
Introduction
Breast cancer is the most diagnosed malignancy worldwide and a leading cause of cancer-related death in women [1]. Recent advancements in diagnostic and therapeutic modalities have led to improved survival and prognosis. Tumor metastasis is one of the driving factors for treatment failure and mortality from cancer with underlying molecular mechanism still poorly understood [2, 3]. One of such alterations includes loss of tumor suppressor gene [4]. Treatment modalities aimed at halting or possibly reversing the molecular pathway leading to metastasis hold promise for effectively treating cancers.
Breast cancers can be broadly divided as per their hormone receptor (HR) and human epidermal growth factor receptor 2 (HER2) status into HR positive, HER2 positive and triple negative breast cancer (TNBC) [5]. In addition to surgery, radiation therapy, endocrine therapy and hormone therapy, tumor-tailored treatment can be provided with therapies targeting HER2, PIK3CA, TRK, CDK4/6, BRCA1/2, and VEGF and PDL1 receptors. Research is underway for multiple other promising therapies [6, 7].
5′Methylthioadenosine phosphorylase (MTAP) is a key enzyme in the polyamine pathway and aids in catabolism of 5′Deoxy-5′-Methythioadenosine (MTA) leading to formation of methionine and adenine. MTAP gene is located at 9P21 surrounded by miR-31 and CDK2NA and has been reported to serve as a tumor suppressor gene [8–10]. MTAP deletion leads to low levels of adenine leading to cellular dependence on de novo purine synthesis and accumulation of MTA which in turn inhibits PRMT5 [11]. Most tumor cells have MTAP, P16 and other tumor suppressor genes located on 9P21 such as CDKN2A and CDKN2B making it a poor target for therapeutic regimens [11]. MTA accumulation in MTAP deleted cells creates a hypomorphic PRMT5 state that is sensitized towards further PRMT5 inhibition making PRMT5 inhibitors a potential therapy for MTAP deleted cancers [8]. PRMT5 inhibition leads to reduced histone methylation of which eventually leads to decrease FOXP1 expression (Figure 1). This not only creates sensitivity to PRMT5 targeting, but also leads to cell apoptosis and decreased metastasis [12, 13]. MTAP downregulation also promotes tumor metastasis by activating the GSK3B/slug/E-cadherin axis in esophageal squamous cell carcinoma [14]. In breast cancer, MTAP downregulation activates ornithine decarboxylase (ODC) which in turn leads to formation of putrescine which promotes tumor migration, invasion and angiogenesis [15]. Cytotoxicity assays with inhibitors of de novo adenine synthesis, 5-fluorouracil (5-FU), methotrexate (MTX) and 5′aza-deoxycytidine (AZA) after MTAP gene knockdown in breast cancer cell lines have shown an increased sensitivity to 5-FU [4].
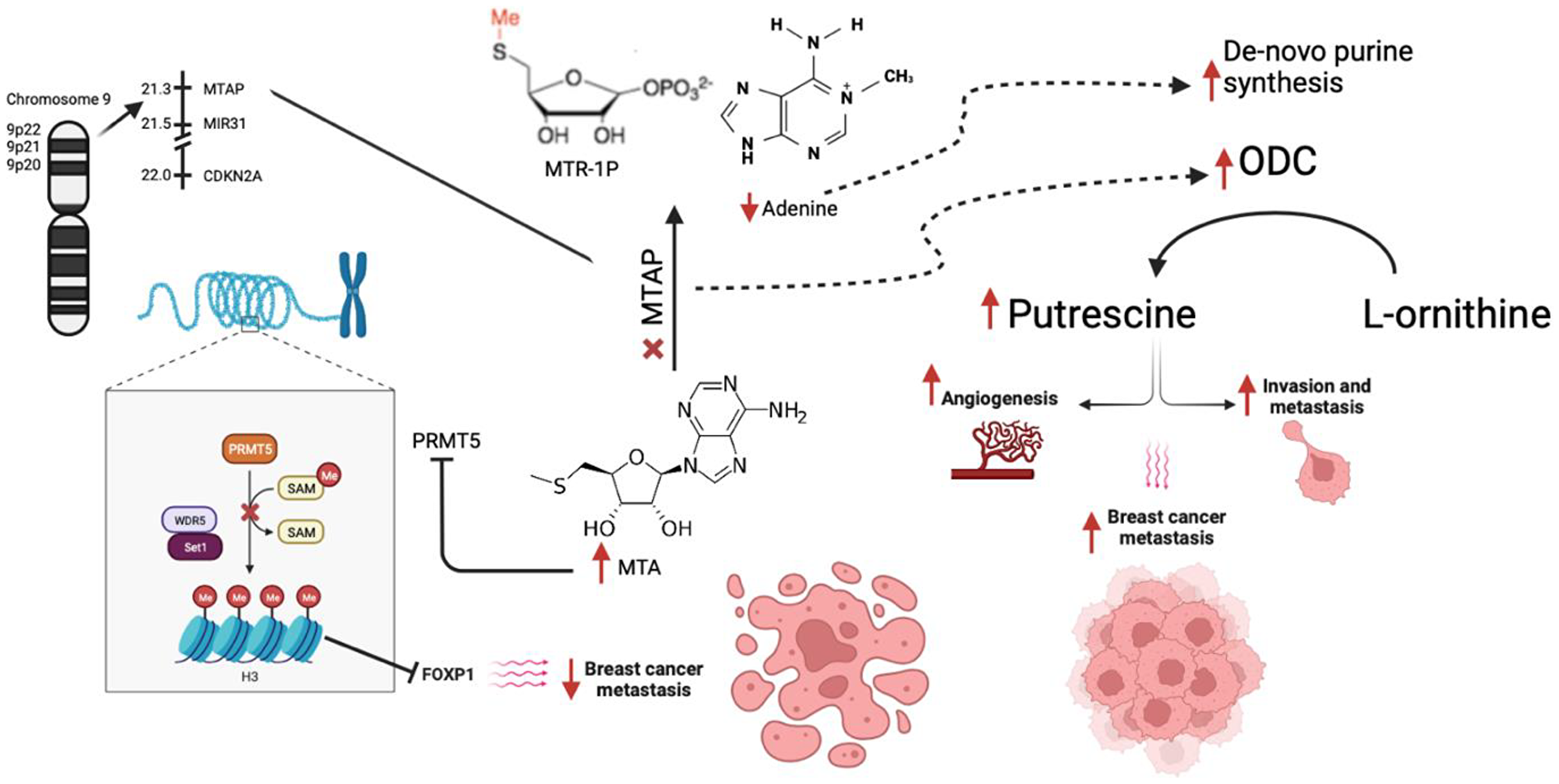
Figure 1: Mechanism of action of MTAP along with downstream effects from its loss. Abbreviations: MTAP: methylthioadenosine phosphorylase; MTR1P: 5-methylthioribose-1-phosphate; ODC: Ornithine decarboxylase; PRMT5: Protein Arginine Methyltransferase 5.
Results
Overall, 7301 cases of metastatic breast cancer underwent hybrid capture based comprehensive genomic profiling (CGP). 208 patients out of 7301 (2.84%) were noted to have MTAP loss (Table 1). The median age of patients with MTAP loss was 54.5 years compared to 57.8 years in MTAP intact (P = 0.002). Tumors with MTAP loss were noted to have lesser ER expression (50%) compared to MTAP intact (70%) (p < 0.001). Similarly, HER2 expression was less frequently noted in MTAP loss tumors as well (1.92% vs 7.8% in MTAP intact, p < 0.05). Triple negative status was noted more frequently noted in MTAP loss (47.28%) than MTAP intact (27%) (p < 0.05).
Table 1: Targetable and non-targetable GA along with number of cases in our cohort and their characteristics
| Cases with MTAP Intact | Cases with MTAP Loss | ||
|---|---|---|---|
| Number of Cases | 7093 | 208 | |
| Mean Age* | 57.8 | 54.5 | |
| ER+/PR+Status by IHC** | 70.0%/49.0% | 50.00%/29.90% | |
| HER2+Amplification by CGP* | 7.80% | 1.92% | |
| TNBC Status* | 27.00% | 47.28% | |
| Driver Alterations/sample** | 5.7 | 8.81 | |
| Non-targetable GA (%) | |||
| TP53* | 51.70 | 61.30 | |
| CDKN2A** | 3.10 | 100.00 | |
| CDKN2B** | 1.30 | 96.70 | |
| RB1* | 7.20 | 1.40 | |
| CDH1* | 14.30 | 0.90 | |
| Targetable GA (%) | |||
| PTEN* | 13.10 | 21.70 | |
| PIK3CA** | 36.8 | 23.60 | |
| NF1 | 6.40 | 9.90 | |
| BRCA1** | 3.70 | 9.90 | |
| ERBB2 amplification* | 7.80 | 1.92 | |
| ERBB2 sequence mutation* | 11.20 | 6.60 | |
| EGFR* | 2.60 | 5.20 | |
| Immuno-Oncology Drug Biomarkers | |||
| MSI High | Frequency | 0.03% | 0.05% |
| Cases Tested | 7077 | 205 | |
| CD274 (PD-L1) Amp | 1.10% | 2.80% | |
| STK11 Inactivating GA | 1.50% | 4.20% | |
| Median TMB | 2.5 | 2.5 | |
| TMB >10%/>20% | 7.84%/7.40% | 5.32%/0.96% | |
| PD-L1 Positive IC Expression (Dako 22C3) | Low (1–49%)* | 11.45% | 42.90% |
| High (> 50%) | 2.86% | 0.00% | |
Among currently non-targetable mutations, MTAP loss tumors had higher frequency of TP53 (61.30 % vs. 51.70, p < 0.05), CDKN2A (100% vs. 3.10%, p < 0.001) and CDKN2B (96.70% vs. 1.30%, p < 0.001) MTAP intact tumors had higher frequency of RB1 (7.20% vs. 1.40%, p < 0.05) and CDH1 (14.30% vs. 0.90%, p < 0.05).
Among targetable mutations, MTAP loss tumors had higher frequency of PTEN (21.70% vs. 13.10%, p < 0.05), BRCA1 (9.90% vs. 3.70%, p < 0.001) and EGFR mutation (5.20% vs. 2.60%, p < 0.05). MTAP intact tumors had higher frequency of PIK3CA (36.8% vs. 23.60%, p < 0.001), ERBB2 amplification (7.80% vs. 1.92%, p < 0.05) and ERBB2 sequence mutation (11.20% vs. 6.60%, p < 0.05). The expression of NF1 was not statistically significant with 9.90% in MTAP loss vs. 6.40 in MTAP intact.
The tumors were also tested for checkpoint inhibitor biomarkers. The analysis included 205 out of 208 MTAP loss and 7077 out of 7093 MTAP intact tumors. The PDL-1 low status analyzed by Dako 22C3 was statistically higher in MTAP loss compared to MTAP intact tumors (42.90% vs. 11.45%, p < 0.05). PDL-1 high status (>50%) was not noted in the MTAP loss tumors and 2.86% in MTAP intact, however this finding was not statistically significant. These results can be seen in Figure 2.
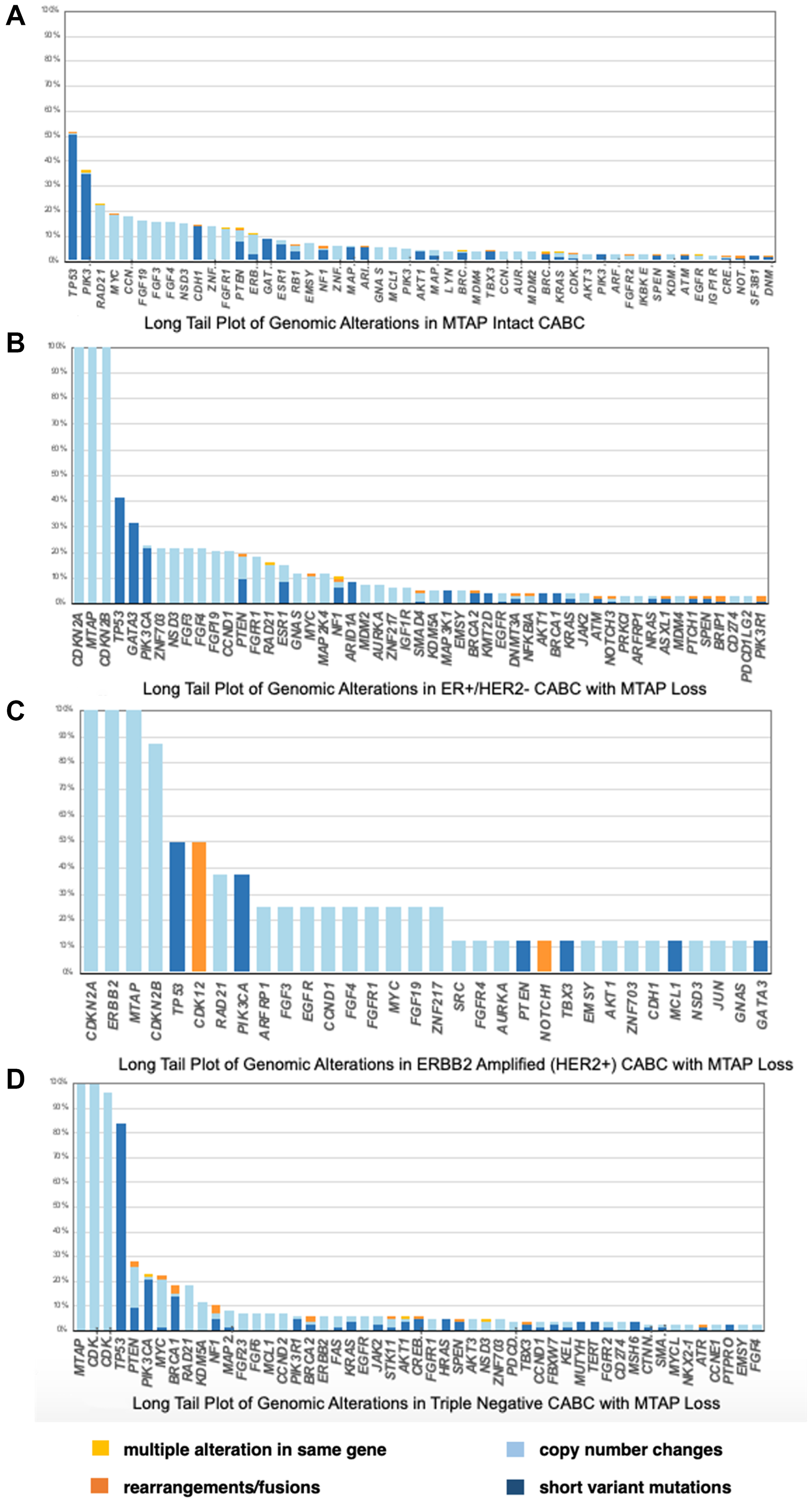
Figure 2: (A) Long Tail Plot of Genomic Alterations in MTAP Intact clinically advanced breast cancer. (B) Long Tail Plot of Genomic Alterations in ER+/HER2− clinically advanced breast cancer with MTAP Loss. (C) Long Tail Plot of Genomic Alterations in ERBB2 Amplified (HER2+) clinically advanced breast cancer with MTAP Loss. (D) Long Tail Plot of Genomic Alterations in Triple Negative clinically advanced breast cancer with MTAP Loss.
DISCUSSION
MTAP is an important enzyme found in almost all tissues in the body. It is an important enzyme in the methionine salvage pathway, responsible for regenerating methionine and adenine, which is in turn essential for the cell cycle [13]. Following the concepts of synthetic lethality, when MTAP is lost in a tumor cell, MTA will build up inside the cell leading to more suppression of PRMT5, thereby increasing their vulnerability to inhibition [16, 17]. PRMT5 inhibitor molecules like GSK332659513, PRT81114 and JNJ-6461917815 are currently under investigation in advanced solid malignancies including breast cancer. Safety data on GSK3326595, were recently reported by the phase 1 METEOR-1 trial (NCT02783300) which included breast cancer patients. Phase 2 portion of the study is currently underway [18]. PRMT5 is an inhibitor of tumor suppressor genes and thus enables the unchecked proliferation of cancer cells. PRMT5 induces methylation of p53 and disrupts its ability to cause death of malignant cells. It also promotes cyclin kinase-dependent neoplastic growth. The clinical paradigm of MTAP deficient cells, by building up MTA, which is a potent inhibitor of PRTM5, was studied as early as 1981 [18]. Another evolving target of interest are the methionine adenosyltransferases MAT1a and MAT2a. They are important cofactors in the polyamine biosynthesis cycle and play an essential role in the growth and survival of cells. In vivo models have shown that MAT2a knockdown reduced the growth and development of MTAP deficient tumor cells [19]. IDE397, a small molecule inhibitor of MAT2A, is under investigation as a part of a Phase 1 trial for advanced solid tumors with MTAP deletion [20].
In our cohort of MBC, the frequency of MTAP loss was 2.84% (208/7301). In the COSMIC gene database, MTAP copy number variation loss was reported in 2.55% (38/1492) breast cancer samples [21]. In the AACR GENIE portal, MTAP deletion was reported in 2.9% (138/15210) breast cancer specimens [4, 22]. These findings are consistent with our analysis. Although literature on MTAP loss in breast cancer is scarce, MTAP and CDKN2A loss co-occur with concordance in up to 90% of the cases, enabling indirect estimation of MTAP loss. A study evaluating this, estimated MTAP loss at around 16% (19/119) from frozen section specimens. Larger datasets as mentioned above revealed a lower number, suggesting that the small size could be the limitation of this study [4].
We provide one of the first large analyses of the spectrum of GA occurring in MTAP deleted MBC with the hope that this would enable identifying potential therapeutic agents in the future.
Even though breast cancer is becoming more common worldwide, its prognosis has improved thanks to advances in early detection and treatment. Currently, distant metastasis has the largest influence on a breast cancer patient’s prognosis. The five-year survival rate of breast cancer patients without metastasis is over 80% [23], but that of patients with metastasis is only around 25% [2, 3]. Yet, the molecular basis for the spread of breast cancer remains poorly understood.
In many human malignancies, such as leukemia [24], lymphoma [25], lung cancer [26], pancreatic cancer [27], and melanoma [28, 29], MTAP is frequently suppressed or absent, making it a potential target for cancer treatment. However, uncertainty still exists regarding MTAP’s clinical and biological impact on breast cancer metastasis.
Similarly, Zhang et al. revealed for the first time a positive correlation between reduced MTAP expression and tumor recurrence in breast cancer patients, indicating that MTAP may be crucial to the malignant development of breast cancer [15]. This study showed that, in an orthotopic breast cancer model using BT20 cells, MTAP downregulation could greatly accelerate both tumor development and metastasis. MTAP expression also differs by breast cancer type: studies have shown that TNBC cells express significantly less MTAP than the more differentiated group made up of Luminal-A breast tumors, this would open the door for novel therapeutical strategies for the treatment of TNBC where endocrine or targeted therapy are usually ineffective [30].
The results of cytotoxicity assays using inhibitors of de novo adenine synthesis (5-FU, AZA, and MTX) after MTAP gene knockdown showed an increased sensitivity, primarily to 5-FU [4]. Vieira de Oliveira also evaluated MTAP expression in two groups of breast cancer patient samples, including fresh tumors and paired normal breast tissue, as well as formalin-fixed paraffin embedded (FFPE) core breast cancer samples diagnosed as Luminal-A tumors and TNBC. Although the difference in MTAP expression between fresh tumors and normal tissue was not statistically significant, MTAP expression was significantly higher in Luminal-A breast tumors compared to TNBC. This suggests that a lack of MTAP expression is associated with more aggressive breast tumors and may support the development of new therapeutic approaches based on MTAP status in TNBC. In our study, BRCA1 mutation was more frequent in MTAP loss MBC (10% vs. 4%; p < 0.0001) which was likely associated with the increased TNBC cases.
There is growing evidence that MTAP can control tumor invasion and migration through many signaling mechanisms. In esophageal cancer, MTAP depletion can activate the GSK3/Slug/E-cadherin axis, promoting migration and invasion [14]. In colorectal cancer, downregulation of MTAP can also influence the epithelial-to-mesenchymal shift and stimulate tumor growth and metastasis [30]. When MTAP is downregulated in melanoma, 5′-methylthioadenosine (MTA) builds up and promotes tumor spread by preventing protein methylation and activating the extracellular signal-regulated kinase (ERK) signal [28]. Emerging studies show that MTAP overexpression dramatically changes the amounts of polyamine metabolites (particularly putrescine) in breast cancer cells [15]. These results are confirmed by the fact that re-expressing MTAP causes loss of anchorage-independent growth in vitro and loss of tumor development in vivo in MCF-7 breast cells that have had MTAP deleted [31].
MTA is a byproduct of the production of polyamines, and MTAP is the only metabolic enzyme that breaks it down into adenine and methylthioribose-1-phosphate (MTR-1-P) [32]. ODC, being the rate limiting enzyme in putrescine formation, is regarded as an independent predictor of a poor clinical outcome in breast cancer [24, 33–35]. ODC and polyamine metabolism have been linked to the proliferation and spread of tumor cells, according to numerous research [36–39]. Overexpression of ODC was strikingly linked with lymph node metastases, lymphovascular invasion in esophageal and breast cancers [40, 41]. MTAP may help prevent the growth and spread of the disease by controlling the ODC activity and putrescine level in breast cancer cells, and by limiting tumor angiogenesis by reducing ODC activity and downregulating the levels of angiogenesis mediators matrix metalloproteinase-2 (MMP2) and Vascular Endothelial Growth Factor D (VEGFD) in breast cancer cells [15].
MTAP in various malignancies
Apart from the previously mentioned malignancies such as melanoma, esophageal and colorectal carcinoma, MTAP has a role to play in different malignancies.
Hellerband et al. [42] detected a decreased or even undetectable MTAP expression in three hepatocellular carcinoma lines and strong cytoplasmatic immunosignals were detectable in surrounding non-tumorous hepatocytes. These findings highlight that the downregulation or loss of MTAP expression in hepatocytes occurs during malignant transformation. Furthermore, Kirovski et al. [42] revealed that downregulation of MTAP in hepatocellular carcinoma increases MTA levels in hepatocellular carcinoma and can potentially be involved in HCC progression.
In osteosarcomas, Miyazaki et al. found that [43] MTAP deficiency was caused by MTAP gene deletion or promoter methylation in most MTAP-negative samples. In in vitro experiment, the MTAP-negative parental cell line was found to be more sensitive to inhibitors of de novo AMP synthesis, compared to the MTAP-positive transfectoma. The authors suggested that the MTAP deficiency frequently observed in osteosarcoma can be targeted with inhibitors of de novo purine synthesis, as a potential chemotherapy strategy for MTAP-negative osteosarcoma patients [43].
Zimling, Jorgensen, and Santoni-Rugiu conducted a study where they analyzed MTAP reactivity in 99 cases of malignant pleural mesothelioma (MPM). They found that 65% of the tumors showed decreased MTAP reactivity. The authors suggested that this decrease in MTAP expression, along with other common markers, could be a valuable diagnostic tool for MPMs. Similarly, the reduced expression of MTAP in triple-negative breast cancer could serve as both a diagnostic and therapeutic marker. Low MTAP expression has been linked to a poor prognosis in glioblastoma [44], gastric cancer [45], and non-small cell lung cancer [46], according to earlier research.
Utilizing MTAP in treatment
Cytotoxic chemotherapy, radiotherapy, hormonal therapy, and immunotherapy have been shown to be effective in the treatment of breast cancer [47]. However, some breast cancer types, and especially TNBC, have no ongoing or maintenance treatment available. This might be due to the metabolic flexibility of cancer cells, which enables compensatory adaptations. It is believed that only a small number of tumor-specific metabolic vulnerabilities have been successfully targeted [48], and that many potential targeted therapies are under investigation, including therapies targeting MTAP deficiency [49].
The rationale behind MTAP targeted therapy is that adenine and methionine cannot be salvaged from endogenous MTA in MTAP-deficient cells. As a result, methionine deprivation and inhibitors of de novo purine synthesis are more toxic to MTAP-deficient cells than to MTAP-positive ones [50, 51]. The difficulty has been in developing a targeted therapy that takes advantage of MTAP deficiency and its resulting alterations in metabolism.
Different strategies based on MTAP status have been proposed that utilize inhibitors of de novo purine synthesis and the enzyme substrate MTA to specifically target and eliminate MTAP-negative cells [52–54].
According to several studies, MTAP-negative tumor cells are up to 20 times more susceptible to purine biosynthesis inhibitors such as MTX, 6-mercaptopurine, azaserine (a powerful inhibitor of the first step in purine biosynthesis), and L-alanosine, than MTAP-positive cells are [50, 55, 56]. The study by Hori et al., which transfected MTAP complementary DNA (cDNA) into a lung cancer cell line lacking MTAP, may have been the most convincing one demonstrating the link between MTAP deficiency and sensitivity to purine and methionine depletion. MTAP deficient cells proved to be more sensitive to purine synthesis inhibitors 5,10-dideazafolate, L-alanosine, and to methionine depletion. The MTAP-containing cell lines, but not the MTAP-deficient cell lines, were entirely rescued from these inhibitors and methionine restriction by adding MTA [56].
Other strategies to take advantage of MTAP-deficiency are also under investigation: MTA and adenine analogs such as 2,6-diaminopurine, 6-methylpurine, 2-fluoroadenine, 6-thioguanine (6-TG), and 5-FU that must undergo phosphoribosylation to transform into its toxic nucleosides are being studied to treat MTAP-deficient malignancies.
Future direction
When MTA is supplied to healthy host cells, MTAP produces a significant amount of adenine. After that, adenine successfully competes with these co-administered drugs for phosphoribosylation by 5-phosphoribosyl-1-pyrophosphate (PRPP). For the drug to have harmful activity, it must be transformed to its toxic nucleotide. However, tumor cells lacking MTAP are unable to convert MTA into adenine. Because of this, PRPP levels are sufficient, and the co-administered drug can easily be transformed to its harmful nucleoside [57]. The significant difference in MTAP activity between tumor and host cells ensures a high level of treatment selectivity, making it a promising therapy for MTAP deficient malignancies in general, and MTAP deficient breast cancer.
MTAP loss is associated with ER-, HER2- and TNBC status, features a distinctive GL with potential to impact both targeted and immunotherapies and enables emerging clinical trials testing MTA2 and PRMT5 inhibitors for patients with clinically advanced breast cancer.
Materials and Methods
The central laboratory (Foundation Medicine, Cambridge, MA, USA) used for comprehensive genomic profiling (CGP) is Clinical Laboratory Improvement Amendments (CLIA)-certified and accredited by the College of American Pathologists. Approval for this study, including a waiver of informed consent, was obtained from the Western Institutional Review Board (Protocol No. 20152817). A minimum of 50 ng of DNA was extracted from 7,301 cases of clinically advanced ductal and lobular breast cancers. Samples used for sequencing featured a minimum of 20% tumor nuclei. After DNA extraction and DNA library preparation, adaptor-ligation based hybrid capture was performed for all coding exons from 324 cancer-related genes plus select introns from 28 genes frequently rearranged in cancer. The Illumina HiSeq instrument was used for DNA sequencing to a mean exon coverage depth of >550X [58, 59]. Tumor mutational burden (TMB) was determined using 0.9 to 1.1 Mb of sequenced DNA [60]. Microsatellite instability (MSI) status was determined on 95 loci [61]. Given that no normal DNA sample was included from each patient, a computational approach was utilized to distinguish somatic vs. germline origin of genomic alterations [62]. PD-L1 expression was determined by immunohistochemistry using 5-micron tissue sections. Following the CDx assay guidelines a tumor proportion score (TPS) was determined for each sample stained with the DAKO 22C2 CDx assay. TPS = (positive tumor cells/total tumor cell) × 100. TPS of 0% was defined as negative, low-level staining defined as 1–49% TPS, and high-level staining defined as ≥50% TPS.
Differences in sample medians were assessed using the unpaired Mann–Whitney–Wilcoxon test. Differences among categorical variables were assessed using chi square test with Yates correction. Statistical tests were 2-sided and used a significance threshold of p < 0.05. Reported p values were not adjusted for multiple testing.
Abbreviations
5-FU: 5-fluorouracil; 6-TG: 6-thioguanine; AZA: 5’aza-deoxycytidine; cDNA: Complementary DNA; HR: hormone receptor; HER2: human epidermal growth factor receptor 2; ERK: Extracellular Signal-Regulated Kinase; MMP2: Matrix Metalloproteinase-2; MTA: 5′-Methylthioadenosine; MTAP: Methylthioadenosine Phosphorylase; MTR-1-P: Methylthioribose-1-Phosphate; MTX: Methotrexate; ODC: Ornithine decarboxylase; PRPP: 5-phosphoribosyl-1-pyrophosphate; TNBC: Triple-Negative Breast Cancer.
CONFLICTS OF INTEREST
Authors have no conflicts of interest to declare.
Ethical statement and consent
Approval for this study, including a waiver of informed consent, was obtained from the Western Institutional Review Board (Protocol No. 20152817).
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021; 71:209–49. https://doi.org/10.3322/caac.21660. [PubMed].
2. Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011; 147:275–92. https://doi.org/10.1016/j.cell.2011.09.024. [PubMed].
3. Gupta GP, Massagué J. Cancer metastasis: building a framework. Cell. 2006; 127:679–95. https://doi.org/10.1016/j.cell.2006.11.001. [PubMed].
4. de Oliveira SF, Ganzinelli M, Chilà R, Serino L, Maciel ME, Urban Cde A, de Lima RS, Cavalli IJ, Generali D, Broggini M, Damia G, Ribeiro EM. Characterization of MTAP Gene Expression in Breast Cancer Patients and Cell Lines. PLoS One. 2016; 11:e0145647. https://doi.org/10.1371/journal.pone.0145647. [PubMed].
5. Gu G, Dustin D, Fuqua SA. Targeted therapy for breast cancer and molecular mechanisms of resistance to treatment. Curr Opin Pharmacol. 2016; 31:97–103. https://doi.org/10.1016/j.coph.2016.11.005. [PubMed].
6. Sukumar J, Gast K, Quiroga D, Lustberg M, Williams N. Triple-negative breast cancer: promising prognostic biomarkers currently in development. Expert Rev Anticancer Ther. 2021; 21:135–48. https://doi.org/10.1080/14737140.2021.1840984. [PubMed].
7. Kunte S, Abraham J, Montero AJ. Novel HER2-targeted therapies for HER2-positive metastatic breast cancer. Cancer. 2020; 126:4278–88. https://doi.org/10.1002/cncr.33102. [PubMed].
8. Mavrakis KJ, McDonald ER 3rd, Schlabach MR, Billy E, Hoffman GR, deWeck A, Ruddy DA, Venkatesan K, Yu J, McAllister G, Stump M, deBeaumont R, Ho S, et al. Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5. Science. 2016; 351:1208–13. https://doi.org/10.1126/science.aad5944. [PubMed].
9. Tang B, Kadariya Y, Chen Y, Slifker M, Kruger WD. Expression of MTAP inhibits tumor-related phenotypes in HT1080 cells via a mechanism unrelated to its enzymatic function. G3 (Bethesda). 2014; 5:35–44. https://doi.org/10.1534/g3.114.014555. [PubMed].
10. Jing W, Zhu H, Liu W, Zhai X, Tian H, Yu J. MTAP-deficiency could predict better treatment response in advanced lung adenocarcinoma patients initially treated with pemetrexed-platinum chemotherapy and bevacizumab. Sci Rep. 2020; 10:843. https://doi.org/10.1038/s41598-020-57812-2. [PubMed].
11. Basu I, Cordovano G, Das I, Belbin TJ, Guha C, Schramm VL. A transition state analogue of 5’-methylthioadenosine phosphorylase induces apoptosis in head and neck cancers. J Biol Chem. 2007; 282:21477–86. https://doi.org/10.1074/jbc.M702287200. [PubMed].
12. Chiang K, Zielinska AE, Shaaban AM, Sanchez-Bailon MP, Jarrold J, Clarke TL, Zhang J, Francis A, Jones LJ, Smith S, Barbash O, Guccione E, Farnie G, et al. PRMT5 Is a Critical Regulator of Breast Cancer Stem Cell Function via Histone Methylation and FOXP1 Expression. Cell Rep. 2017; 21:3498–513. https://doi.org/10.1016/j.celrep.2017.11.096. [PubMed].
13. Marjon K, Cameron MJ, Quang P, Clasquin MF, Mandley E, Kunii K, McVay M, Choe S, Kernytsky A, Gross S, Konteatis Z, Murtie J, Blake ML, et al. MTAP Deletions in Cancer Create Vulnerability to Targeting of the MAT2A/PRMT5/RIOK1 Axis. Cell Rep. 2016; 15:574–87. https://doi.org/10.1016/j.celrep.2016.03.043. [PubMed].
14. Cheng XY, Liu Z, Shang L, Cai HQ, Zhang Y, Cai Y, Xu X, Hao JJ, Wang MR. Deletion and downregulation of MTAP contribute to the motility of esophageal squamous carcinoma cells. Onco Targets Ther. 2017; 10:5855–62. https://doi.org/10.2147/OTT.S151953. [PubMed].
15. Zhang Y, Zhang TT, Gao L, Tan YN, Li YT, Tan XY, Huang TX, Li HH, Bai F, Zou C, Pei XH, Tan BB, Fu L. Downregulation of MTAP promotes Tumor Growth and Metastasis by regulating ODC Activity in Breast Cancer. Int J Biol Sci. 2022; 18:3034–47. https://doi.org/10.7150/ijbs.67149. [PubMed].
16. Dominici C, Sgarioto N, Yu Z, Sesma-Sanz L, Masson JY, Richard S, Raynal NJ. Synergistic effects of type I PRMT and PARP inhibitors against non-small cell lung cancer cells. Clin Epigenetics. 2021; 13:54. https://doi.org/10.1186/s13148-021-01037-1. [PubMed].
17. Ashok Kumar P, Graziano SL, Danziger N, Pavlick D, Severson EA, Ramkissoon SH, Huang RSP, Decker B, Ross JS. Genomic landscape of non-small-cell lung cancer with methylthioadenosine phosphorylase (MTAP) deficiency. Cancer Med. 2023; 12:1157–66. https://doi.org/10.1002/cam4.4971. [PubMed].
18. Feustel K, Falchook GS. Protein Arginine Methyltransferase 5 (PRMT5) Inhibitors in Oncology Clinical Trials: A review. J Immunother Precis Oncol. 2022; 5:58–67. https://doi.org/10.36401/JIPO-22-1. [PubMed].
19. De Fusco C, Schimpl M, Börjesson U, Cheung T, Collie I, Evans L, Narasimhan P, Stubbs C, Vazquez-Chantada M, Wagner DJ, Grondine M, Sanders MG, Tentarelli S, et al. Fragment-Based Design of a Potent MAT2a Inhibitor and in Vivo Evaluation in an MTAP Null Xenograft Model. J Med Chem. 2021; 64:6814–26. https://doi.org/10.1021/acs.jmedchem.1c00067. [PubMed].
20. Guo J, Yang Y, Buettner R, Rosen ST. Targeting the methionine-methionine adenosyl transferase 2A- S -adenosyl methionine axis for cancer therapy. Curr Opin Oncol. 2022; 34:546–51. https://doi.org/10.1097/CCO.0000000000000870. [PubMed].
21. Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, Cole CG, Ward S, Dawson E, Ponting L, Stefancsik R, Harsha B, Kok CY, et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017; 45:D777–83. https://doi.org/10.1093/nar/gkw1121. [PubMed].
22. AACR Project GENIE Consortium. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017; 7:818–31. https://doi.org/10.1158/2159-8290.CD-17-0151. [PubMed].
23. Allemani C, Matsuda T, Di Carlo V, Harewood R, Matz M, Nikšić M, Bonaventure A, Valkov M, Johnson CJ, Estève J, Ogunbiyi OJ, Azevedo E Silva G, Chen WQ, et al, and CONCORD Working Group. Global surveillance of trends in cancer survival 2000-14 (CONCORD-3): analysis of individual records for 37 513 025 patients diagnosed with one of 18 cancers from 322 population-based registries in 71 countries. Lancet. 2018; 391:1023–75. https://doi.org/10.1016/S0140-6736(17)33326-3. [PubMed].
24. Batova A, Diccianni MB, Nobori T, Vu T, Yu J, Bridgeman L, Yu AL. Frequent deletion in the methylthioadenosine phosphorylase gene in T-cell acute lymphoblastic leukemia: strategies for enzyme-targeted therapy. Blood. 1996; 88:3083–90. [PubMed].
25. Bertino JR, Lubin M, Johnson-Farley N, Chan WC, Goodell L, Bhagavathi S. Lack of expression of MTAP in uncommon T-cell lymphomas. Clin Lymphoma Myeloma Leuk. 2012; 12:306–9. https://doi.org/10.1016/j.clml.2012.07.001. [PubMed].
26. Watanabe F, Takao M, Inoue K, Nishioka J, Nobori T, Shiraishi T, Kaneda M, Sakai T, Yada I, Shimpo H. Immunohistochemical diagnosis of methylthioadenosine phosphorylase (MTAP) deficiency in non-small cell lung carcinoma. Lung Cancer. 2009; 63:39–44. https://doi.org/10.1016/j.lungcan.2008.04.019. [PubMed].
27. Subhi AL, Tang B, Balsara BR, Altomare DA, Testa JR, Cooper HS, Hoffman JP, Meropol NJ, Kruger WD. Loss of methylthioadenosine phosphorylase and elevated ornithine decarboxylase is common in pancreatic cancer. Clin Cancer Res. 2004; 10:7290–96. https://doi.org/10.1158/1078-0432.CCR-04-0972. [PubMed].
28. Limm K, Ott C, Wallner S, Mueller DW, Oefner P, Hellerbrand C, Bosserhoff AK. Deregulation of protein methylation in melanoma. Eur J Cancer. 2013; 49:1305–13. https://doi.org/10.1016/j.ejca.2012.11.026. [PubMed].
29. Stevens AP, Spangler B, Wallner S, Kreutz M, Dettmer K, Oefner PJ, Bosserhoff AK. Direct and tumor microenvironment mediated influences of 5’-deoxy-5’-(methylthio)adenosine on tumor progression of malignant melanoma. J Cell Biochem. 2009; 106:210–19. https://doi.org/10.1002/jcb.21984. [PubMed].
30. Zhong Y, Lu K, Zhu S, Li W, Sun S. Characterization of methylthioadenosin phosphorylase (MTAP) expression in colorectal cancer. Artif Cells Nanomed Biotechnol. 2018; 46:2082–87. https://doi.org/10.1080/21691401.2017.1408122. [PubMed].
31. Christopher SA, Diegelman P, Porter CW, Kruger WD. Methylthioadenosine phosphorylase, a gene frequently codeleted with p16(cdkN2a/ARF), acts as a tumor suppressor in a breast cancer cell line. Cancer Res. 2002; 62:6639–44. [PubMed].
32. Appleby TC, Erion MD, Ealick SE. The structure of human 5’-deoxy-5’-methylthioadenosine phosphorylase at 1.7 A resolution provides insights into substrate binding and catalysis. Structure. 1999; 7:629–41. https://doi.org/10.1016/s0969-2126(99)80084-7. [PubMed].
33. Nowotarski SL, Woster PM, Casero RA Jr. Polyamines and cancer: implications for chemotherapy and chemoprevention. Expert Rev Mol Med. 2013; 15:e3. https://doi.org/10.1017/erm.2013.3. [PubMed].
34. Manni A, Washington S, Craig L, Cloud M, Griffith JW, Verderame MF, Texter LJ, Mauger D, Demers LM, Harms JF, Welch DR. Effects of alpha-difluoromethylornithine on local recurrence and pulmonary metastasis from MDA-MB-435 breast cancer xenografts in nude mice. Clin Exp Metastasis. 2003; 20:321–25. https://doi.org/10.1023/a:1024055522067. [PubMed].
35. Manni A, Washington S, Griffith JW, Verderame MF, Mauger D, Demers LM, Samant RS, Welch DR. Influence of polyamines on in vitro and in vivo features of aggressive and metastatic behavior by human breast cancer cells. Clin Exp Metastasis. 2002; 19:95–105. https://doi.org/10.1023/a:1014536909007. [PubMed].
36. Li J, Meng Y, Wu X, Sun Y. Polyamines and related signaling pathways in cancer. Cancer Cell Int. 2020; 20:539. https://doi.org/10.1186/s12935-020-01545-9. [PubMed].
37. Asai Y, Itoi T, Sugimoto M, Sofuni A, Tsuchiya T, Tanaka R, Tonozuka R, Honjo M, Mukai S, Fujita M, Yamamoto K, Matsunami Y, Kurosawa T, et al. Elevated Polyamines in Saliva of Pancreatic Cancer. Cancers (Basel). 2018; 10:43. https://doi.org/10.3390/cancers10020043. [PubMed].
38. Guo Y, Ye Q, Deng P, Cao Y, He D, Zhou Z, Wang C, Zaytseva YY, Schwartz CE, Lee EY, Evers BM, Morris AJ, Liu S, She QB. Spermine synthase and MYC cooperate to maintain colorectal cancer cell survival by repressing Bim expression. Nat Commun. 2020; 11:3243. https://doi.org/10.1038/s41467-020-17067-x. [PubMed].
39. Gerner EW, Meyskens FL Jr. Polyamines and cancer: old molecules, new understanding. Nat Rev Cancer. 2004; 4:781–92. https://doi.org/10.1038/nrc1454. [PubMed].
40. Mafune K, Tanaka Y, Mimori K, Mori M, Takubo K, Makuuchi M. Increased expression of ornithine decarboxylase messenger RNA in human esophageal carcinoma. Clin Cancer Res. 1999; 5:4073–78. [PubMed].
41. Cañizares F, Salinas J, de las Heras M, Diaz J, Tovar I, Martinez P, Peñafiel R. Prognostic value of ornithine decarboxylase and polyamines in human breast cancer: correlation with clinicopathologic parameters. Clin Cancer Res. 1999; 5:2035–41. [PubMed].
42. Kirovski G, Stevens AP, Czech B, Dettmer K, Weiss TS, Wild P, Hartmann A, Bosserhoff AK, Oefner PJ, Hellerbrand C. Down-regulation of methylthioadenosine phosphorylase (MTAP) induces progression of hepatocellular carcinoma via accumulation of 5’-deoxy-5’-methylthioadenosine (MTA). Am J Pathol. 2011; 178:1145–52. https://doi.org/10.1016/j.ajpath.2010.11.059. [PubMed].
43. Miyazaki S, Nishioka J, Shiraishi T, Matsumine A, Uchida A, Nobori T. Methylthioadenosine phosphorylase deficiency in Japanese osteosarcoma patients. Int J Oncol. 2007; 31:1069–76. [PubMed].
44. Li Z, Jin Y, Zou Q, Shi X, Wu Q, Lin Z, He Q, Huang G, Qi S. Integrated genomic and transcriptomic analysis suggests KRT18 mutation and MTAP are key genetic alterations related to the prognosis between astrocytoma and glioblastoma. Ann Transl Med. 2021; 9:713. https://doi.org/10.21037/atm-21-1317. [PubMed].
45. Kim J, Kim MA, Min SY, Jee CD, Lee HE, Kim WH. Downregulation of methylthioadenosin phosphorylase by homozygous deletion in gastric carcinoma. Genes Chromosomes Cancer. 2011; 50:421–33. https://doi.org/10.1002/gcc.20867. [PubMed].
46. Su CY, Chang YC, Chan YC, Lin TC, Huang MS, Yang CJ, Hsiao M. MTAP is an independent prognosis marker and the concordant loss of MTAP and p16 expression predicts short survival in non-small cell lung cancer patients. Eur J Surg Oncol. 2014; 40:1143–50. https://doi.org/10.1016/j.ejso.2014.04.017. [PubMed].
47. Bauer KR, Brown M, Cress RD, Parise CA, Caggiano V. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: a population-based study from the California cancer Registry. Cancer. 2007; 109:1721–28. https://doi.org/10.1002/cncr.22618. [PubMed].
48. Luengo A, Gui DY, Vander Heiden MG. Targeting Metabolism for Cancer Therapy. Cell Chem Biol. 2017; 24:1161–80. https://doi.org/10.1016/j.chembiol.2017.08.028. [PubMed].
49. Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011; 121:2750–67. https://doi.org/10.1172/JCI45014. [PubMed].
50. Chen ZH, Olopade OI, Savarese TM. Expression of methylthioadenosine phosphorylase cDNA in p16-, MTAP- malignant cells: restoration of methylthioadenosine phosphorylase-dependent salvage pathways and alterations of sensitivity to inhibitors of purine de novo synthesis. Mol Pharmacol. 1997; 52:903–11. https://doi.org/10.1124/mol.52.5.903. [PubMed].
51. Tisdale MJ. Methionine synthesis from 5’-methylthioadenosine by tumour cells. Biochem Pharmacol. 1983; 32:2915–20. https://doi.org/10.1016/0006-2952(83)90396-9. [PubMed].
52. Kamatani N, Nelson-Rees WA, Carson DA. Selective killing of human malignant cell lines deficient in methylthioadenosine phosphorylase, a purine metabolic enzyme. Proc Natl Acad Sci U S A. 1981; 78:1219–23. https://doi.org/10.1073/pnas.78.2.1219. [PubMed].
53. Kindler HL, Burris HA 3rd, Sandler AB, Oliff IA. A phase II multicenter study of L-alanosine, a potent inhibitor of adenine biosynthesis, in patients with MTAP-deficient cancer. Invest New Drugs. 2009; 27:75–81. https://doi.org/10.1007/s10637-008-9160-1. [PubMed].
54. Gibson RJ, Keefe DM, Clarke JM, Regester GO, Thompson FM, Goland GJ, Edwards BG, Cummins AG. The effect of keratinocyte growth factor on tumour growth and small intestinal mucositis after chemotherapy in the rat with breast cancer. Cancer Chemother Pharmacol. 2002; 50:53–58. https://doi.org/10.1007/s00280-002-0460-4. [PubMed].
55. Li W, Su D, Mizobuchi H, Martin DS, Gu B, Gorlick R, Cole P, Bertino JR. Status of methylthioadenosine phosphorylase and its impact on cellular response to L-alanosine and methylmercaptopurine riboside in human soft tissue sarcoma cells. Oncol Res. 2004; 14:373–79. https://doi.org/10.3727/0965040041292332. [PubMed].
56. Hori H, Tran P, Carrera CJ, Hori Y, Rosenbach MD, Carson DA, Nobori T. Methylthioadenosine phosphorylase cDNA transfection alters sensitivity to depletion of purine and methionine in A549 lung cancer cells. Cancer Res. 1996; 56:5653–58. [PubMed].
57. Lubin M, Lubin A. Selective killing of tumors deficient in methylthioadenosine phosphorylase: a novel strategy. PLoS One. 2009; 4:e5735. https://doi.org/10.1371/journal.pone.0005735. [PubMed].
58. Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, Schnall-Levin M, White J, Sanford EM, An P, Sun J, Juhn F, Brennan K, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013; 31:1023–31. https://doi.org/10.1038/nbt.2696. [PubMed].
59. Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, Kok CY, Jia M, De T, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015; 43:D805–11. https://doi.org/10.1093/nar/gku1075. [PubMed].
60. Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, Schrock A, Campbell B, Shlien A, Chmielecki J, Huang F, He Y, Sun J, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017; 9:34. https://doi.org/10.1186/s13073-017-0424-2. [PubMed].
61. Trabucco SE, Gowen K, Maund SL, Sanford E, Fabrizio DA, Hall MJ, Yakirevich E, Gregg JP, Stephens PJ, Frampton GM, Hegde PS, Miller VA, Ross JS, et al. A Novel Next-Generation Sequencing Approach to Detecting Microsatellite Instability and Pan-Tumor Characterization of 1000 Microsatellite Instability-High Cases in 67,000 Patient Samples. J Mol Diagn. 2019; 21:1053–66. https://doi.org/10.1016/j.jmoldx.2019.06.011. [PubMed].
62. Sun JX, He Y, Sanford E, Montesion M, Frampton GM, Vignot S, Soria JC, Ross JS, Miller VA, Stephens PJ, Lipson D, Yelensky R. A computational approach to distinguish somatic vs. germline origin of genomic alterations from deep sequencing of cancer specimens without a matched normal. PLoS Comput Biol. 2018; 14:e1005965. https://doi.org/10.1371/journal.pcbi.1005965. [PubMed].