Res Off J Am Assoc Cancer Res. 2012 May 15; 18:2922–9.
26. Sarkar R, Hunter IA, Rajaganeshan R, Perry SL, Guillou P, Jayne DG. Expression of cyclin D2 is an independent predictor of the development of hepatic metastasis in colorectal cancer. Colorectal Dis. 2010 Apr 1; 12:316–23.
27. Tanami H, Tsuda H, Okabe S, Iwai T, Sugihara K, Imoto I, Inazawa J. Involvement of cyclin D3 in liver metastasis of colorectal cancer, revealed by genome-wide copy-number analysis. Lab Invest. 2005 Jun 27; 85:1118–29.
28. Karlsson E, Waltersson MA, Bostner J, Pérez-Tenorio G, Olsson B, Hallbeck A-L, Stål O. High-resolution genomic analysis of the 11q13 amplicon in breast cancers identifies synergy with 8p12 amplification, involving the mTOR targets S6K2 and 4EBP1. Genes Chromosomes Cancer. 2011 Oct; 50:775–87.
29. Kwek SS, Roy R, Zhou H, Climent J, Martinez-Climent JA, Fridlyand J, Albertson DG. Co-amplified genes at 8p12 and 11q13 in breast tumors cooperate with two major pathways in oncogenesis. Oncogene. 2009 Apr 30; 28:1892–903.
30. Spellman P, Gray J. A new treasure in the breast cancer gene hunt. Nat Med. 2011 Apr; 17:422–3.
31. Sircoulomb F, Nicolas N, Ferrari A, Finetti P, Bekhouche I, Rousselet E, Lonigro A, Adélaïde J, Baudelet E, Esteyriès S, Wicinski J, Audebert S, Charafe-Jauffret E, Jacquemier J, Lopez M, Borg J-P, Sotiriou C, Popovici C, Bertucci F, Birnbaum D, Chaffanet M, Ginestier C. ZNF703 gene amplification at 8p12 specifies luminal B breast cancer. EMBO Mol Med. 2011 Mar; 3:153–66.
32. Turner N, Pearson A, Sharpe R, Lambros M, Geyer F, Lopez-Garcia MA, Natrajan R, Marchio C, Iorns E, Mackay A, Gillett C, Grigoriadis A, Tutt A, Reis-Filho JS, Ashworth A. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 2010 Mar 1; 70:2085–94.
33. Koziczak M, Hynes NE. Cooperation between Fibroblast Growth Factor Receptor-4 and ErbB2 in Regulation of Cyclin D1 Translation. J Biol Chem. 2004 Nov 26; 279:50004–11.
34. Tashiro E, Maruki H, Minato Y, Doki Y, Weinstein IB, Imoto M. Overexpression of cyclin D1 contributes to malignancy by up-regulation of fibroblast growth factor receptor 1 via the pRB/E2F pathway. Cancer Res. 2003 Jan 15; 63:424–31.
35. Huang G, Krig S, Kowbel D, Xu H, Hyun B, Volik S, Feuerstein B, Mills GB, Stokoe D, Yaswen P, Collins C. ZNF217 suppresses cell death associated with chemotherapy and telomere dysfunction. Hum Mol Genet. 2005 Nov 1; 14:3219–25.
36. Vendrell JA, Thollet A, Nguyen NT, Ghayad SE, Vinot S, Bièche I, Grisard E, Josserand V, Coll J-L, Roux P, Corbo L, Treilleux I, Rimokh R, Cohen PA. ZNF217 is a marker of poor prognosis in breast cancer that drives epithelial-mesenchymal transition and invasion. Cancer Res. 2012 Jul 15; 72:3593–606.
37. Krig SR, Miller JK, Frietze S, Beckett LA, Neve RM, Farnham PJ, Yaswen PI, Sweeney CA. ZNF217, a candidate breast cancer oncogene amplified at 20q13, regulates expression of the ErbB3 receptor tyrosine kinase in breast cancer cells. Oncogene. 2010 Oct 7; 29:5500–10.
38. Shin H-W, Kobayashi H, Kitamura M, Waguri S, Suganuma T, Uchiyama Y, Nakayama K. Roles of ARFRP1 (ADP-ribosylation factor-related protein 1) in post-Golgi membrane trafficking. J Cell Sci. 2005 Sep 1; 118:4039–48.
39. Nikonova AS, Astsaturov I, Serebriiskii IG, Dunbrack RL Jr, Golemis EA. Aurora A kinase (AURKA) in normal and pathological cell division. Cell Mol Life Sci CMLS. 2013 Feb; 70:661–87.
40. Agarwal NK, Chen C-H, Cho H, Boulbès DR, Spooner E, Sarbassov DD. Rictor regulates cell migration by suppressing RhoGDI2. Oncogene. 2013 May 16; 32:2521–6.
41. Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer. 2009 Sep; 9:631–43.
42. Kilker RL, Hartl MW, Rutherford TM, Planas-Silva MD. Cyclin D1 expression is dependent on estrogen receptor function in tamoxifen-resistant breast cancer cells. J Steroid Biochem Mol Biol. 2004 Sep; 92:63–71.
43. Smalley KSM, Lioni M, Dalla Palma M, Xiao M, Desai B, Egyhazi S, Hansson J, Wu H, King AJ, Van Belle P, Elder DE, Flaherty KT, Herlyn M, Nathanson KL. Increased cyclin D1 expression can mediate BRAF inhibitor resistance in BRAF V600E-mutated melanomas. Mol Cancer Ther. 2008 Sep; 7:2876–83.
44. Shimura T, Noma N, Oikawa T, Ochiai Y, Kakuda S, Kuwahara Y, Takai Y, Takahashi A, Fukumoto M. Activation of the AKT/cyclin D1/Cdk4 survival signaling pathway in radioresistant cancer stem cells. Oncogenesis. 2012 Jun 4; 1:e2.
45. Kenny FS, Hui R, Musgrove EA, Gee JM, Blamey RW, Nicholson RI, Sutherland RL, Robertson JF. Overexpression of cyclin D1 messenger RNA predicts for poor prognosis in estrogen receptor-positive breast cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 1999 Aug; 5:2069–76.
46. Al-Kuraya K, Schraml P, Torhorst J, Tapia C, Zaharieva B, Novotny H, Spichtin H, Maurer R, Mirlacher M, Köchli O, Zuber M, Dieterich H, Mross F, Wilber K, Simon R, Sauter G. Prognostic relevance of gene amplifications and coamplifications in breast cancer. Cancer Res. 2004 Dec 1; 64:8534–40.
47. palblociclib press release. Available from: http://www.pfizer.com/news/press-release/press-release-detail/pfizer_announces_positive_top_line_results_from_paloma_1_evaluating_palbociclib_plus_letrozole_in_women_with_
advanced_breast_cancer.
48. Giacinti C, Giordano A. RB and cell cycle progression. Oncogene. 2006 Aug 28; 25:5220–7.