
Introduction
Recently, an increased expression of the dihydrolipoamide S-succinyltransferase (DLST) gene has been shown to predict poor outcome of neuroblastoma and triple-negative breast cancer (TNBC) [1, 2]. In contrast, knockdown of DLST led to apoptosis in T-cell acute lymphoblastic leukemia and in TNBC [1, 3] suggesting a DLST dependence for tumor cell viability and, consequently highlighting DLST as a potential target for cancer therapy. With respect to its cellular function, DLST converts α-ketoglutarate to succinyl-CoA during oxidative decarboxylation and produces NADH for the oxidative phosphorylation (OXPHOS). Specifically, DLST is an E2 subcomponent of the mitochondrial oxoglutarate dehydrogenase complex (OGDC), catalyzing the overall conversion of 2-oxoglutarate to succinyl-CoA during the tricarboxylic acid (TCA) cycle. There is evidence that certain cancer cells use the TCA cycle extensively for energy production, offering potential therapeutic opportunities by targeting the TCA cycle in cancer cells [4]. In line with this, DLST-overexpressing cell lines showed increased OXPHOS gene signatures [2]. The OXPHOS inhibitor IACS-010759 targets the electron transport chain and leads to a reduction in tumor weight, size, and proliferation of neuroblastoma cells in zebrafish and mouse xenografts [2]. Other FDA-approved drugs, used to treat non-oncologic indications such as metformin, arsenic trioxide, and atovaquone, have also been identified as OXPHOS inhibitors in vivo [5]. Thus, additional vulnerabilities of DLST-activated tumors within and beyond OXPHOS inhibition and drug repurposing are conceivable.
Therefore, we set out to identify new candidates for the treatment of DLST-activated tumors. With the advent of complex genetic datasets of roughly 1000 cell lines in the Cancer Cell Line Encyclopedia (CCLE) and on drug resistance in the Genomics of Drug Sensitivity in Cancer project (GDSC), analyses of drug sensitivity have become possible on a larger scale [6, 7]. We took advantage of these data to test if a potential DLST activation, caused by overexpression or copy number amplification, is associated with drug sensitivity and whether additional OXPHOS inhibitors may serve as therapeutics for tumors with high levels of DLST.
Results
To identify drugs that inhibit viability of specifically DLST-activated tumor cells, we compared cell lines with supposedly activating changes of DLST (DNA amplification, high mRNA levels) to cell lines without DLST changes. In the light of the described DLST activation in neuroblastoma [2], we aimed at detecting an effect specific to 70 cancer cell lines derived from the nervous system. However, only one of these had an increased DLST copy number and none of them showed high mRNA levels, hence, a test of nervous system tumors was not possible (Supplementary Table 1).
Therefore, we extended our analysis across all entities available, of which 55 of 872 (6.31%) cell lines showed these supposedly activating genetic alterations in DLST. We tested resistance and sensitivity for the full set of 250 individual drugs and found that DLST-activated cell lines were more often sensitive to 7 drugs compared to control cell lines without such changes (OR<1, Figure 1, Supplementary Tables 1–3). In Table 1 all drugs associated with sensitivity together with their function and current applications of these drugs in neuroblastoma and other cancers are given. In this literature research we found that the drugs lapatinib and gemcitabine were approved, also as combination therapy, in various cancer types, such as breast, gastric, and lung cancer. Others showed promising anticancer activity in vivo (LY317615, CP724714, and obatoclax mesylate) and in vitro (IPA3, BMS708163) (Table 1). Neuroblastoma tumor size was also reduced in mouse models for gemcitabine and the drug combination of lapatinib and YM155. Under obatoclax mesylate cell lines derived from neuroblastoma showed significantly decreased apoptosis. We next analyzed which compounds had been previously involved in processes related to OXPHOS and found only obatoclax mesylate to reduce OXPHOS in leukemia stem cells. Therefore, we aimed at identifying the signaling pathway involvement of the remaining 6 identified compounds and found 4 out of 6 were protein kinase inhibitors of the extracellular signal-regulated kinases/mitogen-activated protein kinase (ERK/MAPK) signaling pathway: lapatinib, IPA3, LY317615, and CP724714. Of note, we tested 250 individual drugs and after correcting for multiple testing (Bonferroni method), none of the drugs were significantly associated with sensitivity of DLST-activated cell lines.
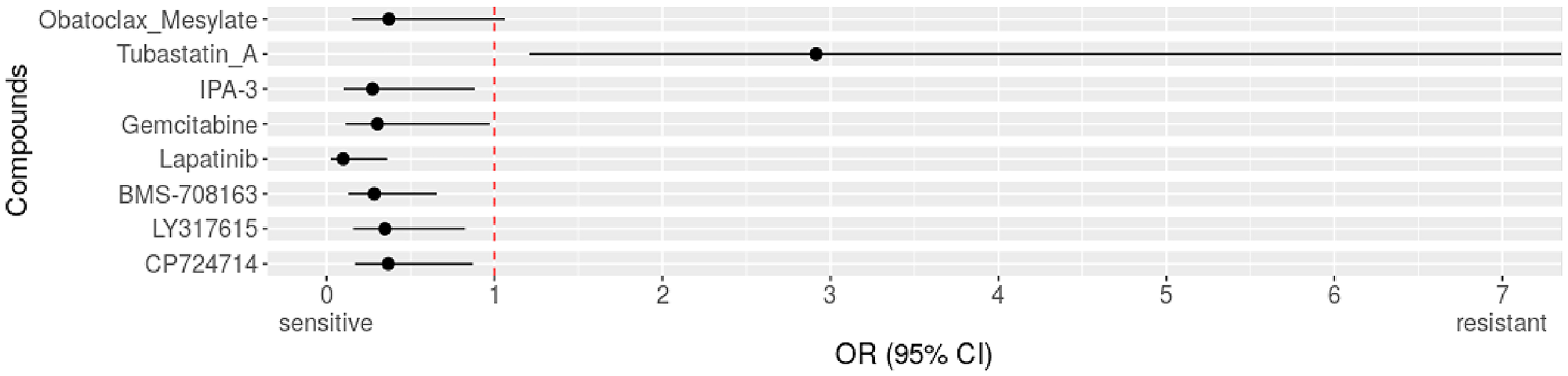
Figure 1: Identified drugs for DLST-activated cell lines across cancer types. Compared to DLST wild types across entities. Forest plot with odds ratio (OR) for resistance of altered cell lines to drugs. OR>1 indicates resistance, OR<1 indicates sensitivity.
Table 1: Drugs associated with sensitivity of DLST-activated cell lines and their relation to OXPHOS and ERK/MAPK pathway
| Drug (p-value) | OR | Mut: s (%), r | Wt: s (%), r | Function | Current applications in cancer types | Related to OXPHOS; ERK/MAPK pathway |
|---|---|---|---|---|---|---|
| Lapatinib (p = 0.0005) | 0.1003 (0.02391–0.3629) | 7 (63.64%), 4 | 28 (14.51%), 165 | inhibition of EGFR and HER2 if high ERB2 expression | drug combination (i.e., lapatinib plus YM155) decreased neuroblastoma tumor size in an in vivo model [18], approved in combination with capecitabine in patients with advanced HER2-positive breast cancer [19] | lapatinib increases ROS levels in models that are sensitive to erbB1/2 blockade [20]; upstream activator of ERK/MAPK pathway [11] |
| IPA3 (p = 0.0263) | 0.2742 (0.1024–0.883) | 5 (13.88%), 31 | 21 (4.30%), 467 | inhibitor of Pak1 | melanoma and colon carcinoma cell lines that carry mutations in NRAS and KRAS are more sensitive to IPA3 [21] | downregulation of Pak2 associated with a shift toward OXPHOS, PAK1 knockout resulted in increased glycolysis [22]; upstream activator of ERK/MAPK pathway [13] |
| BMS 708163 (p = 0.0034) also: Avagacestat | 0.2844 (0.1319–0.6562) | 10 (28.57%), 25 | 52 (10.25%), 455 | γ-secretase inhibitor of Aβ40 and Aβ42 | tested in lung cancer after resistance to gefitinib, induced high level of apoptosis [23] | no relevance found in OXPHOS; no relevance found in ERK/MAPK pathway |
| Gemcitabine (p = 0.0363) | 0.3033 (0.1144–0.9706) | 5 (13.88%), 31 | 23 (4.74%), 462 | standard chemotherapy | Neuroblastoma is highly sensitive to gemcitabine tested (in vitro and in vivo) [24], approved in non-small cell lung cancer, pancreatic, bladder, and breast cancer [25] | treated PDAC cells with phenformin (MRC I inhibitor) + standard chemotherapy (gemcitabine), this treatment is synergistic specifically in high OXPHOS cells [26]; no relevance found in ERK/MAPK pathway |
| LY317615 (p = 0.0142) also: Enzastaurin | 0.3476 (0.1594–0.8258) | 9 (25%), 27 | 53 (10.45%), 454 | PKCβ (protein kinase C-β) selective inhibitor | most promising in B-cell lymphomas [27], could be effective in acute myeloid leukemia therapy [14] | no relevance found in OXPHOS; suppressor of ERK [14] |
| CP724714 (p = 0.0276) | 0.368 (0.1594–0.8258) | 9 (24.32%), 28 | 54 (10.65%), 453 | ErbB2 (HER2) inhibitor | has been tested for the treatment of ErbB2-positive malignancies [28], induces G1 cell cycle block in erbB2-overexpressing human breast carcinoma cells [29] | exacerbated OXPHOS dependency frequently characterizes resistance against chemotherapy or tyrosine kinase inhibitors [30]; upstream activator of ERK/MAPK pathway [12] |
| Obatoclax Mesylate (p = 0.0484) | 0.3721 (0.1524–1.061) | 6 (16.67%), 30 | 34 (7.02%) 450 | inhibitor of the Bcl-2 family of proteins (conserved family that share BH domains) | obatoclax significantly increased apoptosis and autophagy in neuroblastoma cells [31], obatoclax-induced apoptosis was associated with leukemic cell differentiation [32] | inhibition of BCL2 reduce OXPHOS in human leukemia stem cells [8]; no relevance found in ERK/MAPK pathway |
We hypothesized that in an inverse scenario of the described associations above, drugs associated with sensitivity in DLST-activated cell lines would be associated with resistance in DLST-deactivated cell lines (33 of 872, 3.78%). This assumption was true for lapatinib, CP724714, and obatoclax mesylate, which showed an OR for resistance >1 in cell lines with DLST deactivation, however, lacking statistical significance (Supplementary Table 4).
As stated above, the primary analyses identified only one OXPHOS inhibitor but several protein kinase inhibitors were associated with sensitivity. Because this points to an involvement of DLST in the ERK/MAPK pathway, we asked if the sensitivity of DLST-activated cell lines to MAPK inhibitors could be due to concomitant changes in the ERK/MAPK pathway. Such common driver mutations occur in NRAS, KRAS and HRAS. However, DLST-activated cell lines did not show alterations in any of the RAS genes more frequently than all other samples (OR = 1.24, p = 0.23). We, therefore, investigated whether DLST expression was associated with mutation or expression of common genes involved in the ERK/MAPK pathway. DLST expression was not associated with driver mutations in BRAFV600 (p = 0.856), HRAS (p = 0.791) or NRAS (p = 0.609) (Supplementary Figure 1). However, cell lines with driver mutations in KRAS had higher DLST expression than controls (p = 0.033, Figure 2A). Also, coexpression of DLST and ERBB2 (Spearman coef = 0.08, p = 8.46e-3), HRAS (Spearman coef = 0.08, p = 6.10e-3), KRAS (Spearman coef = 0.16, p = 1.25e-7, Figure 2B), and PAK1 (Spearman coef = 0.09, p = 3.5e-3) was observed.
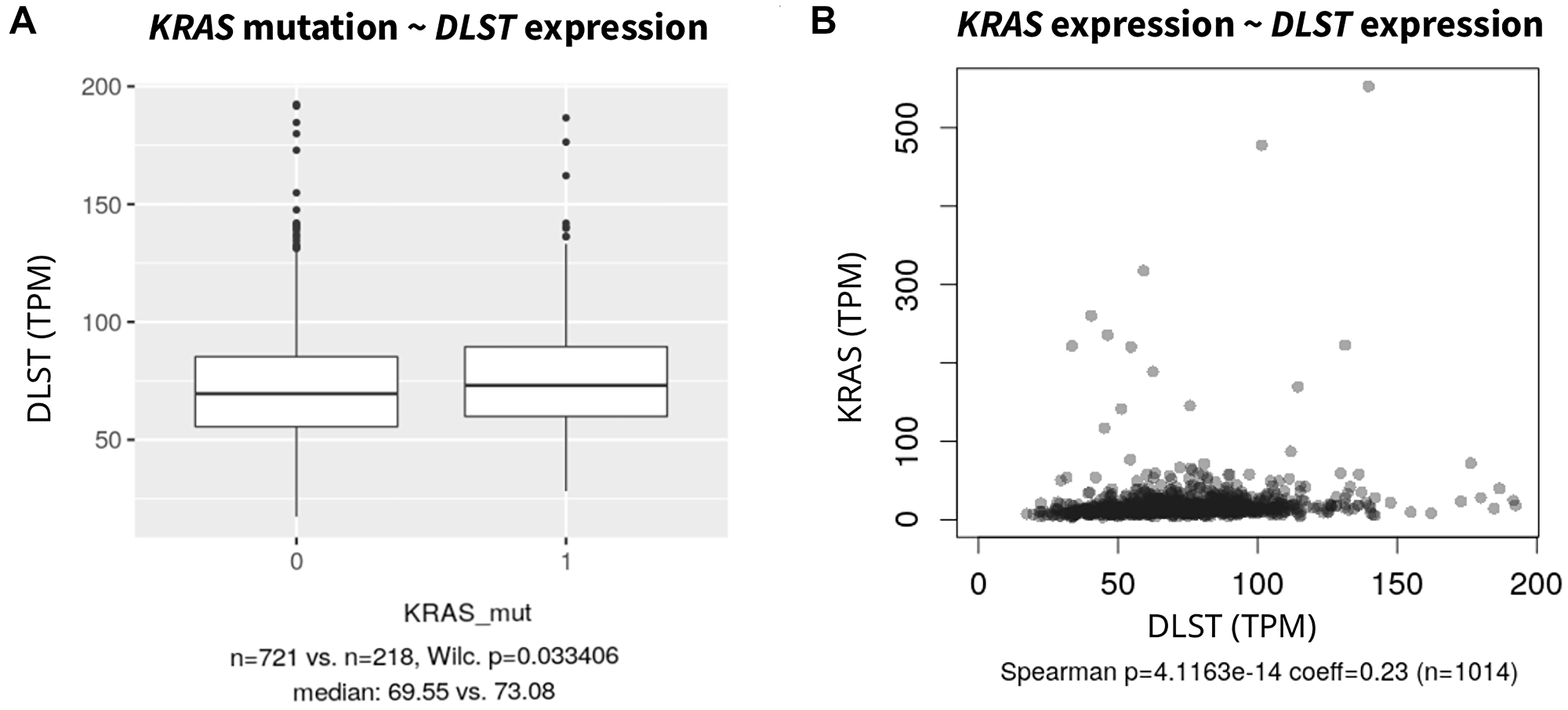
Figure 2: (A) Association of DLST expression in cell lines with KRAS driver mutations against KRAS wild type and (B) Spearman correlation with KRAS expression (without one outlier; KRAS expression >1500). For other common driver mutations such as NRAS, HRAS and BRAF see Supplementary Figure 1.
DISCUSSION
New therapy options are still being explored for the treatment of neuroblastoma including potential molecular pathways and genetic aberrations. While our results indicate that DLST amplification or high mRNA levels are rather rare in cell lines derived from the nervous system, they may nevertheless have an impact on cancer growth as DLST overexpression had led to increased tumor burden and higher frequency of disseminated disease in a zebrafish model of MYCN-driven neuroblastoma [2]. Moreover, beyond this entity, high DLST expression was associated with poor overall survival also in triple-negative breast cancer patients [1] and may warrant studying of DLST activation in further cancer entities.
Recently, the elevated expression of DLST in cancer was associated with aggressiveness in neuroblastoma and with the OXPHOS pathway specifically. Studies showed that OXPHOS can be upregulated to support the growth, survival, and migration of cancer cells, and upregulated DLST expression may pose a mechanism to increase OXPHOS [2, 5]. Given that OXPHOS inhibition slowed tumor growth in zebrafish and mouse xenografts of neuroblastoma cells [2], OXPHOS inhibitors could , therefore, constitute a potential vulnerability of DLST-altered tumors. Three OXPHOS inhibitors (antimycin, oligomycin, IACS-010759) were suggested to inhibit growth of neuroblastoma cells and partly induced apoptosis in zebrafish and mouse xenografts of high-risk neuroblastoma [2]. In the light of a putative association of DLST-activated tumors with elevated OXPHOS, it is of particular interest that in our analysis of a large number of cancer cell lines, DLST-activated cell lines were sensitive to obatoclax mesylate more often than control cell lines. This inhibitor of the Bcl-2-family proteins has previously shown to reduce OXPHOS in human leukemia stem cells [8]. Moreover, besides the candidate drug obatoclax mesylate, other BCL-2 inhibitors, which were not included in the GDSD dataset, such as venetoclax and navitoclax showed promising results in neuroblastoma-derived cell lines and tumors [9]. These data suggest that obatoclax mesylate and other Bcl-2 inhibitors could be efficient in tumors with DLST-activating alterations and should be further investigated in this context [9].
In addition to the described role of DLST in OXPHOS and the identified OXPHOS inhibitor, we found a group of protein kinase inhibitors potentially suitable for therapy of such tumors (Figure 3). Protein kinase inhibitors are used for efficient cancer therapy to modulate cell proliferation, survival, and transformation [10]. All protein kinase inhibitors presented here target the ERK/MAPK pathway, a signaling pathway that is aberrantly activated in cancer. Lapatinib and CP724714 reduce ERK/MAPK pathway activity via inhibition of the upstream epidermal growth factor receptor HER2/ERBB2 [11, 12]. IPA3 binds to the inactive PAK group I molecule, which can also activate the ERK/MAPK signaling pathway [13] and LY317615 suppresses the activation of ERK [14]. As in general, the ERK/MAPK pathway activation is a frequent event in cancer, we asked whether the sensitivity of DLST-activated cell lines to MAPK inhibitors could be due concomitant changes in the ERK/MAPK pathway. Indeed, there was an increased DLST expression in cell lines with KRAS mutations (Figure 3) as well as co-expression of DLST and several genes involved in the ERK/MAPK pathway, which may explain the observed sensitivity to inhibitors of the ERK/MAPK pathway. Furthermore, a link between DLST and ERK/MAPK signaling pathways was previously suggested, as circDLST was shown to sponge with microRNA, activating the NRAS/MEK/ERK signaling in gastric cancer [15]. Thus, DLST potentially constitutes a proxy for ERK/MAPK pathway activation. Our data provides a potential starting point for future research in understanding the role of DLST in cancer and the power of ERK/MAPK-inhibitors in DLST-activated cancer. These findings further highlight how understanding the underlying molecular networks can help to achieve rational care. Initially, the Notch inhibitor BMS 708163 was not linked to either the OXPHOS or MAPK/ERK pathway. However, in agreement with our results, Anderson et al. recently described that cell lines with overexpressed MYC, which is transcriptionally regulated by Notch, have the ability to stabilize DLST [3].
Figure 3: Analysis of DLST-activated cell lines revealed sensitivity to protein kinase inhibiting the ERK/MAPK pathway.
The observed associations reported here have to be seen in the context of the manifold genetic changes that cancer cell lines have accumulated. However, the pooling of hundreds of cell lines in groups may ameliorate the confounding effects of individual genetic variation to some extent and may thus allow more general, mechanistic insights. Nevertheless, the proposed candidate drugs could be subject to further rigorous tests using knock-out and knock-in experiments of cell lines, xenografts in animal models or association studies of DLST-levels in tumors and patient outcome to determine the relevance of DLST in cancer and its potential utility in predictions of clinical outcome.
Materials and Methods
To test differences in drug sensitivity in DLST genetically and expressionally altered cancer cell lines, we used genetic data of 1,739 cell lines in the Cancer Cell Line Encyclopedia dataset (CCLE, accessed 06/2022 at https://www.cbioportal.org/study/summary?id=ccle_broad_2019, Supplementary Table 5). We integrated these genetic data with drug resistance data on 250 drugs of the Genomics of Drug Sensitivity in Cancer project GDSC [6, 7]. Combining the drug resistance data from the GDSC database with the genetic data from the CCLE samples at https://tools.hornlab.org/GDSC/ [16], resulted in a dataset of 872 cell lines. The documented various types of genetic changes in the CCLE dataset provide information on copy number variation, mRNA levels, point mutations, and gene fusions. Here, we focused on supposedly activating changes of DLST defined by copy number amplification or high mRNA expression of DLST (Supplementary Tables 1–3) because, as part of the mitochondrial oxoglutarate dehydrogenase complex (OGDC) and rate limiting enzyme of the TCA cycle, we assumed DLST levels to have direct impact on OGDC activity. We then also analyzed supposedly deactivating changes defined as homozygous deletion, low mRNA, frameshift or nonsense variants (Supplementary Tables 6, 7, 4). Low and high mRNA expression levels were defined with a minimum z-score of -2 and 2, respectively, based on RNA-seq RPKM units compared to the expression distribution of diploid samples in cBioPortal [17]. To determine whether the defined group of cell lines was rather resistant or sensitive to the tested drugs, we calculated odds ratios (OR) for resistance by comparing the counts of resistant and sensitive cell lines in the genetically changed (DLST-activated) and the control cell line group without any DLST changes (DLST wild types) (two-sided Fisher’s exact and chi-square tests). Bonferroni corrected p-value thresholds were < 0.0023 testing 22 pathways (each pathway containing pooled sets of drugs assigned to this pathway) and p < 0.0002 testing 250 individual drugs.
Initially, we analyzed all cell lines across entities and then specifically a subgroup of 70 cell lines that had been documented to derive from the central nervous system (n = 54) or from neuroblastoma (autonomous ganglia, n = 16, Supplementary Table 1). We further tested DLST expression in cell lines across entities (CCLE 2019 cohort) with BRAFV600, NRAS, KRAS, and HRAS driver mutations and tested co-expression with genes involved in the ERK/MAPK pathway (ERBB2, HRAS, NRAS, KRAS, PAK1, via cBioPortal).
Abbreviations
BCL: B-cell lymphoma; CCLE: cancer cell line encyclopedia; DLST: dihydrolipoamide S-succinyltransferase; EGFR: epidermal growth factor receptor; ERBB2: erb-b2 receptor tyrosine kinase 2; ERK: extracellular signal-regulated kinase; FDA: food and drug administration; GDSC: genomics of drug sensitivity in cancer; HER2: human epidermal growth factor 2; IC: half-maximal inhibitory concentration; MAPK: mitogen activated protein kinase; MRC: mitochondrial respiratory complex; MYCN: N-myc proto-oncogene; OGDC: oxoglutarate dehydrogenase complex; OR: odds ratio; OXPHOS: oxidative phosphorylation; PAK1: P21 activated kinase 1; PDAC: Pancreatic ductal adenocarcinoma; RPKM: reads per kilobase per million; TCA: the citric acid cycle; TNBC: triple-negative breast cancer; TPM: Transcripts per million.
ACKNOWLEDGMENTS
We thank Udo Stenzel for help with data analysis.
CONFLICTS OF INTEREST
Authors have no conflicts of interest to declare.
FUNDING
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation (HO 6389/2-2, ‘KFO 337’ - 405344257, HO 6389/3-1). The funders were not involved in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
1. Shen N, Korm S, Karantanos T, Li D, Zhang X, Ritou E, Xu H, Lam A, English J, Zong WX, Liu CT, Shirihai O, Feng H. DLST-dependence dictates metabolic heterogeneity in TCA-cycle usage among triple-negative breast cancer. Commun Biol. 2021; 4:1289. https://doi.org/10.1038/s42003-021-02805-8. [PubMed].
2. Anderson NM, Qin X, Finan JM, Lam A, Athoe J, Missiaen R, Skuli N, Kennedy A, Saini AS, Tao T, Zhu S, Nissim I, Look AT, et al. Metabolic Enzyme DLST Promotes Tumor Aggression and Reveals a Vulnerability to OXPHOS Inhibition in High-Risk Neuroblastoma. Cancer Res. 2021; 81:4417–30. https://doi.org/10.1158/0008-5472.CAN-20-2153. [PubMed].
3. Anderson NM, Li D, Peng HL, Laroche FJ, Mansour MR, Gjini E, Aioub M, Helman DJ, Roderick JE, Cheng T, Harrold I, Samaha Y, Meng L, et al. The TCA cycle transferase DLST is important for MYC-mediated leukemogenesis. Leukemia. 2016; 30:1365–74. https://doi.org/10.1038/leu.2016.26. [PubMed].
4. Anderson NM, Mucka P, Kern JG, Feng H. The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell. 2018; 9:216–37. https://doi.org/10.1007/s13238-017-0451-1. [PubMed].
5. Ashton TM, McKenna WG, Kunz-Schughart LA, Higgins GS. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin Cancer Res. 2018; 24:2482–90. https://doi.org/10.1158/1078-0432.CCR-17-3070. [PubMed].
6. Iorio F, Knijnenburg TA, Vis DJ, Bignell GR, Menden MP, Schubert M, Aben N, Gonçalves E, Barthorpe S, Lightfoot H, Cokelaer T, Greninger P, van Dyk E, et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell. 2016; 166:740–54. https://doi.org/10.1016/j.cell.2016.06.017. [PubMed].
7. Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER 3rd, Barretina J, Gelfand ET, Bielski CM, Li H, Hu K, Andreev-Drakhlin AY, Kim J, et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature. 2019; 569:503–8. https://doi.org/10.1038/s41586-019-1186-3. [PubMed].
8. Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, Ashton JM, Pei S, Grose V, O’Dwyer KM, Liesveld JL, Brookes PS, Becker MW, Jordan CT. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. 2013; 12:329–41. https://doi.org/10.1016/j.stem.2012.12.013. [PubMed].
9. Greengard EG. Molecularly Targeted Therapy for Neuroblastoma. Children (Basel). 2018; 5:142. https://doi.org/10.3390/children5100142. [PubMed].
10. Bhullar KS, Lagarón NO, McGowan EM, Parmar I, Jha A, Hubbard BP, Rupasinghe HPV. Kinase-targeted cancer therapies: progress, challenges and future directions. Mol Cancer. 2018; 17:48. https://doi.org/10.1186/s12943-018-0804-2. [PubMed].
11. Burris HA 3rd. Dual kinase inhibition in the treatment of breast cancer: initial experience with the EGFR/ErbB-2 inhibitor lapatinib. Oncologist. 2004 (Suppl 3); 9:10–15. https://doi.org/10.1634/theoncologist.9-suppl_3-10. [PubMed].
12. Schroeder RL, Stevens CL, Sridhar J. Small molecule tyrosine kinase inhibitors of ErbB2/HER2/Neu in the treatment of aggressive breast cancer. Molecules. 2014; 19:15196–212. https://doi.org/10.3390/molecules190915196. [PubMed].
13. Dummler B, Ohshiro K, Kumar R, Field J. Pak protein kinases and their role in cancer. Cancer Metastasis Rev. 2009; 28:51–63. https://doi.org/10.1007/s10555-008-9168-1. [PubMed].
14. Ruvolo PP, Zhou L, Watt JC, Ruvolo VR, Burks JK, Jiffar T, Kornblau S, Konopleva M, Andreeff M. Targeting PKC-mediated signal transduction pathways using enzastaurin to promote apoptosis in acute myeloid leukemia-derived cell lines and blast cells. J Cell Biochem. 2011; 112:1696–707. https://doi.org/10.1002/jcb.23090. [PubMed].
15. Zhang J, Hou L, Liang R, Chen X, Zhang R, Chen W, Zhu J. CircDLST promotes the tumorigenesis and metastasis of gastric cancer by sponging miR-502-5p and activating the NRAS/MEK1/ERK1/2 signaling. Mol Cancer. 2019; 18:80. https://doi.org/10.1186/s12943-019-1015-1. [PubMed].
16. Boeschen M, Le Duc D, Stiller M, von Laffert M, Schöneberg T, Horn S. Interactive webtool for analyzing drug sensitivity and resistance associated with genetic signatures of cancer cell lines. J Cancer Res Clin Oncol. 2022. [Epub ahead of print]. https://doi.org/10.1007/s00432-022-04503-2. [PubMed].
17. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012; 2:401–4. https://doi.org/10.1158/2159-8290.CD-12-0095. [PubMed].
18. Radic-Sarikas B, Halasz M, Huber KVM, Winter GE, Tsafou KP, Papamarkou T, Brunak S, Kolch W, Superti-Furga G. Lapatinib potentiates cytotoxicity of YM155 in neuroblastoma via inhibition of the ABCB1 efflux transporter. Sci Rep. 2017; 7:3091. https://doi.org/10.1038/s41598-017-03129-6. [PubMed].
19. Voigtlaender M, Schneider-Merck T, Trepel M. Lapatinib. Recent Results Cancer Res. 2018; 211:19–44. https://doi.org/10.1007/978-3-319-91442-8_2. [PubMed].
20. Aird KM, Allensworth JL, Batinic-Haberle I, Lyerly HK, Dewhirst MW, Devi GR. ErbB1/2 tyrosine kinase inhibitor mediates oxidative stress-induced apoptosis in inflammatory breast cancer cells. Breast Cancer Res Treat. 2012; 132:109–19. https://doi.org/10.1007/s10549-011-1568-1. [PubMed].
21. Singhal R, Kandel ES. The response to PAK1 inhibitor IPA3 distinguishes between cancer cells with mutations in BRAF and Ras oncogenes. Oncotarget. 2012; 3:700–8. https://doi.org/10.18632/oncotarget.587. [PubMed].
22. Kořánová T, Dvořáček L, Grebeňová D, Röselová P, Obr A, Kuželová K. PAK1 and PAK2 in cell metabolism regulation. J Cell Biochem. 2022; 123:375–89. https://doi.org/10.1002/jcb.30175. [PubMed].
23. Xie M, He J, He C, Wei S. γ Secretase inhibitor BMS-708163 reverses resistance to EGFR inhibitor via the PI3K/Akt pathway in lung cancer. J Cell Biochem. 2015; 116:1019–27. https://doi.org/10.1002/jcb.25056. [PubMed].
24. Ogawa M, Hori H, Ohta T, Onozato K, Miyahara M, Komada Y. Sensitivity to gemcitabine and its metabolizing enzymes in neuroblastoma. Clin Cancer Res. 2005; 11:3485–93. https://doi.org/10.1158/1078-0432.CCR-04-1781. [PubMed].
25. Toschi L, Finocchiaro G, Bartolini S, Gioia V, Cappuzzo F. Role of gemcitabine in cancer therapy. Future Oncol. 2005; 1:7–17. https://doi.org/10.1517/14796694.1.1.7. [PubMed].
26. Masoud R, Reyes-Castellanos G, Lac S, Garcia J, Dou S, Shintu L, Abdel Hadi N, Gicquel T, El Kaoutari A, Diémé B, Tranchida F, Cormareche L, Borge L, et al. Targeting Mitochondrial Complex I Overcomes Chemoresistance in High OXPHOS Pancreatic Cancer. Cell Rep Med. 2020; 1:100143. https://doi.org/10.1016/j.xcrm.2020.100143. [PubMed].
27. Chen YB, LaCasce AS. Enzastaurin. Expert Opin Investig Drugs. 2008; 17:939–44. https://doi.org/10.1517/13543784.17.6.939. [PubMed].
28. Munster PN, Britten CD, Mita M, Gelmon K, Minton SE, Moulder S, Slamon DJ, Guo F, Letrent SP, Denis L, Tolcher AW. First study of the safety, tolerability, and pharmacokinetics of CP-724,714 in patients with advanced malignant solid HER2-expressing tumors. Clin Cancer Res. 2007; 13:1238–45. https://doi.org/10.1158/1078-0432.CCR-06-1539. [PubMed].
29. Jani JP, Finn RS, Campbell M, Coleman KG, Connell RD, Currier N, Emerson EO, Floyd E, Harriman S, Kath JC, Morris J, Moyer JD, Pustilnik LR, et al. Discovery and pharmacologic characterization of CP-724,714, a selective ErbB2 tyrosine kinase inhibitor. Cancer Res. 2007; 67:9887–93. https://doi.org/10.1158/0008-5472.CAN-06-3559. [PubMed].
30. Sica V, Bravo-San Pedro JM, Stoll G, Kroemer G. Oxidative phosphorylation as a potential therapeutic target for cancer therapy. Int J Cancer. 2020; 146:10–17. https://doi.org/10.1002/ijc.32616. [PubMed].
31. Cournoyer S, Addioui A, Belounis A, Beaunoyer M, Nyalendo C, Le Gall R, Teira P, Haddad E, Vassal G, Sartelet H. GX15-070 (Obatoclax), a Bcl-2 family proteins inhibitor engenders apoptosis and pro-survival autophagy and increases Chemosensitivity in neuroblastoma. BMC Cancer. 2019; 19:1018. https://doi.org/10.1186/s12885-019-6195-y. [PubMed].
32. Opydo-Chanek M, Cichoń I, Rak A, Kołaczkowska E, Mazur L. The pan-Bcl-2 inhibitor obatoclax promotes differentiation and apoptosis of acute myeloid leukemia cells. Invest New Drugs. 2020; 38:1664–76. https://doi.org/10.1007/s10637-020-00931-4. [PubMed].